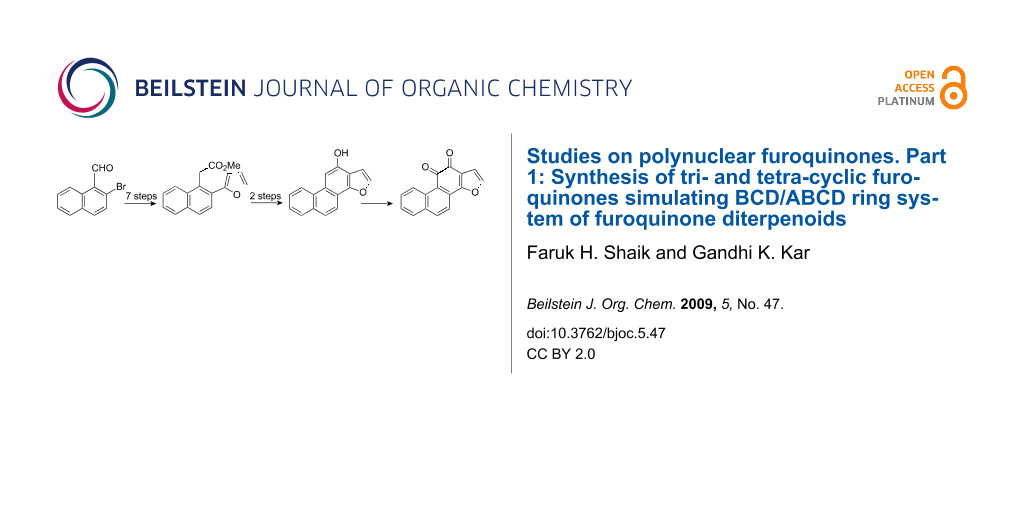
Abstract
Synthesis of phenanthro[1,2-b]furan-10,11-dione, the core nucleus present in Tanshinone-I is described in 8–10 steps starting from 2-bromo-3,4-dihydro-1-naphthaldehyde. The bromoaldehyde was converted to methyl 2-(2-bromo-1-naphthyl)acetate or 2-(2-bromo-1-naphthyl)acetonitrile following the protocol of functional group transformations. Subsequent Suzuki coupling of this ester/nitrile derivative with furan-2-boronic acid produced [2-(2-furyl)-1-naphthyl]acetic ester/nitrile which on hydrolysis furnished the corresponding acid derivative. Cyclization of the acid followed by oxidation of the phenol, with Fremy’s salt, produced the tetra-cyclic furoquinone, phenanthro[1,2-b]furan-10,11-dione. This method has also been extended for the synthesis of the tricyclic furoquinone, naphtho[1,2-b]furan-4,5-dione.

Graphical Abstract
Introduction
Chemistry of furoquinones [1-8], especially the tetra cyclic furoquinones isolated from Chinese red rooted sage Salvia miltiorrhiza Bunge known as “Dan Shen” in Chinese traditional medicine, attracted the attention of synthetic chemists as well as medicinal chemists due to the various biological activities displayed by such class of compounds. Dan Shen is used clinically for treatment of viral hepatitis, cardiac and vascular disorder, hypertension, miscarriage and menstrual disorder [3,9] etc. It has been reported to show potential anticancer activity [10] (against human breast cancer), cytotoxic and antiplatelet aggregation activities [1,11]. It also acts as an antibacterial [1], antioxidant and anti-inflammatory agent [12-14] as well as induces apoptosis in human leukemia cell lines [15,16]. It is believed that broad spectrum of activities of Dan Shen are mainly associated with the presence of tetra cyclic furoquinone diterpenoids like Tanshinone I [17,18], Tanshinone IIA and IIB [19-26], Cryptotanshinone [20,22], Nortanshinone [23,27], Tanshindiol [23,27] etc. (Figure 1). Even the tricyclic furoquinones (Figure 1) have also been reported to possess cancer chemo preventive activity [28].
![[1860-5397-5-47-1]](/bjoc/content/figures/1860-5397-5-47-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Some biologically active tetra cyclic furoquinone derivatives (isolated from Dan Shen) and synthetic tricyclic furoquinones.
Figure 1: Some biologically active tetra cyclic furoquinone derivatives (isolated from Dan Shen) and syntheti...
In all of these compounds the key structural unit present is a furophenanthraquinone or a furonaphthoquinone as well as their di/tetra/hexahydro analog.
We have not come across any synthesis of phenanthro[1,2-b]furo-10,11-dione (13) (the nuclear core structure of Tanshinone-I) though a number of syntheses of naturally occurring tetra cyclic furoquinone diterpenoids have been reported in literature [18-27] in the last few decades. In comparison only very few syntheses of tricyclic furoquinone 18 [29,30] and its derivatives [31-33] have been reported. Encouraged by the broad spectrum biological activity of furoquinones, we aimed to synthesize novel polynuclear furoquinones. Herein we report the synthesis of tetra cyclic furoquinones, phenanthro[1,2-b]furan-10,11-dione (13) and the tricyclic furoquinone, naphtho[1,2-b]furan-4,5-dione (18), simulating respectively the A-B-C-D/B-C-D ring of Tanshinone-I, through a novel pathway.
Results and Discussion
Our aim is to develop a general route for the synthesis of tricyclic/tetracyclic furoquinones with structural diversity. Our approach is shown in Scheme 1 and Scheme 2. In the synthesis of the tetra cyclic furoquinone 13, an easily available starting material 2-bromo-3,4-dihydro-1-naphthaldehyde 1 [34] was utilized as A, B ring precursor and commercially available furan 2-boronic acid as D-ring precursor. In the event of the synthesis, the C ring was constructed to reach the target molecule in 8-10 steps. The key steps in our synthesis deal with the formation of aryl-furyl C–C bond via Suzuki reaction [35] and the generation of the quinone functionality by oxidation of a phenolic intermediate (Figure 2).
![[1860-5397-5-47-2]](/bjoc/content/figures/1860-5397-5-47-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Key strategies for development of C-ring with quinone functionality.
Figure 2: Key strategies for development of C-ring with quinone functionality.
Retrosynthesis of the molecule showed that the required phenolic compound can easily be achieved in 7–8 steps starting from 2-bromo-3,4-dihydro-1-naphthaldehyde (a substrate easily available by Vilsmeier–Haack reaction on 2-tetralone) and furan-2-boronic acid (a commercially available material) (Figure 3).
![[1860-5397-5-47-3]](/bjoc/content/figures/1860-5397-5-47-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Retro synthesis of tetra cyclic furoquinone 13.
Figure 3: Retro synthesis of tetra cyclic furoquinone 13.
When 2-bromo-3,4-dihydro-1-naphthaldehyde (easily obtained in 68% yield by the reaction of 2-tetralone and PBr3/DMF in CHCl3 at room temperature) was aromatized with DDQ in refluxing benzene, 2-bromo-1-naphthaldehyde (2) was produced in excellent yield. Reduction (NaBH4/EtOH) of the aldehyde 2 produced the alcohol 3 as a colorless solid, in 94% yield, which on reaction with PBr3/CCl4 produced the bromide 4 as a light yellow solid. The bromide was then converted (KCN/DMF) to the nitrile derivative 5 which on hydrolysis (KOH/EtOH-H2O, reflux) followed by esterification with CH2N2 furnished methyl 2-(2-bromo-1-naphthyl)acetate 7 in overall good yield. The bromo ester was then subjected to Suzuki reaction with furan-2-boronic acid.
![[1860-5397-5-47-i1]](/bjoc/content/inline/1860-5397-5-47-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reagents and conditions: i) DDQ (1.5 equiv), dry benzene, reflux, argon atmosphere, 37 h, 83%. ii) NaBH4, EtOH, room temperature, 2 h, 94%. iii) PBr3, CCl4, 60 °C, 1 h, 82%. iv) KCN, DMF, room temperature, overnight then 1 h at 50 °C, 68%. v) KOH, EtOH, H2O, reflux, 23 h, 74%. vi) CH2N2, ether, 0 °C- room temperature, 95%. vii) furan-2-boronic acid (1.2 equiv.), Et3N, DMF, Pd(PPh3)4 (2 mol %), 110 °C, argon atmosphere, 53 h, 77%. viii) KOH (2 equiv), EtOH, H2O, reflux, 15 h, 85%. ix) furan-2-boronic acid (1.2 equiv), Et3N, DMF, Pd(PPh3)4 (2 mol %), 110 °C, argon atmosphere, 37 h, 66%. x) KOH, EtOH, H2O, reflux, 45 h, 48%. xi) TFAA, TFA, room temperature, overnight, 71%, xii) Fremy’s salt (3 equiv), MeOH, 1/6 M Na2HPO4 solution. 0–5 °C, overnight, 48%. (All yields are for purified products only.)
Scheme 1: Reagents and conditions: i) DDQ (1.5 equiv), dry benzene, reflux, argon atmosphere, 37 h, 83%. ii) ...
Reaction of compound 7 with furan-2-boronic acid in the presence of Et3N and Pd(PPh3)4 (cat.) in DMF under argon atmosphere produced methyl [2-(2-furyl)-1-naphthyl]acetate 8 (77%) which when hydrolyzed furnished [2-(2-furyl)-1-naphthyl]acetic acid (9) in 85% yield. The acid 9 was also synthesized via hydrolysis of the nitrile derivative 10 which in turn was obtained in 66% yield, by direct Suzuki reaction of 5 with furan-2-boronic acid. However in this case as the intermediate amide 11 produced was resistant to further hydrolysis, the reaction required prolonged reflux and also the yield was relatively poor. Even after reflux of the nitrile derivative 10 in KOH/EtOH-H2O for 45 h 20% of the amide 11 was recovered along with 48% of the desired carboxylic acid. Change of solvent and conditions (e.g., replacement of ethanol with other high boiling alcohols or use of THF as co-solvent and higher temperature) produced no better result. The next step was the introduction of the phenol functionality. In this case our early attempt to prepare the phenol 12 by PPA cyclization of the acid 9 was unsuccessful. We however successfully prepared the phenol as a light yellow solid by cyclization of the acid 9 with trifluoroacetic acid (TFA) and trifluoroacetic anhydride (TFAA) at room temperature in 71% yield. The phenol 12 was finally oxidized (with Fremy’s salt) [36-38] to the o-quinone 13 to complete the synthesis of phenanthro[1,2-b]furan-10,11-dione. The compounds were characterized by usual spectroscopic and analytical methods (1H NMR, 13C NMR, IR, HRMS etc.).
![[1860-5397-5-47-4]](/bjoc/content/figures/1860-5397-5-47-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Assignment of chemical shifts (1H NMR and 13C NMR) of compound 13.
Figure 4: Assignment of chemical shifts (1H NMR and 13C NMR) of compound 13.
The developed pathway was then applied for the synthesis of tricyclic furoquinone 18 (naphtho[1,2-b]furan-4,5-dione) starting from methyl (2-bromophenyl)acetate 14 and furan-2-boronic acid (Scheme 2).
![[1860-5397-5-47-i2]](/bjoc/content/inline/1860-5397-5-47-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reagents and conditions: i) furan-2-boronic acid (1.2 equiv), Et3N, DMF, Pd(PPh3)4 (2 mol %), 110 °C, N2 atmosphere, 7 h, 84%, ii) KOH, EtOH, H2O, reflux, 9 h, 90% iii) TFAA, TFA, room temperature, overnight, 54%, iv) Fremy’s salt (~2.5 equiv), MeOH, 1/6 M Na2HPO4 solution, 0–5 °C, overnight, 56%. (All yields are for purified products only.)
Scheme 2: Reagents and conditions: i) furan-2-boronic acid (1.2 equiv), Et3N, DMF, Pd(PPh3)4 (2 mol %), 110 °...
When methyl (2-bromophenyl)acetate 14 was subjected to Suzuki reaction with furan-2-boronic acid, produced [2-(2-furyl)phenyl]acetic ester 15 as a pale yellow liquid in 84% yield. Subsequent alkaline hydrolysis of 15 produced the carboxylic acid 16 as a white solid in 90% yield. Synthesis of the furonaphthol 17 was achieved in good yield, by cyclization of the carboxylic acid 16 with TFA and TFAA at room temperature. Finally oxidation of the naphthol derivative 17, with Fremy’s Salt, resulted in the formation of naphtho[1,2-b]furan-4,5-dione (18), as a red solid, in 56% yield. The compounds have been characterized by usual spectroscopic analysis (NMR, IR and HRMS data) as well as by analogy with literature report [30].
In general we have developed a novel pathway for the synthesis of polynuclear furoquinones simulating BCD/ABCD ring of natural furoquinone (Tanshinone-I) and we believe the method has great potential towards the synthesis of various polynuclear furoquinone derivatives bearing electron donating/electron withdrawing functionality within its framework as bromoaldehyde derivatives bearing such groups can easily be obtained from corresponding ketones by Vilsmeier–Haack reaction. Very recently we have reported a general method for the synthesis of β-(2-furyl)-α,β-unsaturated aldehydes [39] via Suzuki reaction of β-bromo-α,β-unsaturated aldehydes. These substrates can be used for the synthesis of various non-natural tricyclic and “U-shaped” tetra cyclic furoquinone derivatives. The work is under progress and the results will be published in due course.
Experimental
Furan-2-boronic acid, 2-tetralone and tetrakis(triphenylphosphine)palladium(0) and DDQ were purchased from Sigma-Aldrich (U.S.A). Trifluoroacetic anhydride was purchased from Alfa Aesar (Lancaster). 2-Bromo-3,4-dihydronaphthalene-1-carbaldehyde was prepared from 2-tetralone by Vilsmeier-Haack reaction. Solvents were dried following standard literature procedure. Fremy’s salt (potassium nitrosodisulfonate) was prepared in the laboratory as per literature procedure [36]. 1H NMR spectra were recorded on Bruker 500 MHz (at Chemgen Pharma, Kolkata) and Bruker 400 MHz (at Chembiotek International, Kolkata) and Bruker 300 MHz (IACS, Kolkata) NMR spectrometer respectively. ESI mass spectra were recorded on a micro mass Q-TOF mass spectrometer (serial no. YA 263) at IACS, Kolkata. IR spectral data were obtained from a JASCO FT/IR680 PLUS Spectrometer.
[2-(Furan-2-yl)-naphthalen-1-yl]acetic acid methyl ester (8)
A mixture of compound 7 (150 mg, 0.54 mmol), furan-2-boronic acid (75 mg, 0.64 mmol) and Et3N (0.5 mL, 3.6 mmol) was degasified with argon for 25 minutes. Now to it the catalyst Pd(PPh3)4 (~15 mg, 2 mol %) was added. The mixture was then heated with stirring under argon at 110 °C till the reaction was completed (~53 h). After cooling to room temperature, the mixture was poured into cold water and extracted with ether. Organic layer was washed successively with NaHCO3 solution, 5% brine and dried (Na2SO4) and solvent removed. Crude product thus obtained was then purified by column chromatography [silica gel/ pet. ether (60–80 °C) and ethyl acetate mixture, 10:1]. Compound 8 was obtained as a white solid (110 mg) in 77% yield, m.p., 109–111 °C. IR(KBr) νmax : 1737.6 cm−1; 1H NMR (400 MHz, CDCl3) δ: 3.72 (3H, s), 4.33 (2H, s), 6.53 (1H, dd, J = 1.7 and 3 Hz), 6.67 (1H, d, J = 3 Hz), 7.50 (1H, t, J = 7.2 Hz), 7.56 (1H, d, J = 7.2 Hz), 7.57 (1H, br s), 7.72 (1H, d, J = 8.6 Hz), 7.82 (1H, d, J = 9.4 Hz), 7.84 (1H, d, J = 9.3 Hz), 8.01 (1H, d, J = 8.4 Hz), ppm.
[2-(Furan-2-yl)-naphthalen-1-yl]acetic acid (9)
A mixture of compound 8 (100 mg, 0.38 mmol), KOH (43 mg, 0.76 mmol), 2 mL water and 2 mL ethanol was refluxed on a water bath for 15 h. Excess EtOH was distilled out as much as possible and then the residue was diluted with 2–3 mL of water. Neutral part was extracted with ether. Aqueous alkaline part was cooled in ice and acidified with HCl. Separated solid was thoroughly extracted with ethyl acetate. After usual work up, 80 mg (85%) of the acid 9 was obtained as white solid, m.p., 197–199 °C. IR (KBr) νmax 1692.2 cm−1. (Treatment of the acid 9 with diazomethane in ether produced the methyl ester derivative identical with compound 8.)
Phenanthro[1,2-b]furan-11-ol (12)
A mixture of compound 9 (100 mg, 0.39 mmol), 3 mL trifluoroacetic anhydride, 0.9 mL trifluoroacetic acid was stirred at room temperature overnight protecting from moisture. The mixture was then poured in ice cold saturated NaHCO3 solution and stirred well and extracted thoroughly with CH2Cl2. The organic layer was washed with aq. NaHCO3 solution and finally with H2O and dried (Na2SO4). Removal of solvent afforded the crude product which was purified by column chromatography [silica gel/pet. ether (60–80 °C) and ethyl acetate mixture, 19:1] to furnish 65 mg (71%) of 12 as a light yellow solid, m.p., 175–177 °C. IR (KBr) νmax 3235, 3302 cm−1; 1H NMR (500 MHz, CDCl3) δ: 5.47 (1H, s), 7.04 (1H, br s), 7.58 (1H, t, J = 7.4 Hz), 7.63 (1H, t, J = 7.5 Hz), 7.75 (1H, d, J = 8.8 Hz), 7.78 (1H, br s), 7.85 (1H, s), 7.91 (1H, d, J = 7.8 Hz), 8.22 (1H, d, J = 8.8 Hz), 8.55 (1H, d, J = 8.2 Hz) ppm.
Phenanthro[1,2-b]furan-10,11-dione (13)
To a stirred solution of potassium nitrosodisulfonate (Fremy’s salt) (170 mg, 0.65 mmol) in 12 mL 1/6 (M) Na2HPO4 solution taken in a 50 mL round bottomed flask, a solution of the furonaphthol derivative 12 (50 mg, 0.21 mmol) in 6 mL methanol was added drop wise. Stirring was continued at 0–5 °C for 2 h and then left overnight in freeze. The dark red solid separated was filtered and purified by column chromatography [silica gel/ pet. ether (60–80 °C) and ethyl acetate, 10:1]. An analytical sample was prepared further by recrystallisation from pet ether–ethyl acetate mixture. Yield, 25 mg (48%), m.p., 178–180 °C. IR (KBr) νmax 1686, 1667 cm−1; 1H NMR (500 MHz, CDCl3) δ: 6.87 (1H, br s), 7.53 (1H, d, J = 7.5 Hz), 7.55 (1H, br s), 7.70 (1H, br t, J = 7.5 Hz), 7.81–7.85 (2H, a doublet and a triplet merged together), 8.12 (1H, d, J = 8.5 Hz), 9.40 (1H, d, J = 8.5 Hz) ppm; 13C NMR (125 MHz, CDCl3) δ: 109.11, 119.16, 120.79, 122.88, 126.66, 127.66, 129.20, 130.07, 131.20, 132.47, 134.65, 137.33, 145.54, 161.31, 174.53, 183.07 ppm; HRMS (ESI, 70 eV): m/z = 271.0372 [M++Na] (calculated mass for C16H8O3Na: 271.0371 [M++Na]).
Naphtho[1,2-b]furan-4,5-dione (18)
To a stirred solution of potassium nitrosodisulfonate (Fremy’s salt) (170 mg, 0.63 mmol) in 5 mL 1/6 (M) Na2HPO4 solution taken in a 25 mL round bottomed flask, a solution of the furonaphthol 17 (50 mg, 0.27 mmol) in 2 mL methanol was added drop wise. Stirring was continued at 0–5 °C for 2 h and then left overnight in freeze. The brick red solid separated was filtered to obtain 35 mg of crude product which on purification by column chromatography (silica gel/pet. ether–ethyl acetate, 5:1) afforded 30 mg (56%) of the title compound (18). An analytical sample was prepared by further recrystallisation from pet. ether–EtOAc mixture; m.p., 205–206 °C (lit. [30] m.p 213–215 °C ). IR(KBr) νmax 1674.9 cm−1 (br, strong); 1H NMR (500 MHz, CDCl3) δ: 6.88 (1H, d, J = 1.8 Hz), 7.48 (1H, t, J = 7.5 Hz), 7.51 (1H, d, J = 1.8 Hz), 7.67 (1H, t, J = 7.5 Hz), 7.74 (1H, d, J = 7.5 Hz), 8.1 (1H, d, J = 7.7 Hz) ppm; 13C NMR (125 MHz, CDCl3) δ: 108.94, 121.56, 122.41, 128.49, 128.81, 130.36, 130.64, 135.48, 145.13, 160.64, 174.54, 180.56 ppm. HRMS (ESI, 70 eV): m/z = 199.0399 [M++H] (calculated mass for C12H7O3: 199.0391 [M++H]).[lit. [30]: 1H NMR (300 MHz, CDCl3) δ: 8.09 (1H, d, J = 7.9 Hz), 7.73–7.62 (2H, m), 7.49 (1H, d, J = 2.0 Hz), 7.45 (1H, m), 6.86 (1H, d, J = 2.0 Hz)].
Supporting Information
Supporting information features experimental procedures, analytical data and NMR spectra for some selected compounds.
| Supporting Information File 1: Experimental procedures | ||
| Format: DOC | Size: 46.5 KB | Download |
| Supporting Information File 2: 1H and 13C NMR Spectra. | ||
| Format: DOC | Size: 1.7 MB | Download |
References
-
Fang, C. N.; Chang, P. L.; Hsu, T. P. Acta Chim. Sin. 1976, 34, 197–209.
Return to citation in text: [1] [2] [3] -
Chang, H. M.; Chui, K. Y.; Tan, F. W. L.; Yang, Y.; Zhong, Z. P.; Lee, C. M.; Sham, H. L.; Wong, H. N. C. J. Med. Chem. 1991, 34, 1675–1692. doi:10.1021/jm00109a022
Return to citation in text: [1] -
Chang, H. M.; But, P. P., Eds. Pharmacology and Application of Chinese Materia Medica; World Scientific Publishing Co. Pvt. Ltd.: Singapore, 1986; Vol. 1, pp 255–268.
Return to citation in text: [1] [2] -
del Corral, J. M. M.; Castro, M. A.; Oliveira, A. B.; Gualberto, S. A.; Cuevas, C.; Feliciano, A. S. Bioorg. Med. Chem. 2006, 14, 7231–7240. doi:10.1016/j.bmc.2006.06.053
Return to citation in text: [1] -
Bian, W.; Chen, F.; Bai, L.; Zhang, P.; Qin, W. Acta Biochim. Biophys. Sin. 2008, 40, 1–6. doi:10.1111/j.1745-7270.2008.00370.x
Return to citation in text: [1] -
Nair, V.; Treesa, P. M.; Maliakal, D.; Rath, N. P. Tetrahedron 2001, 57, 7705–7710. doi:10.1016/S0040-4020(01)00700-1
Return to citation in text: [1] -
King, T. J.; Read, G. J. Chem. Soc. 1961, 5090–5094. doi:10.1039/jr9610005090
Return to citation in text: [1] -
Chang, H. M.; Cheng, K. P.; Choang, T. F.; Chow, H. F.; Chui, K. Y.; Hon, P. M.; Tan, F. W. L.; Yang, Y.; Zhong, Z. P.; Lee, C. M.; Sham, H. L.; Chan, C. F.; Cui, Y. X.; Wong, H. N. C. J. Org. Chem. 1990, 55, 3537–3543. doi:10.1021/jo00298a029
Return to citation in text: [1] -
Duke, J. A.; Avensu, E. S. Medicinal Plants of China; Reference Publications: Algonac, MI, 1985; Vol. 2, p 381.
Return to citation in text: [1] -
Wang, X.; Wei, Y.; Yuan, S.; Liu, G.; Lu, Y.; Zhang, J.; Wang, W. Int. J. Cancer 2005, 116, 799–807. doi:10.1002/ijc.20880
Return to citation in text: [1] -
Mosaddik, M. A. Phytomedicine 2003, 10, 682–685.
Return to citation in text: [1] -
Wang, A. M.; Sha, S. H.; Lesniak, W.; Schacht, J. Antimicrob. Agents Chemother. 2003, 47, 1836–1841. doi:10.1128/AAC.47.6.1836-1841.2003
Return to citation in text: [1] -
Zhou, L.; Zuo, Z.; Chow, M. S. S. J. Clin. Pharmacol. 2005, 45, 1345–1359. doi:10.1177/0091270005282630
Return to citation in text: [1] -
Fu, J.; Huang, H.; Liu, J.; Pi, R.; Chen, J.; Liu, P. Eur. J. Pharmacol. 2007, 568, 213–221. doi:10.1016/j.ejphar.2007.04.031
Return to citation in text: [1] -
Yoon, Y.; Kim, Y. O.; Jeon, W. K.; Park, H. J.; Sung, H. J. J. Ethnopharmacol. 1999, 68, 121–127. doi:10.1016/S0378-8741(99)00059-8
Return to citation in text: [1] -
Sung, H. J.; Choi, S. M.; Yoon, Y.; An, K. S. Exp. Mol. Med. 1999, 31, 174–178.
Return to citation in text: [1] -
Nakao, M.; Fukushima, T. J. Pharm. Soc. Jpn. 1934, 54, 154.
See for first extraction of Tanshinone from “Dan Shen”.
Return to citation in text: [1] -
Danheiser, R. L.; Casebier, D. S.; Loebach, J. L. Tetrahedron Lett. 1992, 33, 1149–1152. doi:10.1016/S0040-4039(00)91882-3
Return to citation in text: [1] [2] -
Takikura, K. J. Pharm. Soc. Jpn. 1941, 61, 475–482.
Return to citation in text: [1] [2] -
Kakisawa, H.; Inouye, Y. J. Chem. Soc., Chem. Commun. 1968, 1327–1328.
Return to citation in text: [1] [2] [3] -
Baille, A.; Thomson, R. H. J. Chem. Soc. 1968, 48–52.
Return to citation in text: [1] [2] -
Kakisawa, H.; Tateishi, M.; Kusumi, T. Tetrahedron Lett. 1968, 34, 3783–3786. doi:10.1016/S0040-4039(00)75540-7
Return to citation in text: [1] [2] [3] -
Inouye, Y.; Kakisawa, H. Bull. Chem. Soc. Jpn. 1969, 42, 3318–3323. doi:10.1246/bcsj.42.3318
Return to citation in text: [1] [2] [3] [4] -
Lee, J.; Snyder, J. K. J. Am. Chem. Soc. 1989, 111, 1522–1524. doi:10.1021/ja00186a070
Return to citation in text: [1] [2] -
Danheiser, R. L.; Casebier, D. S.; Firooznia, F. J. Org. Chem. 1995, 60, 8341–8350. doi:10.1021/jo00131a006
Return to citation in text: [1] [2] -
Jiang, Y. Y.; Li, Q.; Lu, W.; Cai, J. C. Tetrahedron Lett. 2003, 44, 2073–2075. doi:10.1016/S0040-4039(03)00191-6
Return to citation in text: [1] [2] -
Lee, J.; Snyder, J. K. J. Org. Chem. 1990, 55, 4995–5008. doi:10.1021/jo00304a008
Return to citation in text: [1] [2] [3] -
Itoigawa, M.; Ito, C.; Tan, H. T. W.; Okuda, M.; Tokuda, H.; Nishino, H.; Furukawa, H. Cancer Lett. 2001, 174, 135–139. doi:10.1016/S0304-3835(01)00707-8
Return to citation in text: [1] -
Brandao, M. A. F.; de Oliveira, A. B.; Snieckus, V. Tetrahedron Lett. 1993, 34, 2437–2440. doi:10.1016/S0040-4039(00)60435-5
Return to citation in text: [1] -
Lee, Y. R.; Kim, B. S.; Jung, Y. U.; Koh, W. S.; Cha, J. S.; Kim, N. W. Synth. Commun. 2002, 32, 3099–3105. doi:10.1081/SCC-120013719
Return to citation in text: [1] [2] [3] [4] -
Huot, R.; Brassard, P. Can. J. Chem. 1974, 52, 88–94. doi:10.1139/v74-012
Return to citation in text: [1] -
Lee, J.; Tang, J.; Snyder, J. K. Tetrahedron Lett. 1987, 28, 3427–3430. doi:10.1016/S0040-4039(00)96317-2
Return to citation in text: [1] -
Shishido, K.; Takata, T.; Omodani, T.; Shibuya, M. Chem. Lett. 1993, 22, 557–560. doi:10.1246/cl.1993.557
Return to citation in text: [1] -
Gilchrist, T. L.; Summersell, R. J. J. Chem. Soc., Perkin Trans. 1 1988, 2595–2601. doi:10.1039/P19880002595
Return to citation in text: [1] -
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007
Return to citation in text: [1] -
Zimmer, H.; Lankin, D. C.; Horgan, S. W. Chem. Rev. 1971, 71, 229–246. doi:10.1021/cr60270a005
Return to citation in text: [1] [2] -
Teuber, H. J. Organic Syntheses; 1988; Vol. 6, p 480.
Return to citation in text: [1] -
Ray, J. K.; Kar, G. K.; Karmakar, A. C. J. Org. Chem. 1991, 56, 2268–2270. doi:10.1021/jo00006a063
Return to citation in text: [1] -
Samanta, K.; Kar, G. K.; Sarkar, A. K. Tetrahedron Lett. 2008, 49, 1461–1464. doi:10.1016/j.tetlet.2008.01.010
Return to citation in text: [1]
| 36. | Zimmer, H.; Lankin, D. C.; Horgan, S. W. Chem. Rev. 1971, 71, 229–246. doi:10.1021/cr60270a005 |
| 37. | Teuber, H. J. Organic Syntheses; 1988; Vol. 6, p 480. |
| 38. | Ray, J. K.; Kar, G. K.; Karmakar, A. C. J. Org. Chem. 1991, 56, 2268–2270. doi:10.1021/jo00006a063 |
| 34. | Gilchrist, T. L.; Summersell, R. J. J. Chem. Soc., Perkin Trans. 1 1988, 2595–2601. doi:10.1039/P19880002595 |
| 35. | Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007 |
| 1. | Fang, C. N.; Chang, P. L.; Hsu, T. P. Acta Chim. Sin. 1976, 34, 197–209. |
| 2. | Chang, H. M.; Chui, K. Y.; Tan, F. W. L.; Yang, Y.; Zhong, Z. P.; Lee, C. M.; Sham, H. L.; Wong, H. N. C. J. Med. Chem. 1991, 34, 1675–1692. doi:10.1021/jm00109a022 |
| 3. | Chang, H. M.; But, P. P., Eds. Pharmacology and Application of Chinese Materia Medica; World Scientific Publishing Co. Pvt. Ltd.: Singapore, 1986; Vol. 1, pp 255–268. |
| 4. | del Corral, J. M. M.; Castro, M. A.; Oliveira, A. B.; Gualberto, S. A.; Cuevas, C.; Feliciano, A. S. Bioorg. Med. Chem. 2006, 14, 7231–7240. doi:10.1016/j.bmc.2006.06.053 |
| 5. | Bian, W.; Chen, F.; Bai, L.; Zhang, P.; Qin, W. Acta Biochim. Biophys. Sin. 2008, 40, 1–6. doi:10.1111/j.1745-7270.2008.00370.x |
| 6. | Nair, V.; Treesa, P. M.; Maliakal, D.; Rath, N. P. Tetrahedron 2001, 57, 7705–7710. doi:10.1016/S0040-4020(01)00700-1 |
| 7. | King, T. J.; Read, G. J. Chem. Soc. 1961, 5090–5094. doi:10.1039/jr9610005090 |
| 8. | Chang, H. M.; Cheng, K. P.; Choang, T. F.; Chow, H. F.; Chui, K. Y.; Hon, P. M.; Tan, F. W. L.; Yang, Y.; Zhong, Z. P.; Lee, C. M.; Sham, H. L.; Chan, C. F.; Cui, Y. X.; Wong, H. N. C. J. Org. Chem. 1990, 55, 3537–3543. doi:10.1021/jo00298a029 |
| 29. | Brandao, M. A. F.; de Oliveira, A. B.; Snieckus, V. Tetrahedron Lett. 1993, 34, 2437–2440. doi:10.1016/S0040-4039(00)60435-5 |
| 30. | Lee, Y. R.; Kim, B. S.; Jung, Y. U.; Koh, W. S.; Cha, J. S.; Kim, N. W. Synth. Commun. 2002, 32, 3099–3105. doi:10.1081/SCC-120013719 |
| 1. | Fang, C. N.; Chang, P. L.; Hsu, T. P. Acta Chim. Sin. 1976, 34, 197–209. |
| 11. | Mosaddik, M. A. Phytomedicine 2003, 10, 682–685. |
| 31. | Huot, R.; Brassard, P. Can. J. Chem. 1974, 52, 88–94. doi:10.1139/v74-012 |
| 32. | Lee, J.; Tang, J.; Snyder, J. K. Tetrahedron Lett. 1987, 28, 3427–3430. doi:10.1016/S0040-4039(00)96317-2 |
| 33. | Shishido, K.; Takata, T.; Omodani, T.; Shibuya, M. Chem. Lett. 1993, 22, 557–560. doi:10.1246/cl.1993.557 |
| 10. | Wang, X.; Wei, Y.; Yuan, S.; Liu, G.; Lu, Y.; Zhang, J.; Wang, W. Int. J. Cancer 2005, 116, 799–807. doi:10.1002/ijc.20880 |
| 28. | Itoigawa, M.; Ito, C.; Tan, H. T. W.; Okuda, M.; Tokuda, H.; Nishino, H.; Furukawa, H. Cancer Lett. 2001, 174, 135–139. doi:10.1016/S0304-3835(01)00707-8 |
| 30. | Lee, Y. R.; Kim, B. S.; Jung, Y. U.; Koh, W. S.; Cha, J. S.; Kim, N. W. Synth. Commun. 2002, 32, 3099–3105. doi:10.1081/SCC-120013719 |
| 3. | Chang, H. M.; But, P. P., Eds. Pharmacology and Application of Chinese Materia Medica; World Scientific Publishing Co. Pvt. Ltd.: Singapore, 1986; Vol. 1, pp 255–268. |
| 9. | Duke, J. A.; Avensu, E. S. Medicinal Plants of China; Reference Publications: Algonac, MI, 1985; Vol. 2, p 381. |
| 18. | Danheiser, R. L.; Casebier, D. S.; Loebach, J. L. Tetrahedron Lett. 1992, 33, 1149–1152. doi:10.1016/S0040-4039(00)91882-3 |
| 19. | Takikura, K. J. Pharm. Soc. Jpn. 1941, 61, 475–482. |
| 20. | Kakisawa, H.; Inouye, Y. J. Chem. Soc., Chem. Commun. 1968, 1327–1328. |
| 21. | Baille, A.; Thomson, R. H. J. Chem. Soc. 1968, 48–52. |
| 22. | Kakisawa, H.; Tateishi, M.; Kusumi, T. Tetrahedron Lett. 1968, 34, 3783–3786. doi:10.1016/S0040-4039(00)75540-7 |
| 23. | Inouye, Y.; Kakisawa, H. Bull. Chem. Soc. Jpn. 1969, 42, 3318–3323. doi:10.1246/bcsj.42.3318 |
| 24. | Lee, J.; Snyder, J. K. J. Am. Chem. Soc. 1989, 111, 1522–1524. doi:10.1021/ja00186a070 |
| 25. | Danheiser, R. L.; Casebier, D. S.; Firooznia, F. J. Org. Chem. 1995, 60, 8341–8350. doi:10.1021/jo00131a006 |
| 26. | Jiang, Y. Y.; Li, Q.; Lu, W.; Cai, J. C. Tetrahedron Lett. 2003, 44, 2073–2075. doi:10.1016/S0040-4039(03)00191-6 |
| 27. | Lee, J.; Snyder, J. K. J. Org. Chem. 1990, 55, 4995–5008. doi:10.1021/jo00304a008 |
| 19. | Takikura, K. J. Pharm. Soc. Jpn. 1941, 61, 475–482. |
| 20. | Kakisawa, H.; Inouye, Y. J. Chem. Soc., Chem. Commun. 1968, 1327–1328. |
| 21. | Baille, A.; Thomson, R. H. J. Chem. Soc. 1968, 48–52. |
| 22. | Kakisawa, H.; Tateishi, M.; Kusumi, T. Tetrahedron Lett. 1968, 34, 3783–3786. doi:10.1016/S0040-4039(00)75540-7 |
| 23. | Inouye, Y.; Kakisawa, H. Bull. Chem. Soc. Jpn. 1969, 42, 3318–3323. doi:10.1246/bcsj.42.3318 |
| 24. | Lee, J.; Snyder, J. K. J. Am. Chem. Soc. 1989, 111, 1522–1524. doi:10.1021/ja00186a070 |
| 25. | Danheiser, R. L.; Casebier, D. S.; Firooznia, F. J. Org. Chem. 1995, 60, 8341–8350. doi:10.1021/jo00131a006 |
| 26. | Jiang, Y. Y.; Li, Q.; Lu, W.; Cai, J. C. Tetrahedron Lett. 2003, 44, 2073–2075. doi:10.1016/S0040-4039(03)00191-6 |
| 23. | Inouye, Y.; Kakisawa, H. Bull. Chem. Soc. Jpn. 1969, 42, 3318–3323. doi:10.1246/bcsj.42.3318 |
| 27. | Lee, J.; Snyder, J. K. J. Org. Chem. 1990, 55, 4995–5008. doi:10.1021/jo00304a008 |
| 36. | Zimmer, H.; Lankin, D. C.; Horgan, S. W. Chem. Rev. 1971, 71, 229–246. doi:10.1021/cr60270a005 |
| 17. |
Nakao, M.; Fukushima, T. J. Pharm. Soc. Jpn. 1934, 54, 154.
See for first extraction of Tanshinone from “Dan Shen”. |
| 18. | Danheiser, R. L.; Casebier, D. S.; Loebach, J. L. Tetrahedron Lett. 1992, 33, 1149–1152. doi:10.1016/S0040-4039(00)91882-3 |
| 23. | Inouye, Y.; Kakisawa, H. Bull. Chem. Soc. Jpn. 1969, 42, 3318–3323. doi:10.1246/bcsj.42.3318 |
| 27. | Lee, J.; Snyder, J. K. J. Org. Chem. 1990, 55, 4995–5008. doi:10.1021/jo00304a008 |
| 30. | Lee, Y. R.; Kim, B. S.; Jung, Y. U.; Koh, W. S.; Cha, J. S.; Kim, N. W. Synth. Commun. 2002, 32, 3099–3105. doi:10.1081/SCC-120013719 |
| 15. | Yoon, Y.; Kim, Y. O.; Jeon, W. K.; Park, H. J.; Sung, H. J. J. Ethnopharmacol. 1999, 68, 121–127. doi:10.1016/S0378-8741(99)00059-8 |
| 16. | Sung, H. J.; Choi, S. M.; Yoon, Y.; An, K. S. Exp. Mol. Med. 1999, 31, 174–178. |
| 30. | Lee, Y. R.; Kim, B. S.; Jung, Y. U.; Koh, W. S.; Cha, J. S.; Kim, N. W. Synth. Commun. 2002, 32, 3099–3105. doi:10.1081/SCC-120013719 |
| 12. | Wang, A. M.; Sha, S. H.; Lesniak, W.; Schacht, J. Antimicrob. Agents Chemother. 2003, 47, 1836–1841. doi:10.1128/AAC.47.6.1836-1841.2003 |
| 13. | Zhou, L.; Zuo, Z.; Chow, M. S. S. J. Clin. Pharmacol. 2005, 45, 1345–1359. doi:10.1177/0091270005282630 |
| 14. | Fu, J.; Huang, H.; Liu, J.; Pi, R.; Chen, J.; Liu, P. Eur. J. Pharmacol. 2007, 568, 213–221. doi:10.1016/j.ejphar.2007.04.031 |
| 20. | Kakisawa, H.; Inouye, Y. J. Chem. Soc., Chem. Commun. 1968, 1327–1328. |
| 22. | Kakisawa, H.; Tateishi, M.; Kusumi, T. Tetrahedron Lett. 1968, 34, 3783–3786. doi:10.1016/S0040-4039(00)75540-7 |
| 39. | Samanta, K.; Kar, G. K.; Sarkar, A. K. Tetrahedron Lett. 2008, 49, 1461–1464. doi:10.1016/j.tetlet.2008.01.010 |
© 2009 Shaik and Kar; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)