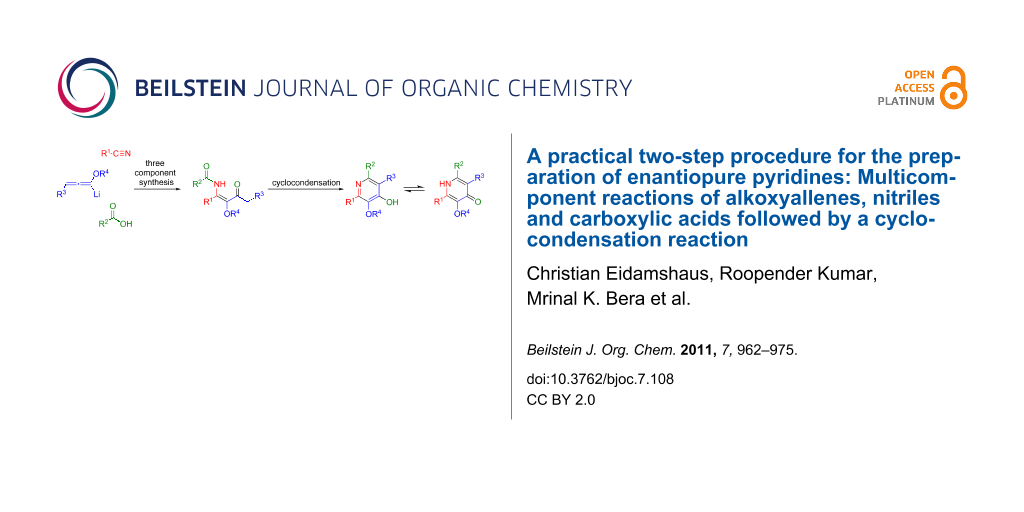
Abstract
A practical approach to highly functionalized 4-hydroxypyridine derivatives with stereogenic side chains in the 2- and 6-positions is described. The presented two-step process utilizes a multicomponent reaction of alkoxyallenes, nitriles and carboxylic acids to provide β-methoxy-β-ketoenamides which are transformed into 4-hydroxypyridines in a subsequent cyclocondensation. The process shows broad substrate scope and leads to differentially substituted enantiopure pyridines in good to moderate yields. The preparation of diverse substituted lactic acid derived pyrid-4-yl nonaflates is described. Additional evidence for the postulated mechanism of the multicomponent reaction is presented.

Graphical Abstract
Introduction
The pyridine core is ubiquitous in pharmacologically active agents, agrochemicals and natural products [1-5]. The HMG-CoA reductase inhibitors Glenvastatin and Cerivastatin are exemplarily mentioned as pharmaceuticals that feature the pyridine nucleus [6-10]. Natural products that contain a pyridine ring include the 3-alkylpyridine alkaloid niphatesine C and the fuzanin family [11,12]. Moreover, the ability to form coordination compounds makes pyridines ideal ligands for transition metal-catalyzed processes and for the construction of supramolecular architectures [13]. Pyridines with chiral side chains are widely employed as ligands in asymmetric transformations, for instance, in the asymmetric hydrogenation of olefins, in enantioselective additions of metal organyls to aldehydes and enones, as well as in palladium-catalyzed allylic substitution reactions [14-20]. Thus, the synthesis of specifically functionalized pyridines is of considerable interest, and many approaches toward this heterocyclic structure have been disclosed in the literature [21]. In addition to classical pyridine syntheses such as the Kröhnke reaction, many new approaches have recently been developed [22-26]. Despite the wide range of conceptually different syntheses, only few methods for introducing chirality into pyridine side chains have been described: Enantioselective reduction of pyridine carbonyl compounds, the addition of lithiated pyridine derivatives to chiral ketones or the resolution of racemates being the most common approaches. The preparation of chiral pyridine derivatives starting from simple enantiopure precursors is less common [27,28].
Recently, we reported a new synthesis of pyridines based on the trimethylsilyl trifluoromethanesulfonate (TMSOTf) induced cyclocondensation reaction of β-ketoenamides [29-34]. This cyclocondensation step can be rationalized as a 6π-electrocyclization of the disilylated intermediate 5 to provide dihydropyridine 6. Elimination of trimethylsilanol and subsequent O-desilylation affords the 4-hydroxypyridine 7 (Scheme 1). The desired β-ketoenamides 4 are either accessible by acylation of enaminoketones or by a multicomponent reaction of lithiated alkoxyallenes, nitriles and carboxylic acids [35,36].
![[1860-5397-7-108-i1]](/bjoc/content/inline/1860-5397-7-108-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of β-ketoenamides and subsequent cyclocondensation to 4-hydroxypyridines. a) Et2O, −40 °C to r.t. 16 h, b) TMSOTf, NEt3, CH2Cl2 or ClCH2CH2Cl reflux.
Scheme 1: Preparation of β-ketoenamides and subsequent cyclocondensation to 4-hydroxypyridines. a) Et2O, −40 ...
In the past, we devoted considerable interest to the synthesis of substituted pyridine derivatives by this route and investigated their use in subsequent transformations [37-39]. Broadening the scope of the process to functionalized, chiral starting materials is the subject of this report. Additionally, herein we disclose the full experimental data for compounds reported in a preliminary communication [40].
Results and Discussion
In continuation of our previous work, we addressed the question whether chiral starting materials react in the above sequence without loss of enantiopurity [40]. Chiral carboxylic acids are readily available and their use would allow for a rapid access to pyridines with side chains bearing stereogenic centers. In recent years we studied intensively the multicomponent reactions of lithiated alkoxyallenes with nitriles and carboxylic acids and could demonstrate that precursor compounds with alkyl, alkenyl or aryl substituents are smoothly converted into β-ketoenamides and subsequently transformed into the desired 4-hydroxypyridines. The mechanism of the multicomponent reaction is depicted in Scheme 2. In the first step, a lithiated alkoxyallene such as 10 adds to a nitrile to yield an iminoallenyl anion 11. Protonation of 11 then gives a resonance stabilized cation 12 which can be attacked in β-position by a carboxylate to afford an enol ester 14. The β-ketoenamide 16 is then formed by transfer of the acyl group to the amino group and subsequent tautomerization.
![[1860-5397-7-108-i2]](/bjoc/content/inline/1860-5397-7-108-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Mechanistic rational for the formation of β-ketoenamides 16.
Scheme 2: Mechanistic rational for the formation of β-ketoenamides 16.
In some cases we observed the formation of minor amounts of 4-hydroxypyridines along with the β-ketoenamides. Depending on the substitution pattern of the β-ketoenamide, a condensation to the corresponding pyridine can spontaneously occur. In most cases a second step is necessary and the cyclocondensation must be induced or completed by treatment with TMSOTf and an amine base in a suitable solvents at elevated temperatures. In order to minimize the operational effort, the process can be performed as quasi-one-pot procedure without purification of the intermediary β-ketoenamide.
Scope and limitations
Following the protocol mentioned before, we successfully prepared a series of 4-hydroxypyridines with chiral functional groups present in a side chain. As can be seen in Table 1 not merely chiral aliphatic carboxylic acids and nitriles such as 17 and 31 can be transformed into 4-hydroxypyridines, but rather complex substrates with appropriately protected functional groups. For instance, when lithiated methoxyallene was added to pivalonitrile and reacted with O-silylated mandelic acid 21, pyridine derivative 22 was obtained in good yield over two steps. Furthermore, readily available N,N-dibenzylated amino acids, such as those derived from valine and phenylalanine, 23 and 25 gave the respective pyridines 24 and 26 in 45% and 50% yield, respectively. Carboxylic acids featuring aromatic units and branched side chains including quaternary α-carbon atoms were also tolerated. Besides chiral carboxylic acids, enantiopure nitriles were also successfully converted into pyridine derivatives in comparable yields. As an example, (S)-2-methylbutyronitrile (31) could be converted into compound 40 in good yield. The use of carboxylic acids and nitriles with structurally identical substituents allows a rapid access to pyridine derivatives such as 32 which almost has C2-symmetry. Compound 32 is derived from nitrile 31 and carboxylic acid 17 and was obtained in high yield after two steps. Products 34 and 35 were prepared from enantiopure acid 29 and racemic O-TBS-mandelonitrile 33. The diastereomeric pyridines obtained from this reaction are easily separable by column chromatography to give 34 and 35 in moderate yields. If not commercially available, the desired nitriles were prepared from the corresponding acids by an amide formation/dehydration sequence according to literature procedures [41,42]. Not all transformations proceeded in very good yields, however, it should be noted that in only a few cases attempts to optimize the conditions have been undertaken. Hence, there may be room for improvement of yields in cases where the standard conditions led only to moderate yields.
Table 1: Scope of the synthesis of 4-hydroxypyridine derivatives from lithiated methoxyallene, nitriles and carboxylic acids.
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-108-i7.svg?max-width=637&scale=1.0)
|
|||
|
Carboxylic Acid
R2CO2H |
Nitrile
R1-CN |
Producta | Yieldb |
|---|---|---|---|
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-108-i8.svg?max-width=637&scale=1.0)
|
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-108-i9.svg?max-width=637&scale=1.0)
|
![[Graphic 4]](/bjoc/content/inline/1860-5397-7-108-i10.svg?max-width=637&scale=1.0)
|
24% |
![[Graphic 5]](/bjoc/content/inline/1860-5397-7-108-i11.svg?max-width=637&scale=1.0)
|
![[Graphic 6]](/bjoc/content/inline/1860-5397-7-108-i12.svg?max-width=637&scale=1.0)
|
![[Graphic 7]](/bjoc/content/inline/1860-5397-7-108-i13.svg?max-width=637&scale=1.0)
|
30% |
![[Graphic 8]](/bjoc/content/inline/1860-5397-7-108-i14.svg?max-width=637&scale=1.0)
|
![[Graphic 9]](/bjoc/content/inline/1860-5397-7-108-i15.svg?max-width=637&scale=1.0)
|
![[Graphic 10]](/bjoc/content/inline/1860-5397-7-108-i16.svg?max-width=637&scale=1.0)
|
50% |
![[Graphic 11]](/bjoc/content/inline/1860-5397-7-108-i17.svg?max-width=637&scale=1.0)
|
![[Graphic 12]](/bjoc/content/inline/1860-5397-7-108-i18.svg?max-width=637&scale=1.0)
|
![[Graphic 13]](/bjoc/content/inline/1860-5397-7-108-i19.svg?max-width=637&scale=1.0)
|
45% |
![[Graphic 14]](/bjoc/content/inline/1860-5397-7-108-i20.svg?max-width=637&scale=1.0)
|
![[Graphic 15]](/bjoc/content/inline/1860-5397-7-108-i21.svg?max-width=637&scale=1.0)
|
![[Graphic 16]](/bjoc/content/inline/1860-5397-7-108-i22.svg?max-width=637&scale=1.0)
|
50% |
![[Graphic 17]](/bjoc/content/inline/1860-5397-7-108-i23.svg?max-width=637&scale=1.0)
|
![[Graphic 18]](/bjoc/content/inline/1860-5397-7-108-i24.svg?max-width=637&scale=1.0)
|
![[Graphic 19]](/bjoc/content/inline/1860-5397-7-108-i25.svg?max-width=637&scale=1.0)
|
45% |
![[Graphic 20]](/bjoc/content/inline/1860-5397-7-108-i26.svg?max-width=637&scale=1.0)
|
![[Graphic 21]](/bjoc/content/inline/1860-5397-7-108-i27.svg?max-width=637&scale=1.0)
|
![[Graphic 22]](/bjoc/content/inline/1860-5397-7-108-i28.svg?max-width=637&scale=1.0)
|
24% |
![[Graphic 23]](/bjoc/content/inline/1860-5397-7-108-i29.svg?max-width=637&scale=1.0)
|
![[Graphic 24]](/bjoc/content/inline/1860-5397-7-108-i30.svg?max-width=637&scale=1.0)
|
![[Graphic 25]](/bjoc/content/inline/1860-5397-7-108-i31.svg?max-width=637&scale=1.0)
|
85% |
![[Graphic 26]](/bjoc/content/inline/1860-5397-7-108-i32.svg?max-width=637&scale=1.0)
|
![[Graphic 27]](/bjoc/content/inline/1860-5397-7-108-i33.svg?max-width=637&scale=1.0)
|
![[Graphic 28]](/bjoc/content/inline/1860-5397-7-108-i34.svg?max-width=637&scale=1.0)
|
26%
(as a separable 1:1 mixture of diastereomers) |
| CF3CO2H |
![[Graphic 29]](/bjoc/content/inline/1860-5397-7-108-i35.svg?max-width=637&scale=1.0)
|
![[Graphic 30]](/bjoc/content/inline/1860-5397-7-108-i36.svg?max-width=637&scale=1.0)
|
37% |
| CF3CO2H |
![[Graphic 31]](/bjoc/content/inline/1860-5397-7-108-i37.svg?max-width=637&scale=1.0)
|
![[Graphic 32]](/bjoc/content/inline/1860-5397-7-108-i38.svg?max-width=637&scale=1.0)
|
28% |
| CF3CO2H |
![[Graphic 33]](/bjoc/content/inline/1860-5397-7-108-i39.svg?max-width=637&scale=1.0)
|
![[Graphic 34]](/bjoc/content/inline/1860-5397-7-108-i40.svg?max-width=637&scale=1.0)
|
56% |
![[Graphic 35]](/bjoc/content/inline/1860-5397-7-108-i41.svg?max-width=637&scale=1.0)
|
![[Graphic 36]](/bjoc/content/inline/1860-5397-7-108-i42.svg?max-width=637&scale=1.0)
|
![[Graphic 37]](/bjoc/content/inline/1860-5397-7-108-i43.svg?max-width=637&scale=1.0)
|
– |
| CF3CO2H |
![[Graphic 38]](/bjoc/content/inline/1860-5397-7-108-i44.svg?max-width=637&scale=1.0)
|
![[Graphic 39]](/bjoc/content/inline/1860-5397-7-108-i45.svg?max-width=637&scale=1.0)
|
– |
aOnly the predominant tautomer in CDCl3 is depicted; bAll yields are based on the nitrile.
Unfortunately, all attempts to incorporate proline-derived moieties failed. N-Benzylproline (41) turned out to be almost insoluble in ethereal solvents, which might explain why the desired β-ketoenamide was not formed [43]. To increase the solubility of the proline component, we changed the protective group from benzyl to the more lipophilic trityl group [44]. Surprisingly, the use of trityl-protected proline did not give the β-ketoenamide 47 as main product (Scheme 3). Instead, a diastereomeric 1:1 mixture of the β-keto-enolester 48 was isolated in 49% yield together with minor amounts of the expected product 47. The formation of 48 is additional evidence for our previously suggested mechanism (Scheme 2). We assume that the bulkiness of the trityl group hampers the transfer of the acyl group from intermediate 46 to 47. Upon the addition of water, the enamine moiety of 46 was hydrolyzed to furnish enol ester 48.
![[1860-5397-7-108-i3]](/bjoc/content/inline/1860-5397-7-108-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of proline derivative 45 and formation of β-ketoenamide 47 and enolester 48.
Scheme 3: Reaction of proline derivative 45 and formation of β-ketoenamide 47 and enolester 48.
The pyridines in Table 1 are depicted in their predominant tautomeric form as found in CDCl3 at ambient temperature. Interestingly, the pyridone/pyridinol equilibrium seems to depend on the substituents at the C-2 or C-6 side chains. In general, a hydrogen bond-acceptor seems to stabilize the pyridone tautomer, whereas the pyridinol tautomer is favored when a hydrogen bond-donor is present. Compound 37 exists exclusively as pyridone tautomer, but after desilylation the resulting product, with a free hydroxy group in the side chain, strongly prefers the pyridinol tautomer. It seems reasonable to assume that the pyridone tautomer is stabilized through an internal hydrogen bond between a silyl ether or a tertiary amine moiety of the side chain as observed for compounds 22 and 24. Moreover, we found that the equilibrium is strongly influenced by the solvent. In CDCl3 pyridine 40 exclusively exists in its pyridone form, but in methanol-d4 the equilibrium shifts completely to the pyridinol tautomer.
Subsequent transformations of the prepared pyridine derivatives
To prove the enantiopurity of the pyridines derived from carboxylic acids and nitriles, which are prone to racemization, i.e., substrates with tertiary stereogenic centers in α-position, compounds 18, 22 and 40 were transformed into esters 50, 49 and 51, respectively (Table 2). Treatment of the pyridones with Mosher acid chloride in a mixture of pyridine and dichloromethane as solvent afforded the desired esters in good yields. Comparison with the diastereomeric compounds obtained from racemic starting materials unambiguously shows that the sequence proceeds without noticeable racemization, since 49, 50 and 51 were obtained in diastereomeric pure form (Table 2) as judged by 1H NMR analysis (estimated error 3–5%). For instance, the signal of the methoxy group at C-3 of diastereomeric 49’ (obtained by starting with racemic mandelic acid) appears at 3.43 ppm in the 1H NMR-spectrum, whereas this signal for 49 occurs at 3.49 ppm (Figure 1). In addition, the tert-butyl group of the OTBS groups of the two diastereoisomers 49 and 49’ show signals at different frequencies.
Table 2: Esterification of different pyridinol derivatives with the 3,3,3-trifluoro-2-methoxy-2-phenylpropanoic acid.
![[Graphic 40]](/bjoc/content/inline/1860-5397-7-108-i46.svg?max-width=637&scale=1.0)
|
||
| Pyridine | Product | Yield |
|---|---|---|
| 22 |
![[Graphic 41]](/bjoc/content/inline/1860-5397-7-108-i47.svg?max-width=637&scale=1.0)
|
68%
dr >95:5a |
| 18 |
![[Graphic 42]](/bjoc/content/inline/1860-5397-7-108-i48.svg?max-width=637&scale=1.0)
|
55%
dr >95:5a |
| 40 |
![[Graphic 43]](/bjoc/content/inline/1860-5397-7-108-i49.svg?max-width=637&scale=1.0)
|
67%
dr >95:5a |
aDetermined by 1H NMR spectroscopic analysis of the crude products.
![[1860-5397-7-108-1]](/bjoc/content/figures/1860-5397-7-108-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 1H NMR spectra of 49 and the mixture of diastereoisomers 49 and 49’.
Figure 1: 1H NMR spectra of 49 and the mixture of diastereoisomers 49 and 49’.
To explore the chemistry of the synthesized pyridine derivatives, we investigated the selective functionalization of the 4-hydroxy group. Pyridone 20 was nonaflated according to previously established conditions to provide 52 in 56% yield (Scheme 4). As we have already demonstrated, pyrid-4-yl nonaflates are excellent coupling partners in palladium-catalyzed transformations such as Suzuki, Stille, Heck and Sonogashira reactions [45].
![[1860-5397-7-108-i4]](/bjoc/content/inline/1860-5397-7-108-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of pyrid-4-yl nonaflate 52.
Scheme 4: Synthesis of pyrid-4-yl nonaflate 52.
However, in contrast to the smooth nonaflation, the selective O-alkylation of the synthesized pyridines turned out to be more challenging (Scheme 5). Whereas pyridone 22 could be O-methylated in good yield with methyl iodide in THF, the same conditions converted 30 into 55 in a disappointing 30% yield. Desilylation of 53 and 55 with HF in pyridine gave the desired deprotected pyridine derivatives 54 and 56 in high yields. This type of enantiopure hydroxymethyl-substituted pyridine derivatives is of particular interest as they are known to be efficient catalysts for the asymmetric addition of zinc organyls to aldehydes [14,46].
![[1860-5397-7-108-i5]](/bjoc/content/inline/1860-5397-7-108-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: O-Methylation of pyridine derivatives 22 and 30 followed by desilylation.
Scheme 5: O-Methylation of pyridine derivatives 22 and 30 followed by desilylation.
Preparation of lactic acid derived pyrid-4-yl nonaflates
In the course of our investigations on the scope of the present procedure, we also discovered that easily available O-TBS-protected lactic acid and O-TBS-protected lactic nitrile are excellent reaction partners. We became interested in exploring the scope of this reaction with respect to lactic acid derived starting materials in more detail. In contrast to the previously described procedures, we decided to purify the reaction mixture at the stage of the β-alkoxy-β-ketoenamides 58 obtained by the three-component reaction. Recently we demonstrated that β-alkoxy-β-ketoenamides are not only valuable intermediates in the synthesis of 4-hydroxypyridines 57, but that they can also serve as precursors in the synthesis of 5-alkoxypyrimidines 59 (Scheme 6). When β-alkoxy-β-ketoenamides 58 were treated with an ammonia source such as NH4OAc in MeOH, 5-alkoxypyrimidines with the general structure of 59 were formed in high yields [37,38,47]. By this simple change in the reaction conditions not only pyridine but also pyrimidine derivatives with lactic acid based side chains should be accessible.
![[1860-5397-7-108-i6]](/bjoc/content/inline/1860-5397-7-108-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Formation of 5-alkoxypyrimidines from β-alkoxy-β-ketoenamides.
Scheme 6: Formation of 5-alkoxypyrimidines from β-alkoxy-β-ketoenamides.
O-TBS-protected lactic nitrile 63 was prepared following a literature procedure in four steps starting from enantiopure methyl lactate [48]. The scope of the multicomponent reaction with respect to lactic acid derived precursors is summarized in Table 3.
![[Graphic 44]](/bjoc/content/inline/1860-5397-7-108-i50.svg?max-width=637&scale=1.0)
![[Graphic 45]](/bjoc/content/inline/1860-5397-7-108-i51.svg?max-width=637&scale=1.0)
![[Graphic 46]](/bjoc/content/inline/1860-5397-7-108-i52.svg?max-width=637&scale=1.0)
![[Graphic 47]](/bjoc/content/inline/1860-5397-7-108-i53.svg?max-width=637&scale=1.0)
![[Graphic 48]](/bjoc/content/inline/1860-5397-7-108-i54.svg?max-width=637&scale=1.0)
![[Graphic 49]](/bjoc/content/inline/1860-5397-7-108-i55.svg?max-width=637&scale=1.0)
![[Graphic 50]](/bjoc/content/inline/1860-5397-7-108-i56.svg?max-width=637&scale=1.0)
![[Graphic 51]](/bjoc/content/inline/1860-5397-7-108-i57.svg?max-width=637&scale=1.0)
![[Graphic 52]](/bjoc/content/inline/1860-5397-7-108-i58.svg?max-width=637&scale=1.0)
![[Graphic 53]](/bjoc/content/inline/1860-5397-7-108-i59.svg?max-width=637&scale=1.0)
![[Graphic 54]](/bjoc/content/inline/1860-5397-7-108-i60.svg?max-width=637&scale=1.0)
![[Graphic 55]](/bjoc/content/inline/1860-5397-7-108-i61.svg?max-width=637&scale=1.0)
![[Graphic 56]](/bjoc/content/inline/1860-5397-7-108-i62.svg?max-width=637&scale=1.0)
![[Graphic 57]](/bjoc/content/inline/1860-5397-7-108-i63.svg?max-width=637&scale=1.0)
![[Graphic 58]](/bjoc/content/inline/1860-5397-7-108-i64.svg?max-width=637&scale=1.0)
![[Graphic 59]](/bjoc/content/inline/1860-5397-7-108-i65.svg?max-width=637&scale=1.0)
![[Graphic 60]](/bjoc/content/inline/1860-5397-7-108-i66.svg?max-width=637&scale=1.0)
![[Graphic 61]](/bjoc/content/inline/1860-5397-7-108-i67.svg?max-width=637&scale=1.0)
![[Graphic 62]](/bjoc/content/inline/1860-5397-7-108-i68.svg?max-width=637&scale=1.0)
Table 3 shows that O-TBS-protected lactic acid 60 and O-TBS-protected lactic nitrile 63 gave the desired ketoenamides in moderate to high yields. When lithiated methoxyallene was reacted with pivalonitrile or benzonitrile followed by the addition of O-TBS-protected lactic acid, the corresponding ketoenamides 61 and 62 were isolated in 58% yield. Reaction of lithiated methoxyallene with O-TBS-protected lactic nitrile and benzoic acid furnished 64 in high yield. Heterocyclic moieties were also well tolerated as demonstrated by the efficient reaction of 2-thiophene carboxylic acid. The relatively low yield in the formation of 66 might be explained by the poor solubility of 2-picolinic acid in ethereal solvents rather than for reactivity reasons. In contrast to the other examples, enamide 66 was obtained as a 1:1 mixture of (E)- and (Z)-isomers. This may be due to alternative hydrogen bond formation with the NH unit to the pyridine nitrogen rather than to the carbonyl group. Subsequent cyclocondensation with TMSOTf and NEt3 in 1,2-dichloroethane gave the expected pyridine derivatives, which were directly converted into pyrid-4-yl nonaflates in a second step. The results are depicted in Table 4.
Table 4: Cyclization and nonaflation of lactic acid derived β-alkoxy-β-ketoenamides.
![[Graphic 63]](/bjoc/content/inline/1860-5397-7-108-i69.svg?max-width=637&scale=1.0)
|
||
| β-Alkoxy-β-ketoenamide | Product | Yielda |
|---|---|---|
![[Graphic 64]](/bjoc/content/inline/1860-5397-7-108-i70.svg?max-width=637&scale=1.0)
|
![[Graphic 65]](/bjoc/content/inline/1860-5397-7-108-i71.svg?max-width=637&scale=1.0)
|
56% |
![[Graphic 66]](/bjoc/content/inline/1860-5397-7-108-i72.svg?max-width=637&scale=1.0)
|
![[Graphic 67]](/bjoc/content/inline/1860-5397-7-108-i73.svg?max-width=637&scale=1.0)
|
61% |
![[Graphic 68]](/bjoc/content/inline/1860-5397-7-108-i74.svg?max-width=637&scale=1.0)
|
![[Graphic 69]](/bjoc/content/inline/1860-5397-7-108-i75.svg?max-width=637&scale=1.0)
|
63% |
![[Graphic 70]](/bjoc/content/inline/1860-5397-7-108-i76.svg?max-width=637&scale=1.0)
|
![[Graphic 71]](/bjoc/content/inline/1860-5397-7-108-i77.svg?max-width=637&scale=1.0)
|
52% |
![[Graphic 72]](/bjoc/content/inline/1860-5397-7-108-i78.svg?max-width=637&scale=1.0)
|
![[Graphic 73]](/bjoc/content/inline/1860-5397-7-108-i79.svg?max-width=637&scale=1.0)
|
72% |
![[Graphic 74]](/bjoc/content/inline/1860-5397-7-108-i80.svg?max-width=637&scale=1.0)
|
![[Graphic 75]](/bjoc/content/inline/1860-5397-7-108-i81.svg?max-width=637&scale=1.0)
|
39% |
aYields over two steps based on the ketoenamide.
In all examples the cyclization/nonaflation sequence provided the pyrid-4-yl nonaflates in good yields. Apparently, the reactivity in this sequence is not strongly governed by the structure of the original ketoenamide. Even the configuration of the enamide double bond seems to have no influence on the cyclization, since the (E/Z)-mixture of enamide 66 also gave the corresponding pyridine in good yield. Obviously, the diastereomers are in equilibrium under the cyclization conditions. Of particular interest are the pyrid-4-yl nonaflates 71 and 72, possessing a chiral side chain as well as heteroaromatic units. 2,2’-Bipyridines with structures similar to 72 might show interesting properties when used as ligands in asymmetric transformations. The nonaflate moiety should allow electronic fine tuning of the ligand properties in palladium-catalyzed or nucleophilic substitution reactions.
Conclusion
We have demonstrated that enantiopure functionalized carboxylic acids and nitriles can be used without problems in our previously reported pyridine synthesis. The starting materials were successfully transformed into the corresponding pyridines without loss of enantiopurity to yield enantiopure 4-hydroxypyridine derivatives with stereogenic side chains at C-2 and C-6. The 4-hydroxy group allows further variations. Applications of the prepared pyridines as ligands or catalysts in asymmetric transformations will be studied and will be the subject of future reports.
Experimental
General methods: Reactions were generally performed under an argon atmosphere in flame-dried flasks, and the components were added by syringe. Methanol was purchased in p.a. quality and stored under an argon atmosphere over molecular sieves (4 Å). Triethylamine was distilled from CaH2 and stored over KOH under an atmosphere of argon. Pyridine was used as purchased and stored over KOH under an atmosphere of argon. 1,2-Dichloroethane was purchased in p.a. quality and stored over molecular sieves (4 Å) under an atmosphere of argon. Tetrahydrofuran, diethyl ether, toluene and dichloromethane were obtained from the solvent purification system MB-SPS-800 (M. Braun). Products were purified by flash chromatography on silica gel (230–400 mesh, Merck). Unless otherwise stated, yields refer to analytically pure samples. Internal standards: for 1H NMR CDCl3 (δ = 7.26 ppm), TMS (δ = 0.00 ppm), CD3OD (δ = 3.31 ppm), C6D6 (δ = 7.16 ppm), for 13C NMR CDCl3 (δ = 77.0 ppm), CD3OD (δ = 49.0 ppm), C6D6 (δ = 128.1 ppm). NMR spectra were recorded on Bruker AC 250, ECP 400, AC 500, AVIII 700, or Jeol Eclipse 500 instruments in CDCl3, CD3OD, or C6D6 solution. Integrals are in accord with assignments; coupling constants are given in Hz. IR spectra were measured with a FT-IR spectrometer Nicolet 5 SXC or with a Nexus FT-IR equipped with a Nicolet Smart DuraSamplIR ATR. MS and HRMS analyses were obtained with Finnigan Varian Ionspec QFT-7 (ESI-FT-ICR) and Agilent ESI-TOF 6210 (4 μL/min, 1 bar, 4000 V) instruments. Elemental analyses were obtained with “Elemental-Analyzers“ (Perkin–Elmer or Carlo Erba). Melting points were measured with a Reichert apparatus (Thermovar) and are uncorrected. Optical rotations ([α]D) were determined with a Perkin–Elmer 241 polarimeter at the temperatures given. Commercially available chemicals were used without further purification unless otherwise stated.
Typical procedure for the preparation of 3-methoxy-4-hydroxypyridines without isolation of the intermediate β-alkoxy-β-ketoenamide (Procedure 1)
A solution of n-BuLi (2.5 M in hexanes, 0.31 mL, 0.79 mmol) was added dropwise to a solution of methoxyallene (59 µL, 0.71 mmol) in diethyl ether (5 mL) at −40 °C. After stirring at that temperature for 15 min, pivalonitrile was added (59 mg, 0.71 mmol) and the resulting yellow solution stirred for 4 h at −40 °C. The solution was then cooled to −78 °C and (S)-2-methylbutyric acid (0.23 mL, 2.14 mmol) added. Stirring was continued overnight during which time the mixture was slowly allowed to reach r.t. The reaction was quenched by the addition of sat. aq. NaHCO3 solution (10 mL) and the aqueous phase extracted with diethyl ether (2 × 20 mL). The combined organic layers were washed with brine, dried with Na2SO4, filtered and the solvent was evaporated under reduced pressure. The residue was re-dissolved in CH2Cl2 (14 mL) and TMSOTf (0.41 mL, 2.1 mmol) and NEt3 (0.30 mL, 2.1 mmol) were added. The mixture was heated under reflux under an atmosphere of argon for 2 d. After complete consumption of the starting material (by TLC), the reaction was quenched by the addition of aq. sat. NH4Cl solution (20 mL) and the aqueous layer extracted with CH2Cl2 (2 × 30 mL). The combined organic layers were dried with Na2SO4, filtered and evaporated. The residue was purified by flash column chromatography on silica gel (eluent: hexane/ethyl acetate 1:9) to afford 18 (41 mg, 24%) as colorless crystals.
(S)-6-sec-Butyl-2-tert-butyl-3-methoxypyridin-4-one (18): mp 109–110 °C; [α]D22 +20.9 (c 2.3, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.90 (t, J ≈ 7 Hz, 3H, 4’-H), 1.24 (d, J = 6.9 Hz, 3H, 1’-H), 1.43 (s, 9H, t-Bu), 1.59 (quint, J ≈ 7 Hz, 2H, 3’-H), 2.47 (sext, J ≈ 7 Hz, 1H, 2’-H), 3.94 (s, 3H, OMe), 6.26 (s, 1H, 5-H), 7.74 (s, 1H, NH) ppm; 13C NMR (101 MHz, CDCl3) δ 11.8 (q, C-4’), 19.5 (q, C-1’), 28.4, 29.5 (2, s, t-Bu), 35.1 (t, C-3’), 39.9 (d, C-2’), 58.9 (q, OMe), 114.2 (d, C-5), 146.0, 146.4, 150.7 (3 s, C-2, C-3, C-6), 176.1 (s, C-4) ppm; IR (KBr) ![[Graphic 76]](/bjoc/content/inline/1860-5397-7-108-i82.svg?max-width=637&scale=1.18182) : 3250 (N-H), 2965–2910 (=C-H, C-H), 1620 (C=O), 1580, 1540 (C=C) cm−1; HRMS–ESI (m/z): [M + H]+ calcd for C14H24NO2, 238.1807; found, 238.1803; Anal. calcd for C14H23NO2: C, 70.85; H, 9.77; N, 5.90; found: C, 70.81; H, 9.79; N, 5.38.
: 3250 (N-H), 2965–2910 (=C-H, C-H), 1620 (C=O), 1580, 1540 (C=C) cm−1; HRMS–ESI (m/z): [M + H]+ calcd for C14H24NO2, 238.1807; found, 238.1803; Anal. calcd for C14H23NO2: C, 70.85; H, 9.77; N, 5.90; found: C, 70.81; H, 9.79; N, 5.38.
Typical procedure for the preparation of β-alkoxy-β-ketoenamides (Procedure 2)
A solution of n-BuLi (1.30 mL, 3.28 mmol, 2.5 M in hexanes) was added to a solution of methoxyallene (0.30 mL, 3.28 mmol) in diethyl ether (20 mL) at −50 °C. After stirring for 30 min at −50 °C, the reaction mixture was cooled to −78 °C and (S)-TBS-lactic nitrile (200 mg, 1.14 mmol) in anhydrous diethyl ether (5 mL) was added to the mixture. After stirring for 4 h, a solution of benzoic acid (0.84 g, 6.88 mmol) in anhydrous DMF (10 mL) was added. The mixture was stirred overnight and slowly allowed to reach r.t. The reaction was quenched with sat. aq. NaHCO3 solution (15 mL) and the product extracted with diethyl ether (3 × 40 mL). The combined organic layers were washed with brine, dried with Na2SO4, filtered and the solvent was removed under reduced pressure. The crude product was purified by column chromatography on silica gel (eluent:hexane/EtOAc 3:1) to give 64 as a pale yellow oil (300 mg, 73%).
(S)-N-{1-[1-(tert-Butyldimethylsiloxy)ethyl]-2-methoxy-3-oxo-but-1-enyl}benzamide (64) 1H NMR (500 MHz, CDCl3) δ 0.07, 0.11, 0.88 (3 s, 3H, 3H, 9H, OTBS), 1.46 (d, J = 6.4 Hz, 3H, 2’’-H), 2.32 (s, 3H, 4’-H), 3.51 (s, 3H, OMe), 5.33 (q, J = 6.4 Hz, 1H, 1’’-H), 7.43–7.53, 7.85–7.87 (2 m, 3H, 2H, Ph), 10.10 (br s, 1H, NH) ppm; 13C NMR (125 MHz, CDCl3) δ −4.8, −4.7 (2 q, OTBS), 18.2 (q, C-4’), 21.9 (q, C-2’’), 25.9, 27.2 (2, q, OTBS), 60.9 (q, OMe), 65.6 (d, C-1’’), 127.5, 128.8, 132.2, 134.2, 139.6, 141.9 (3 d, 3 s, Ph, C-1’, C-2’), 165.0 (s, C-1), 200.2 (s, C-3’) ppm; IR (ATR) : 3315 (NH), 2955–2855 (=CH, C-H), 1720–1515 (C=O, C=C) cm−1; HRMS–ESI (m/z): [M + H]+ calcd for C20H31NNaO4Si, 400.1915; found, 400.1930.
Typical procedure for the cyclization of β-alkoxy-β-ketoenamides to 4-hydroxypyridines and subsequent nonaflation (Procedure 3)
Enamide 64 (40 mg, 0.11 mmol) was dissolved in 1,2-dichloroethane (2 mL) and placed in a sealable tube. Triethylamine (48 µL, 0.32 mmol) and TMSOTf (58 µL, 0.32 mmol) were added at r.t., and the resulting mixture was heated at 90 °C for 2 d. After complete consumption of the starting material (TLC), the reaction was quenched with sat. aq. NH4Cl solution (2 mL). After extraction with dichloromethane (3 × 10 mL), the combined organic layers were dried with Na2SO4 filtered and the solvent was removed under reduced pressure. The crude product was purified by flash column chromatography (SiO2, EtOAc/Methanol 10:1) to afford the respective pyridine derivative (36 mg, 94%) as a brown liquid.
The pyridine derivative (33 mg, 0.09 mmol) was dissolved in THF (3 mL) and NaH (6.6 mg, 0.28 mmol) added under an argon atmosphere. Nonafluorobutanesulfonyl fluoride (50 µL, 0.28 mmol) was added dropwise at room temperature. The mixture was stirred at the same temperature for 12 h and quenched by the slow addition of water. The resulting product was extracted with diethyl ether (3 × 10 mL), dried with Na2SO4, filtered and concentrated to dryness. The residue was purified by column chromatography on silica gel (eluent: 2–5% EtOAc in hexane) to afford 70 (39 mg, 67%) as a colorless oil.
(S)-2-[1-(tert-Butyldimethylsiloxy)ethyl]-3-methoxy-6-phenyl-pyridin-4-yl nonaflate (70): [α]D22 −21.2 (c 0.3, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.00, 0.05, 0.87 (3 s, 3H, 3H, 9H, OTBS), 1.60 (d, J = 6.6 Hz, 3H, 2’-H), 3.95 (s, 3H, OMe), 5.30 (q, J = 6.6 Hz, 1H, 1’-H), 7.42–7.48 (m, 3H, Ph), 7.51 (s, 1H, 5-H), 7.97–7.98 (m, 2H, Ph) ppm; 13C NMR (125 MHz, CDCl3) δ −4.6, −4.4 (2 q, OTBS), 18.3 (q, C-2’), 25.9, 30.3 (2, s, OTBS), 62.6 (q, OMe), 68.8 (d, C-1’), 112.3, 126.9, 128.8, 129.5, 137.7, 144.4, 150.1, 153.7, 160.0 (4 d, 5 s, Ph, C-2, C-3, C-4, C-5, C-6) ppm; IR (ATR) : 3310 (NH), 3010–2835 (=CH, C-H), 1685–1510 (C=O, C=C) cm−1; HRMS–ESI (m/z): [M + H]+ calcd for C24H29F9NO5SSi, 642.1387; found, 642.1403.
Typical procedure for the esterification of 4-pyridones with 3,3,3-trifluoro-2-methoxy-2-phenylpropanoic acid (Procedure 4)
Pyridone 22 (22 mg, 0.06 mmol) was dissolved in anhydrous CH2Cl2 (0.3 mL) and anhydrous pyridine (0.3 mL), and (S)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoic acid (17 µL, 0.09 mmol) added. The mixture was stirred under an atmosphere of argon at r.t. for 16 h. After complete consumption of the starting material (TLC), the mixture was diluted with CH2Cl2 (10 mL) and the organic layer was successively washed with sat. NaHCO3 solution, 1 M HCl and H2O (10 mL each). The organic layer was dried with Na2SO4, filtered and the solvent was removed under reduced pressure to afford 49 (25 mg, 68%) as a colorless oil.
(S,S)-2-tert-Butyl-6-[(tert-butyldimethylsiloxy)phenylmethyl]-3-methoxypyridin-4-yl 3,3,3-trifluoro-2-methoxy-2-phenylpropanoate (49): [α]D22 +14.0 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.01, 0.02, 0.96 (s, 3H, 3H, 9H, OTBS), 1.35 (s, 9H, t-Bu), 3.49, 3.68 (2 s, 3H each, OMe), 5.80 (s, 1H, 1’-H), 7.17 (s, 1H, 5-H), 7.19–7.22, 7.27–7.31, 7.48–7.53, 7.61–7.71 (4 m, 10H, Ph) ppm; 19F NMR (376 MHz, CDCl3) δ −71.1 (s, CF3) ppm; IR (neat) : 2955–2930 (C-H), 1775 (C=O), 1570–1450 (C=C), 1170–1105 (=C-H), 780–700 (C-F) cm−1; HRMS–ESI (m/z): [M + H]+ calcd for C33H43F3NO5Si, 618.2857; found, 618.2896.
Acknowledgements
Generous support of this work by the Deutsche Forschungsgemeinschaft (SFB 765), the Studienstiftung des Deutschen Volkes (PhD fellowship to CE) and the Bayer Schering Pharma AG is most gratefully acknowledged. We thank Dr. R. Zimmer for help during the preparation of the manuscript.
References
-
Newkome, G. R. In Chemistry of Heterocyclic Compounds; Newkome, G. R., Ed.; Wiley: New York, 1984; Vol. 15.
Return to citation in text: [1] -
McKillop, A.; Boulton, A. J. In Comprehensive Heterocyclic Chemistry; Katritzky, A. R.; Rees, C. W., Eds.; Pergamon Press: Oxford, 1984; Vol. 2, pp 67 ff.
Return to citation in text: [1] -
Kleemann, A.; Engel, J.; Kutscher, B. Pharmaceutical Substances; Thieme: Stuttgart, 2000.
Return to citation in text: [1] -
Spitzner, D. Science of Synthesis; Thieme: Stuttgart, 2004; Vol. 15.
Return to citation in text: [1] -
Jones, G. In Comprehensive Heterocyclic Chemistry II; McKillop, A., Ed.; Pergamon Press: Oxford, 1996; Vol. 5, pp 167 ff.
Return to citation in text: [1] -
Wess, G.; Kesseler, K.; Baader, E.; Bartmann, W.; Beck, G.; Bergmann, A.; Jendralla, H.; Bock, K.; Holzstein, O.; Kleine, H.; Schnierer, M. Tetrahedron Lett. 1990, 31, 2545–2548. doi:10.1016/0040-4039(90)80121-2
Return to citation in text: [1] -
Miyachi, N.; Yanagawa, Y.; Iwasaki, H.; Ohara, Y.; Hiyama, T. Tetrahedron Lett. 1993, 34, 8267–8270. doi:10.1016/S0040-4039(00)61407-7
Return to citation in text: [1] -
Beck, G. Synlett 2002, 837–850. doi:10.1055/s-2002-31890
Return to citation in text: [1] -
Beck, G.; Kesseler, K.; Baader, E.; Bartmann, W.; Bergmann, A.; Granzer, E.; Jendralla, H.; Von Kerekjarto, B.; Krause, R. J. Med. Chem. 1990, 33, 52–60. doi:10.1021/jm00163a010
Return to citation in text: [1] -
Cattaneo, D.; Baldelli, S.; Merlini, S.; Zenoni, S.; Perico, N.; Remuzzi, G. Expert Opin. Ther. Pat. 2004, 14, 1553–1566. doi:10.1517/13543776.14.11.1553
Return to citation in text: [1] -
O'Hagan, D. Nat. Prod. Rep. 1997, 14, 637–651. doi:10.1039/NP9971400637
Return to citation in text: [1] -
Aida, W.; Ohtsuki, T.; Li, X.; Ishibashi, M. Tetrahedron 2009, 65, 369–373. doi:10.1016/j.tet.2008.10.040
Return to citation in text: [1] -
Lehn, J.-M. Supramolecular Chemistry - Concepts and Perspectives; VCH: Weinheim, 1995.
Return to citation in text: [1] -
Bolm, C.; Schlingloff, G.; Harms, K. Chem. Ber. 1992, 125, 1191–1203. doi:10.1002/cber.19921250529
Return to citation in text: [1] [2] -
Kwong, H.-L.; Yeung, H.-L.; Yeung, C.-T.; Lee, W.-S.; Lee, C.-S.; Wong, W.-L. Coord. Chem. Rev. 2007, 251, 2188–2222. doi:10.1016/j.ccr.2007.03.010
Return to citation in text: [1] -
Bolm, C.; Ewald, M.; Felder, M. Chem. Ber. 1992, 125, 1205–1215. doi:10.1002/cber.19921250530
Return to citation in text: [1] -
Bolm, C.; Ewald, M.; Zehnder, M.; Neuburger, M. A. Chem. Ber. 1992, 125, 453–458. doi:10.1002/cber.19921250224
Return to citation in text: [1] -
Trost, B. M.; Weiss, A. H. Adv. Synth. Catal. 2009, 351, 963–983. doi:10.1002/adsc.200800776
Return to citation in text: [1] -
Drury, W. J.; Zimmermann, N.; Keenan, M.; Hayashi, M.; Kaiser, S.; Goddard, R.; Pfaltz, A. Angew. Chem., Int. Ed. 2003, 116, 72–76. doi:10.1002/anie.200352755
Return to citation in text: [1] -
Roseblade, S. J.; Pfaltz, A. Acc. Chem. Res. 2007, 40, 1402–1411. doi:10.1021/ar700113g
Return to citation in text: [1] -
Henry, G. D. Tetrahedron 2004, 60, 6043–6061. doi:10.1016/j.tet.2004.04.043
Return to citation in text: [1] -
Movassaghi, M.; Hill, M. D.; Ahmad, O. K. J. Am. Chem. Soc. 2007, 129, 10096–10097. doi:10.1021/ja073912a
Return to citation in text: [1] -
Liu, S.; Liebeskind, L. S. J. Am. Chem. Soc. 2008, 130, 6918–6919. doi:10.1021/ja8013743
Return to citation in text: [1] -
Manning, J. R.; Davies, H. M. L. J. Am. Chem. Soc. 2008, 130, 8602–8603. doi:10.1021/ja803139k
Return to citation in text: [1] -
Xin, X.; Wang, Y.; Kumar, S.; Liu, X.; Lin, Y.; Dong, D. Org. Biomol. Chem. 2010, 8, 3078–3082. doi:10.1039/C001117G
Return to citation in text: [1] -
Hill, M. D. Chem.–Eur. J. 2010, 16, 12052–12062. doi:10.1002/chem.201001100
Return to citation in text: [1] -
Rahm, F.; Stranne, R.; Bremberg, U.; Nordstrom, K.; Cernerud, M.; Macedo, E.; Moberg, C. J. Chem. Soc., Perkin Trans. 1 2000, 1983–1990. doi:10.1039/b000269k
Return to citation in text: [1] -
Rahm, F.; Fischer, A.; Moberg, C. Eur. J. Org. Chem. 2003, 4205–4215. doi:10.1002/ejoc.200300368
Return to citation in text: [1] -
Flögel, O.; Dash, J.; Brüdgam, I.; Hartl, H.; Reissig, H.-U. Chem.–Eur. J. 2004, 10, 4283–4290. doi:10.1002/chem.200400322
Return to citation in text: [1] -
Dash, J.; Lechel, T.; Reissig, H.-U. Org. Lett. 2007, 9, 5541–5544. doi:10.1021/ol702468s
Return to citation in text: [1] -
Lechel, T.; Dash, J.; Hommes, P.; Lentz, D.; Reissig, H.-U. J. Org. Chem. 2009, 75, 726–732. doi:10.1021/jo9022183
Return to citation in text: [1] -
Lechel, T.; Reissig, H.-U. Pure Appl. Chem. 2010, 82, 1835–1845. doi:10.1351/PAC-CON-09-09-06
Return to citation in text: [1] -
Brasholz, M.; Reissig, H.-U.; Zimmer, R. Acc. Chem. Res. 2009, 42, 45–56. doi:10.1021/ar800011h
Return to citation in text: [1] -
Lechel, T.; Dash, J.; Brüdgam, I.; Reissig, H.-U. Eur. J. Org. Chem. 2008, 3647–3655. doi:10.1002/ejoc.200800398
Return to citation in text: [1] -
Zimmer, R.; Reissig, H.-U. Donor-Substituted Allenes. In Modern Allene Chemistry; Krause, N.; Hashmi, A. S. K., Eds.; Wiley-VCH: Weinheim, 2004; pp 425–492.
Return to citation in text: [1] -
Reissig, H.-U.; Zimmer, R. Cumulenes and Allenes: Synthesis from Other Allenes. In Science of Synthesis; Krause, N., Ed.; Thieme: Stuttgart, 2007; Vol. 44, pp 301–352.
Return to citation in text: [1] -
Bera, M. K.; Reissig, H.-U. Synthesis 2010, 2129–2138. doi:10.1055/s-0029-1218787
Return to citation in text: [1] [2] -
Zimmer, R.; Lechel, T.; Rancan, G.; Bera, M. K.; Reissig, H.-U. Synlett 2010, 1793–1796. doi:10.1055/s-0030-1258088
Return to citation in text: [1] [2] -
Lechel, T.; Dash, J.; Eidamshaus, C.; Brüdgam, I.; Lentz, D.; Reissig, H.-U. Org. Biomol. Chem. 2010, 8, 3007–3014. doi:10.1039/B925468D
Return to citation in text: [1] -
Eidamshaus, C.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 1162–1166. doi:10.1002/adsc.200800789
Return to citation in text: [1] [2] -
Couty, F.; David, O.; Larmanjat, B.; Marrot, J. J. Org. Chem. 2007, 72, 1058–1061. doi:10.1021/jo062221e
Return to citation in text: [1] -
Aureggi, V.; Frankevicius, V.; Kitching, M. O.; Ley, S. V.; Longbottom, D. A.; Oelke, A. J.; Sedelmeier, G. Org. Synth. 2008, 85, 72–87.
Return to citation in text: [1] -
Traverse, J. F.; Zhao, Y.; Hoveyda, A. H.; Snapper, M. L. Org. Lett. 2005, 7, 3151–3154. doi:10.1021/ol050814q
Return to citation in text: [1] -
Barlos, K.; Papaioannou, D.; Theodoropoulos, D. J. Org. Chem. 1982, 47, 1324–1326. doi:10.1021/jo00346a031
Return to citation in text: [1] -
Högermeier, J.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 2747–2763. doi:10.1002/adsc.200900566
Return to citation in text: [1] -
Liebehentschel, S.; Cvengroš, J.; Jacobi von Wangelin, A. Synlett 2007, 2574–2578. doi:10.1055/s-2007-986630
Return to citation in text: [1] -
Lechel, T.; Möhl, S.; Reissig, H.-U. Synlett 2009, 1059–1062. doi:10.1055/s-0028-1088220
Return to citation in text: [1] -
Massad, S. K.; Hawkins, L. D.; Baker, D. C. J. Org. Chem. 1983, 48, 5180–5182. doi:10.1021/jo00174a006
Return to citation in text: [1]
| 37. | Bera, M. K.; Reissig, H.-U. Synthesis 2010, 2129–2138. doi:10.1055/s-0029-1218787 |
| 38. | Zimmer, R.; Lechel, T.; Rancan, G.; Bera, M. K.; Reissig, H.-U. Synlett 2010, 1793–1796. doi:10.1055/s-0030-1258088 |
| 47. | Lechel, T.; Möhl, S.; Reissig, H.-U. Synlett 2009, 1059–1062. doi:10.1055/s-0028-1088220 |
| 45. | Högermeier, J.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 2747–2763. doi:10.1002/adsc.200900566 |
| 14. | Bolm, C.; Schlingloff, G.; Harms, K. Chem. Ber. 1992, 125, 1191–1203. doi:10.1002/cber.19921250529 |
| 46. | Liebehentschel, S.; Cvengroš, J.; Jacobi von Wangelin, A. Synlett 2007, 2574–2578. doi:10.1055/s-2007-986630 |
| 1. | Newkome, G. R. In Chemistry of Heterocyclic Compounds; Newkome, G. R., Ed.; Wiley: New York, 1984; Vol. 15. |
| 2. | McKillop, A.; Boulton, A. J. In Comprehensive Heterocyclic Chemistry; Katritzky, A. R.; Rees, C. W., Eds.; Pergamon Press: Oxford, 1984; Vol. 2, pp 67 ff. |
| 3. | Kleemann, A.; Engel, J.; Kutscher, B. Pharmaceutical Substances; Thieme: Stuttgart, 2000. |
| 4. | Spitzner, D. Science of Synthesis; Thieme: Stuttgart, 2004; Vol. 15. |
| 5. | Jones, G. In Comprehensive Heterocyclic Chemistry II; McKillop, A., Ed.; Pergamon Press: Oxford, 1996; Vol. 5, pp 167 ff. |
| 14. | Bolm, C.; Schlingloff, G.; Harms, K. Chem. Ber. 1992, 125, 1191–1203. doi:10.1002/cber.19921250529 |
| 15. | Kwong, H.-L.; Yeung, H.-L.; Yeung, C.-T.; Lee, W.-S.; Lee, C.-S.; Wong, W.-L. Coord. Chem. Rev. 2007, 251, 2188–2222. doi:10.1016/j.ccr.2007.03.010 |
| 16. | Bolm, C.; Ewald, M.; Felder, M. Chem. Ber. 1992, 125, 1205–1215. doi:10.1002/cber.19921250530 |
| 17. | Bolm, C.; Ewald, M.; Zehnder, M.; Neuburger, M. A. Chem. Ber. 1992, 125, 453–458. doi:10.1002/cber.19921250224 |
| 18. | Trost, B. M.; Weiss, A. H. Adv. Synth. Catal. 2009, 351, 963–983. doi:10.1002/adsc.200800776 |
| 19. | Drury, W. J.; Zimmermann, N.; Keenan, M.; Hayashi, M.; Kaiser, S.; Goddard, R.; Pfaltz, A. Angew. Chem., Int. Ed. 2003, 116, 72–76. doi:10.1002/anie.200352755 |
| 20. | Roseblade, S. J.; Pfaltz, A. Acc. Chem. Res. 2007, 40, 1402–1411. doi:10.1021/ar700113g |
| 43. | Traverse, J. F.; Zhao, Y.; Hoveyda, A. H.; Snapper, M. L. Org. Lett. 2005, 7, 3151–3154. doi:10.1021/ol050814q |
| 13. | Lehn, J.-M. Supramolecular Chemistry - Concepts and Perspectives; VCH: Weinheim, 1995. |
| 44. | Barlos, K.; Papaioannou, D.; Theodoropoulos, D. J. Org. Chem. 1982, 47, 1324–1326. doi:10.1021/jo00346a031 |
| 11. | O'Hagan, D. Nat. Prod. Rep. 1997, 14, 637–651. doi:10.1039/NP9971400637 |
| 12. | Aida, W.; Ohtsuki, T.; Li, X.; Ishibashi, M. Tetrahedron 2009, 65, 369–373. doi:10.1016/j.tet.2008.10.040 |
| 40. | Eidamshaus, C.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 1162–1166. doi:10.1002/adsc.200800789 |
| 6. | Wess, G.; Kesseler, K.; Baader, E.; Bartmann, W.; Beck, G.; Bergmann, A.; Jendralla, H.; Bock, K.; Holzstein, O.; Kleine, H.; Schnierer, M. Tetrahedron Lett. 1990, 31, 2545–2548. doi:10.1016/0040-4039(90)80121-2 |
| 7. | Miyachi, N.; Yanagawa, Y.; Iwasaki, H.; Ohara, Y.; Hiyama, T. Tetrahedron Lett. 1993, 34, 8267–8270. doi:10.1016/S0040-4039(00)61407-7 |
| 8. | Beck, G. Synlett 2002, 837–850. doi:10.1055/s-2002-31890 |
| 9. | Beck, G.; Kesseler, K.; Baader, E.; Bartmann, W.; Bergmann, A.; Granzer, E.; Jendralla, H.; Von Kerekjarto, B.; Krause, R. J. Med. Chem. 1990, 33, 52–60. doi:10.1021/jm00163a010 |
| 10. | Cattaneo, D.; Baldelli, S.; Merlini, S.; Zenoni, S.; Perico, N.; Remuzzi, G. Expert Opin. Ther. Pat. 2004, 14, 1553–1566. doi:10.1517/13543776.14.11.1553 |
| 41. | Couty, F.; David, O.; Larmanjat, B.; Marrot, J. J. Org. Chem. 2007, 72, 1058–1061. doi:10.1021/jo062221e |
| 42. | Aureggi, V.; Frankevicius, V.; Kitching, M. O.; Ley, S. V.; Longbottom, D. A.; Oelke, A. J.; Sedelmeier, G. Org. Synth. 2008, 85, 72–87. |
| 29. | Flögel, O.; Dash, J.; Brüdgam, I.; Hartl, H.; Reissig, H.-U. Chem.–Eur. J. 2004, 10, 4283–4290. doi:10.1002/chem.200400322 |
| 30. | Dash, J.; Lechel, T.; Reissig, H.-U. Org. Lett. 2007, 9, 5541–5544. doi:10.1021/ol702468s |
| 31. | Lechel, T.; Dash, J.; Hommes, P.; Lentz, D.; Reissig, H.-U. J. Org. Chem. 2009, 75, 726–732. doi:10.1021/jo9022183 |
| 32. | Lechel, T.; Reissig, H.-U. Pure Appl. Chem. 2010, 82, 1835–1845. doi:10.1351/PAC-CON-09-09-06 |
| 33. | Brasholz, M.; Reissig, H.-U.; Zimmer, R. Acc. Chem. Res. 2009, 42, 45–56. doi:10.1021/ar800011h |
| 34. | Lechel, T.; Dash, J.; Brüdgam, I.; Reissig, H.-U. Eur. J. Org. Chem. 2008, 3647–3655. doi:10.1002/ejoc.200800398 |
| 37. | Bera, M. K.; Reissig, H.-U. Synthesis 2010, 2129–2138. doi:10.1055/s-0029-1218787 |
| 38. | Zimmer, R.; Lechel, T.; Rancan, G.; Bera, M. K.; Reissig, H.-U. Synlett 2010, 1793–1796. doi:10.1055/s-0030-1258088 |
| 39. | Lechel, T.; Dash, J.; Eidamshaus, C.; Brüdgam, I.; Lentz, D.; Reissig, H.-U. Org. Biomol. Chem. 2010, 8, 3007–3014. doi:10.1039/B925468D |
| 27. | Rahm, F.; Stranne, R.; Bremberg, U.; Nordstrom, K.; Cernerud, M.; Macedo, E.; Moberg, C. J. Chem. Soc., Perkin Trans. 1 2000, 1983–1990. doi:10.1039/b000269k |
| 28. | Rahm, F.; Fischer, A.; Moberg, C. Eur. J. Org. Chem. 2003, 4205–4215. doi:10.1002/ejoc.200300368 |
| 40. | Eidamshaus, C.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 1162–1166. doi:10.1002/adsc.200800789 |
| 22. | Movassaghi, M.; Hill, M. D.; Ahmad, O. K. J. Am. Chem. Soc. 2007, 129, 10096–10097. doi:10.1021/ja073912a |
| 23. | Liu, S.; Liebeskind, L. S. J. Am. Chem. Soc. 2008, 130, 6918–6919. doi:10.1021/ja8013743 |
| 24. | Manning, J. R.; Davies, H. M. L. J. Am. Chem. Soc. 2008, 130, 8602–8603. doi:10.1021/ja803139k |
| 25. | Xin, X.; Wang, Y.; Kumar, S.; Liu, X.; Lin, Y.; Dong, D. Org. Biomol. Chem. 2010, 8, 3078–3082. doi:10.1039/C001117G |
| 26. | Hill, M. D. Chem.–Eur. J. 2010, 16, 12052–12062. doi:10.1002/chem.201001100 |
| 48. | Massad, S. K.; Hawkins, L. D.; Baker, D. C. J. Org. Chem. 1983, 48, 5180–5182. doi:10.1021/jo00174a006 |
| 35. | Zimmer, R.; Reissig, H.-U. Donor-Substituted Allenes. In Modern Allene Chemistry; Krause, N.; Hashmi, A. S. K., Eds.; Wiley-VCH: Weinheim, 2004; pp 425–492. |
| 36. | Reissig, H.-U.; Zimmer, R. Cumulenes and Allenes: Synthesis from Other Allenes. In Science of Synthesis; Krause, N., Ed.; Thieme: Stuttgart, 2007; Vol. 44, pp 301–352. |
© 2011 Eidamshaus et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)