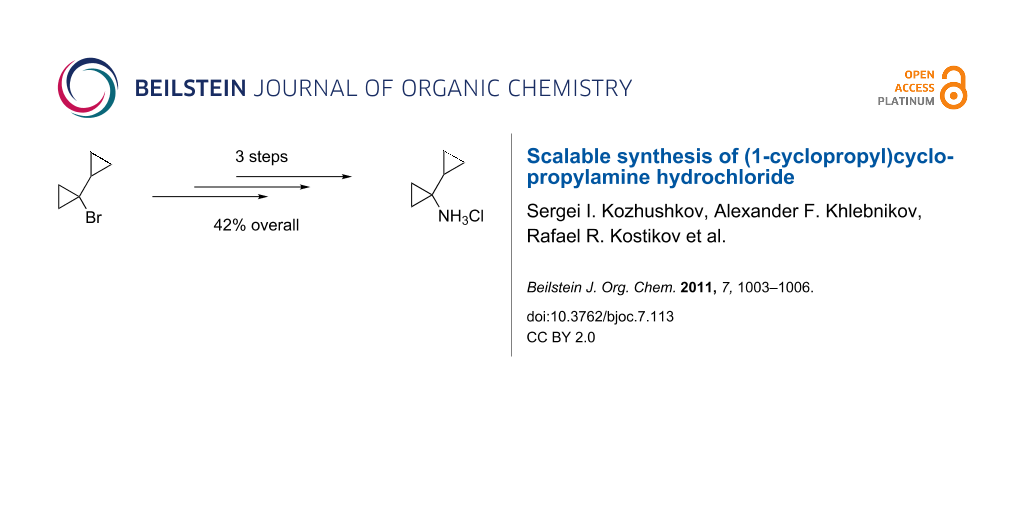
Abstract
1-Cyclopropylcyclopropanecarboxylic acid (2), which is accessible on a large scale (900 mmol) from 1-bromo-1-cyclopropylcyclopropane (1) in 64% yield (89% on a 12.4 mmol scale), has been subjected to a Curtius degradation employing the Weinstock protocol to furnish the N-Boc-protected (1-cyclopropyl)cyclopropylamine 3 (76%). Deprotection of 3 with hydrogen chloride in diethyl ether gave the (1-cyclopropyl)cyclopropylamine hydrochloride (4·HCl) in 87% yield.

Graphical Abstract
Introduction
Several recent patent applications have stirred an increasing interest in research departments of pharmaceutical and agrochemical companies concerning 1- and 2-substituted 1,1'-bicyclopropyl derivatives. Among them, intermediates containing a (1-cyclopropyl)cyclopropylamine moiety appear to be particularly important and desirable for the preparation of biologically active and pharmacologically relevant compounds. For example, a number of derivatives of (1-cyclopropyl)cyclopropylamine (4) have been found to be useful variously for the treatment of hepatitis C [3,4], as pest control agents [5], as inhibitors of methicillin-resistant Staphylococcus aureus [5], as pesticides, insecticides and acaricides [7-13] and more. This amine has been prepared from cyclopropyl cyanide [3-13] by application of the Szymoniak–Kulinkovich reductive cyclopropanation procedure [14,15]. In our hands, however, this patented protocol [3-13] provided poor yields (15–20%) of impure 4 [16], which had to be purified by conversion to the corresponding tert-butyl carbamate and subsequent column chromatography. Thus, this procedure was not easily scalable to 10–50 g quantities. To meet such demands, we have developed an alternative route to 4 from the easily available corresponding carboxylic acid 2 [17,18] by Curtius degradation [19,20].
Results and Discussion
Preparation of the acid 2 from the known 1-bromo-1-cyclopropylcyclopropane (1) [21,22] according to the published procedure [17] was accomplished on a 100 g scale (Scheme 1). However, the yield of the carboxylation on a scale of 12.4 mmol, 900 mmol and 1400 mmol, was 89, 64 and 62%, respectively. This is associated with the longer reaction time employed on a larger scale, during which the intermediate 1-cyclopropyl-1-lithiocyclopropane may be trapped by the by-product tert-butyl bromide, leading to isobutene by dehydrobromination [23,24]. Indeed, the reaction on a 200 mmol scale, but over a period of 3 h, furnished 2 in 46% yield only. According to previous experience, this undesired side reaction can be suppressed by employing two equivalents of tert-butyllithium [23]. Thus, the yield of 2 may be improved even for large scale preparation.
![[1860-5397-7-113-i1]](/bjoc/content/inline/1860-5397-7-113-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of 1-(cyclopropyl)cyclopropylamine hydrochloride (4·HCl).
Scheme 1: Preparation of 1-(cyclopropyl)cyclopropylamine hydrochloride (4·HCl).
Curtius degradation of the acid 2 via the corresponding azide, according to the Weinstock protocol [19,20] as previously employed in different examples [2,25], furnished the N-Boc-protected (1-cyclopropyl)cyclopropylamine 3 in 76% yield. It was essential to carefully dry the solution of the intermediate azide, otherwise the yield of 3 dropped dramatically, and the desired product was accompanied by 1,3-di(bicyclopropyl)urea (5) in up to 50% yield (Scheme 1). The structure of the latter was confirmed by an X-ray crystal structure analysis (Figure 1) [26].
![[1860-5397-7-113-1]](/bjoc/content/figures/1860-5397-7-113-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of 1,3-di(bicyclopropyl)urea (5) in the crystal [26].
Figure 1: Structure of 1,3-di(bicyclopropyl)urea (5) in the crystal [26].
The carbamate 3 was deprotected by treatment with hydrogen chloride in diethyl ether affording the amine hydrochloride 4·HCl in 87% yield. The latter was thus obtained from 1-bromo-1-cyclopropylcyclopropane (1) on a scale of 50 g in 42% overall yield (Scheme 1).
Conclusion
The newly developed procedure allows the preparation of 1-(cyclopropyl)cyclopropylamine (4) in five steps from commercially available methyl cyclopropanecarboxylate, reproducibly, on a 50 g and even larger scale. In this respect it is superior to the previously published and patented access to 4 from cyclopropanecarbonitrile, which in the hands of five different researchers in our laboratory required chromatographic separation of the intermediately prepared N-Boc derivative, which involved the rather costly di-tert-butyl pyrocarbonate and made that an overall three-step procedure.
Experimental
1H and 13C NMR spectra were recorded at 300 MHz [1H] and 62.9 MHz [13C, additional DEPT (Distortionless Enhancement by Polarization Transfer)] on Bruker AM 250 and Varian Mercury Vx300 instruments in CDCl3 and D2O solutions, CHCl3/CDCl3 and DHO as internal references. EI-MS, ESI-MS and HRMS spectra were measured with Finnigan MAT 95 (70 eV), Finnigan LCQ and Bruker Daltonic APEX IV 7T FTICR instruments, respectively. Melting points were determined on a Büchi 510 capillary melting point apparatus, values are uncorrected. TLC analyses were performed on precoated sheets (0.25 mm Sil G/UV254) from Macherey-Nagel). All chemicals were used as received. 1-Bromo-1-cyclopropylcyclopropane (1) was obtained according to the previously published procedure [21]. A 5.0 N solution of HCl in Et2O was prepared by saturation of anhydrous Et2O with gaseous HCl at 0 °C. Anhydrous diethyl ether was obtained by distillation from sodium benzophenone ketyl, acetone by distillation from anhydrous potassium carbonate. Anhydrous tert-butyl alcohol was obtained employing molecular sieves (4 Å) [27]. Organic extracts were dried over MgSO4. All reactions in anhydrous solvents were carried out under an argon atmosphere in flame-dried glassware.
Synthesis of 1-cyclopropylcyclopropanecarboxylic acid (2)
Under mechanical stirring and cooling with pentane/liq. N2, a solution of t-BuLi (1.7 M in pentane, 560 mL, 952.0 mmol) was added dropwise to a solution of 1-bromo-1-cyclopropylcyclopropane (1) (146.0 g, 907.0 mmol) in anhydrous Et2O (2.2 L) at −78 °C within 40 min. After stirring at −78 °C for an additional 25 min, an excess of dry ice was added in several portions (T ≤ −70 °C), and the mixture was allowed to slowly warm up to ambient temperature during a period of 2 h. The reaction was quenched with an ice-cold solution of KOH (60.0 g, 1.070 mol) in H2O (1 L), the aqueous layer was washed with ether (3 × 100 mL), and then acidified with conc. aq. HCl solution at 0–5 °C (ca. 175 mL). The resulting mixture was extracted with ether (4 × 300 mL), the combined organic phases were dried and concentrated under reduced pressure to give the acid 2 (73.2 g, 64%) as colorless crystals, mp 50–51 °C (lit. [17]: mp: 51–52 °C), which was used in the next step without further purification. Its NMR spectra were identical to the published ones [17].
Synthesis of tert-butyl 1-(cyclopropyl)cyclopropylcarbamate (3)
To a mechanically stirred solution of the acid 2 (70.60 g, ca. 560.0 mmol) in anhydrous acetone (1.7 L), was added Et3N (76.2 g, 105.0 mL, 753.0 mmol) dropwise at −5 °C. After additional stirring at this temperature for 15 min, neat ethyl chloroformate (103.7 g, 91.0 mL, 956.0 mmol) was added at the same temperature over a period of 30 min, and the resulting mixture was stirred at this temperature for an additional 2 h. Then a solution of NaN3 (75.0 g, 1.0 mol) in H2O (200 mL) was added over a period of 1.5 h. The reaction mixture was stirred at 0 °C for 1.5 h, concentrated under reduced pressure at 0 °C to about a half of the original volume, poured into ice-cold water (2 L), and the mixture extracted with diethyl ether (4 × 400 ml) and pentane (2 × 350 ml). The combined organic solutions were washed with ice-cold water (2 × 400 mL), dried under stirring with MgSO4 at 0 °C for 1 h and concentrated under reduced pressure at 0 °C/20–30 Torr. The residue was taken up with pentane (300 mL), dried and concentrated under the same conditions. It was then dissolved in anhydrous t-BuOH (200 mL), and this solution was added dropwise to anhydrous t-BuOH (1300 mL) kept at 80 °C under vigorous stirring over a period of 2.5 h. The resulting solution was heated under reflux for an additional 9 h. The main volume of t-BuOH (ca. 1300 mL) was distilled off under ambient pressure in a nitrogen flow. After cooling, the residue mixture was dried at 20 °C/0.1 Torr to give essentially pure carbamate 3 (84.0 g, 76%) as a colorless solid, mp 69–70 °C, Rf 0.38 (hexane/Et2O 5:1), which was used in the next step without further purification. 1H NMR (300 MHz, CDCl3) δ 4.91 (br s, 1H, NH), 1.39 (s, 9H, 3 CH3), 1.30–1.20 (br m, 1H, cPr-H), 0.64–0.57 (br m, 2H, cPr-H), 0.52–0.45 (br m, 2H, cPr-H), 0.37–0.31 (m, 2H, cPr-H), 0.09–0.04 (m, 2H, cPr-H); 13C NMR (62.9 MHz, CDCl3) δ 155.2 (C), 79.0 (C), 34.1 (C), 28.3 (3 CH3), 15.6 (CH), 11.9 (2 CH2), 2.6 (2 CH2); EIMS (70 eV) m/z: 141 (M+ − C4H8), 126, 96, 82, 58, 57, 43; HRMS–ESI (m/z): calcd for C11H19NNaO2, 220.1308; found, 220.1314.
Synthesis of (1-cyclopropyl)cyclopropylamine hydrochloride (4·HCl)
Under stirring, a solution of the carbamate 3 (84.0 g, 425.8 mmol) in Et2O (100 mL) was added to a ca. 5.0 N HCl solution in Et2O (700 mL) in one portion at 0 °C. The reaction mixture was stirred at 0 °C for 4 h and at ambient temperature for 20 h. The formed precipitate was filtered off, washed with Et2O (200 mL) and dried in a vacuum desiccator over P4O10 overnight to give 4·HCl (49.7 g, 87%) as a colorless powder, which slowly decomposes above ca. 135 °C and melts at 196–198 °C (dec.); 1H NMR (300 MHz, D2O) δ 1.30–1.26 (m, 1H, cPr-H), 0.71–0.60 and 0.60–0.55 (m AA'BB', 4H, cPr-H), 0.49–0.42 and 0.13–0.08 (m AA'BB', 4H, cPr-H).
When a solution of the intermediate azide in the preparation of 3 was not sufficiently dried, the thermolysis in t-BuOH along with tert-butylcarbamate 3 gave the 1,3-di(bicyclopropyl)urea (5) in up to 50% yield. Compound 5 was isolated as a colorless solid after deprotection of 3 with HCl/Et2O by evaporation of the mother liquor followed by recrystallization of the residue from hexane/CHCl3; mp 159–161 °C. The structure of 5 was confirmed by X-ray crystal structure analysis [26]. 5: 1H NMR (300 MHz, CDCl3) δ 5.21 (br s, 2H, NH), 1.28–1.16 (m, 2H, 2 CH cPr-H), 0.73–0.61 (m AA'BB', 8H, 4 CH2, cPr-H), 0.44–0.41 and 0.17–0.13 (m AA'BB', 8H, 4 CH2, cPr-H); 13C NMR (62.9 MHz, CDCl3) δ 158.8 (C), 33.9 (2 C), 15.5 (2 CH), 12.6 (2 CH2), 2.8 (6 CH2); EIMS (70 eV) m/z: 219 (M+ − H), 205 (M+ − H−CH2), 191 (M+ − H−C2H4), 124 (M+ − H−NC6H9), 96 (M+ − H−NC6H9−CO), 82 (M+ − H−NC6H9−CH2−CO).
References
-
de Meijere, A.; Kozhushkov, S. I. Mendeleev Commun. 2010, 20, 301–311. doi:10.1016/j.mencom.2010.11.001
-
Kozhushkov, S. I.; Wagner-Gillen, K.; Khlebnikov, A. F.; de Meijere, A. Synthesis 2010, 3967–3973. doi:10.1055/s-0030-1258964
Return to citation in text: [1] -
Pracitto, R.; Kadow, J. F.; Bender, J. A.; Beno, B. R.; Grant-Young, K. A.; Han, Y.; Hewawasam, P.; Nickel, A.; Parcella, K. E.; Yeung, K.-S.; Chupak, L. S. Compounds for the treatment of hepatitis C. U.S. Pat. Appl. 2010/0063068 A1, March 11, 2010.
Chem. Abstr. 2010, 152, 335190.
Return to citation in text: [1] [2] [3] -
Pracitto, R.; Kadow, J. F.; Bender, J. A.; Beno, B. R.; Grant-Young, K. A.; Han, Y.; Hewawasam, P.; Nickel, A.; Parcella, K. E.; Yeung, K.-S.; Chupak, L. S. Compounds for the treatment of hepatitis C. U.S. Pat. Appl. 2010/0184800 A1, July 22, 2010.
Chem. Abstr. 2010, 153, 204354.
Return to citation in text: [1] [2] [3] -
Mita, T.; Furukawa, Y.; Iwasa, M.; Komoda, M. Isoxazoline-substituted benzamide compound and pest control agent. WO Pat. Appl. 2008/108448 A1, Sept 12, 2008.
Chem. Abstr. 2008, 149, 355887.
Return to citation in text: [1] [2] [3] [4] -
Chen, C.-L.; Clark, R. B.; Deng, Y.; He, M.; Plamondon, L.; Sun, C.; Xiao, X.-Y. Tetracycline compounds. WO Pat. Appl. 2010/129057 A2, Nov 11, 2010.
Chem. Abstr. 2010, 153, 600600.
Return to citation in text: [1] [2] -
Vettiger, T.; Stoller, A.; Jackson, D. A. Process for the preparation of anthranilamide derivatives. WO Pat. Appl. 2008/031548 A1, March 20, 2008.
Chem. Abstr. 2008, 148, 355784.
Return to citation in text: [1] [2] [3] -
Vettiger, T.; Stoller, A.; Jackson, D. A. Process for the preparation of anthranilamide derivatives. WO Pat. Appl. 2008/031549 A1, March 20, 2008.
Chem. Abstr. 2008, 148, 355782.
Return to citation in text: [1] [2] [3] -
Loiseleur, O.; Hall, R. G.; Stoller, A. D.; Graig, G. W.; Jeanguenat, A.; Edmunds, A. Condensed anthranilamide derivatives as insecticides. WO Pat. Appl 2009/024341 A2, Feb 26, 2009.
Chem. Abstr. 2009, 150, 260188.
Return to citation in text: [1] [2] [3] -
Hughes, D. J.; Peace, J. E.; Riley, S.; Russell, S.; Swanborough, J. J.; Jeanguenat, A.; Renold, P.; Hall, R. G.; Loiseleur, O.; Trah, S.; Wenger, J. Pesticidal mixtures containing bicyclic anthranilamides. WO Pat. Appl. 2007/009661 A2, Jan 25, 2007.
Chem. Abstr. 2007, 146, 178834.
Return to citation in text: [1] [2] [3] -
Loiseleur, O.; Durieux, P.; Trah, S.; Edmunds, A.; Jeanguenat, A.; Stoller, A.; Hughes, D. J. Pesticides containing a bicyclic bisamide structure. WO Pat. Appl. 2007/093402 A1, Aug 23, 2007.
Chem. Abstr. 2007, 147, 301166.
Return to citation in text: [1] [2] [3] -
Jeanguenat, A.; O'Sullivan, A. C.; Mühlebach, M.; Trah, S.; Hall, R. G. Cyano anthranilamide insecticides. WO Pat. Appl. 2006/111341 A1, Oct 26, 2006.
Chem. Abstr. 2006, 145, 455006.
Return to citation in text: [1] [2] [3] -
O'Sullivan, A. C.; Hughes, D.; Jeanguenat, A.; Mühlebach, M.; Loiseleur, O. Heterocyclic diamide insecticidal agents. WO Pat. Appl. 2006/040113 A2, April 20, 2006.
Chem. Abstr. 2006, 144, 364548.
Return to citation in text: [1] [2] [3] -
Bertus, P.; Szymoniak, J. Chem. Commun. 2001, 1792–1793. doi:10.1039/B105293B
Return to citation in text: [1] -
Bertus, P.; Szymoniak, J. Synlett 2007, 1346–1356. doi:10.1055/s-2007-980342
Return to citation in text: [1] -
According to the SciFinder data bank, compound 4·HCl is commercially available from ChemBridge Corporation and other suppliers, however, at a price of 475 USD/g.
Return to citation in text: [1] -
de Meijere, A.; Khlebnikov, A. F.; Sünnemann, H. W.; Frank, D.; Rauch, K.; Yufit, D. S. Eur. J. Org. Chem. 2010, 3295–3301. doi:10.1002/ejoc.201000209
Return to citation in text: [1] [2] [3] [4] -
Kirmse, W.; Rode, J.; Rode, K. Chem. Ber. 1986, 119, 3672–3693. doi:10.1002/cber.19861191214
Return to citation in text: [1] -
Weinstock, J. J. Org. Chem. 1961, 26, 3511. doi:10.1021/jo01067a604
Return to citation in text: [1] [2] -
Jendralla, H. Chem. Ber. 1980, 113, 3585–3596. doi:10.1002/cber.19801131116
Return to citation in text: [1] [2] -
de Meijere, A.; Kozhushkov, S. I.; Späth, T. Org. Synth. 2002, 78, 142–151.
Org. Synth. 2004, Coll. Vol. 10, 88–97.
Return to citation in text: [1] [2] -
de Meijere, A.; Kozhushkov, S. I.; Spaeth, T.; Zefirov, N. S. J. Org. Chem. 1993, 58, 502–505. doi:10.1021/jo00054a040
Return to citation in text: [1] -
Seebach, D.; Neumann, H. Chem. Ber. 1974, 107, 847–853. doi:10.1002/cber.19741070311
Return to citation in text: [1] [2] -
Bailey, W. F.; Patricia, J. J. J. Organomet. Chem. 1988, 352, 1–46. doi:10.1016/0022-328X(88)83017-1
Return to citation in text: [1] -
Brandl, M.; Kozhushkov, S. I.; Yufit, D. S.; Howard, J. A. K.; de Meijere, A. Eur. J. Org. Chem. 1998, 2785–2795. doi:10.1002/(SICI)1099-0690(199812)1998:12<2785::AID-EJOC2785>3.0.CO;2-R
Return to citation in text: [1] -
CCDC-828827 (5) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB21EZ, UK; fax: (+44)1223-336-033; or deposit@ccdc.cam.ac.uk).
Return to citation in text: [1] [2] [3] -
Williams, D. B. G.; Lawton, M. J. Org. Chem. 2010, 75, 8351–8354. doi:10.1021/jo101589h
Return to citation in text: [1]
| 21. |
de Meijere, A.; Kozhushkov, S. I.; Späth, T. Org. Synth. 2002, 78, 142–151.
Org. Synth. 2004, Coll. Vol. 10, 88–97. |
| 26. | CCDC-828827 (5) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB21EZ, UK; fax: (+44)1223-336-033; or deposit@ccdc.cam.ac.uk). |
| 26. | CCDC-828827 (5) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB21EZ, UK; fax: (+44)1223-336-033; or deposit@ccdc.cam.ac.uk). |
| 3. |
Pracitto, R.; Kadow, J. F.; Bender, J. A.; Beno, B. R.; Grant-Young, K. A.; Han, Y.; Hewawasam, P.; Nickel, A.; Parcella, K. E.; Yeung, K.-S.; Chupak, L. S. Compounds for the treatment of hepatitis C. U.S. Pat. Appl. 2010/0063068 A1, March 11, 2010.
Chem. Abstr. 2010, 152, 335190. |
| 4. |
Pracitto, R.; Kadow, J. F.; Bender, J. A.; Beno, B. R.; Grant-Young, K. A.; Han, Y.; Hewawasam, P.; Nickel, A.; Parcella, K. E.; Yeung, K.-S.; Chupak, L. S. Compounds for the treatment of hepatitis C. U.S. Pat. Appl. 2010/0184800 A1, July 22, 2010.
Chem. Abstr. 2010, 153, 204354. |
| 3. |
Pracitto, R.; Kadow, J. F.; Bender, J. A.; Beno, B. R.; Grant-Young, K. A.; Han, Y.; Hewawasam, P.; Nickel, A.; Parcella, K. E.; Yeung, K.-S.; Chupak, L. S. Compounds for the treatment of hepatitis C. U.S. Pat. Appl. 2010/0063068 A1, March 11, 2010.
Chem. Abstr. 2010, 152, 335190. |
| 4. |
Pracitto, R.; Kadow, J. F.; Bender, J. A.; Beno, B. R.; Grant-Young, K. A.; Han, Y.; Hewawasam, P.; Nickel, A.; Parcella, K. E.; Yeung, K.-S.; Chupak, L. S. Compounds for the treatment of hepatitis C. U.S. Pat. Appl. 2010/0184800 A1, July 22, 2010.
Chem. Abstr. 2010, 153, 204354. |
| 5. |
Mita, T.; Furukawa, Y.; Iwasa, M.; Komoda, M. Isoxazoline-substituted benzamide compound and pest control agent. WO Pat. Appl. 2008/108448 A1, Sept 12, 2008.
Chem. Abstr. 2008, 149, 355887. |
| 6. |
Chen, C.-L.; Clark, R. B.; Deng, Y.; He, M.; Plamondon, L.; Sun, C.; Xiao, X.-Y. Tetracycline compounds. WO Pat. Appl. 2010/129057 A2, Nov 11, 2010.
Chem. Abstr. 2010, 153, 600600. |
| 7. |
Vettiger, T.; Stoller, A.; Jackson, D. A. Process for the preparation of anthranilamide derivatives. WO Pat. Appl. 2008/031548 A1, March 20, 2008.
Chem. Abstr. 2008, 148, 355784. |
| 8. |
Vettiger, T.; Stoller, A.; Jackson, D. A. Process for the preparation of anthranilamide derivatives. WO Pat. Appl. 2008/031549 A1, March 20, 2008.
Chem. Abstr. 2008, 148, 355782. |
| 9. |
Loiseleur, O.; Hall, R. G.; Stoller, A. D.; Graig, G. W.; Jeanguenat, A.; Edmunds, A. Condensed anthranilamide derivatives as insecticides. WO Pat. Appl 2009/024341 A2, Feb 26, 2009.
Chem. Abstr. 2009, 150, 260188. |
| 10. |
Hughes, D. J.; Peace, J. E.; Riley, S.; Russell, S.; Swanborough, J. J.; Jeanguenat, A.; Renold, P.; Hall, R. G.; Loiseleur, O.; Trah, S.; Wenger, J. Pesticidal mixtures containing bicyclic anthranilamides. WO Pat. Appl. 2007/009661 A2, Jan 25, 2007.
Chem. Abstr. 2007, 146, 178834. |
| 11. |
Loiseleur, O.; Durieux, P.; Trah, S.; Edmunds, A.; Jeanguenat, A.; Stoller, A.; Hughes, D. J. Pesticides containing a bicyclic bisamide structure. WO Pat. Appl. 2007/093402 A1, Aug 23, 2007.
Chem. Abstr. 2007, 147, 301166. |
| 12. |
Jeanguenat, A.; O'Sullivan, A. C.; Mühlebach, M.; Trah, S.; Hall, R. G. Cyano anthranilamide insecticides. WO Pat. Appl. 2006/111341 A1, Oct 26, 2006.
Chem. Abstr. 2006, 145, 455006. |
| 13. |
O'Sullivan, A. C.; Hughes, D.; Jeanguenat, A.; Mühlebach, M.; Loiseleur, O. Heterocyclic diamide insecticidal agents. WO Pat. Appl. 2006/040113 A2, April 20, 2006.
Chem. Abstr. 2006, 144, 364548. |
| 19. | Weinstock, J. J. Org. Chem. 1961, 26, 3511. doi:10.1021/jo01067a604 |
| 20. | Jendralla, H. Chem. Ber. 1980, 113, 3585–3596. doi:10.1002/cber.19801131116 |
| 7. |
Vettiger, T.; Stoller, A.; Jackson, D. A. Process for the preparation of anthranilamide derivatives. WO Pat. Appl. 2008/031548 A1, March 20, 2008.
Chem. Abstr. 2008, 148, 355784. |
| 8. |
Vettiger, T.; Stoller, A.; Jackson, D. A. Process for the preparation of anthranilamide derivatives. WO Pat. Appl. 2008/031549 A1, March 20, 2008.
Chem. Abstr. 2008, 148, 355782. |
| 9. |
Loiseleur, O.; Hall, R. G.; Stoller, A. D.; Graig, G. W.; Jeanguenat, A.; Edmunds, A. Condensed anthranilamide derivatives as insecticides. WO Pat. Appl 2009/024341 A2, Feb 26, 2009.
Chem. Abstr. 2009, 150, 260188. |
| 10. |
Hughes, D. J.; Peace, J. E.; Riley, S.; Russell, S.; Swanborough, J. J.; Jeanguenat, A.; Renold, P.; Hall, R. G.; Loiseleur, O.; Trah, S.; Wenger, J. Pesticidal mixtures containing bicyclic anthranilamides. WO Pat. Appl. 2007/009661 A2, Jan 25, 2007.
Chem. Abstr. 2007, 146, 178834. |
| 11. |
Loiseleur, O.; Durieux, P.; Trah, S.; Edmunds, A.; Jeanguenat, A.; Stoller, A.; Hughes, D. J. Pesticides containing a bicyclic bisamide structure. WO Pat. Appl. 2007/093402 A1, Aug 23, 2007.
Chem. Abstr. 2007, 147, 301166. |
| 12. |
Jeanguenat, A.; O'Sullivan, A. C.; Mühlebach, M.; Trah, S.; Hall, R. G. Cyano anthranilamide insecticides. WO Pat. Appl. 2006/111341 A1, Oct 26, 2006.
Chem. Abstr. 2006, 145, 455006. |
| 13. |
O'Sullivan, A. C.; Hughes, D.; Jeanguenat, A.; Mühlebach, M.; Loiseleur, O. Heterocyclic diamide insecticidal agents. WO Pat. Appl. 2006/040113 A2, April 20, 2006.
Chem. Abstr. 2006, 144, 364548. |
| 2. | Kozhushkov, S. I.; Wagner-Gillen, K.; Khlebnikov, A. F.; de Meijere, A. Synthesis 2010, 3967–3973. doi:10.1055/s-0030-1258964 |
| 25. | Brandl, M.; Kozhushkov, S. I.; Yufit, D. S.; Howard, J. A. K.; de Meijere, A. Eur. J. Org. Chem. 1998, 2785–2795. doi:10.1002/(SICI)1099-0690(199812)1998:12<2785::AID-EJOC2785>3.0.CO;2-R |
| 5. |
Mita, T.; Furukawa, Y.; Iwasa, M.; Komoda, M. Isoxazoline-substituted benzamide compound and pest control agent. WO Pat. Appl. 2008/108448 A1, Sept 12, 2008.
Chem. Abstr. 2008, 149, 355887. |
| 23. | Seebach, D.; Neumann, H. Chem. Ber. 1974, 107, 847–853. doi:10.1002/cber.19741070311 |
| 24. | Bailey, W. F.; Patricia, J. J. J. Organomet. Chem. 1988, 352, 1–46. doi:10.1016/0022-328X(88)83017-1 |
| 5. |
Mita, T.; Furukawa, Y.; Iwasa, M.; Komoda, M. Isoxazoline-substituted benzamide compound and pest control agent. WO Pat. Appl. 2008/108448 A1, Sept 12, 2008.
Chem. Abstr. 2008, 149, 355887. |
| 23. | Seebach, D.; Neumann, H. Chem. Ber. 1974, 107, 847–853. doi:10.1002/cber.19741070311 |
| 17. | de Meijere, A.; Khlebnikov, A. F.; Sünnemann, H. W.; Frank, D.; Rauch, K.; Yufit, D. S. Eur. J. Org. Chem. 2010, 3295–3301. doi:10.1002/ejoc.201000209 |
| 18. | Kirmse, W.; Rode, J.; Rode, K. Chem. Ber. 1986, 119, 3672–3693. doi:10.1002/cber.19861191214 |
| 21. |
de Meijere, A.; Kozhushkov, S. I.; Späth, T. Org. Synth. 2002, 78, 142–151.
Org. Synth. 2004, Coll. Vol. 10, 88–97. |
| 22. | de Meijere, A.; Kozhushkov, S. I.; Spaeth, T.; Zefirov, N. S. J. Org. Chem. 1993, 58, 502–505. doi:10.1021/jo00054a040 |
| 17. | de Meijere, A.; Khlebnikov, A. F.; Sünnemann, H. W.; Frank, D.; Rauch, K.; Yufit, D. S. Eur. J. Org. Chem. 2010, 3295–3301. doi:10.1002/ejoc.201000209 |
| 16. | According to the SciFinder data bank, compound 4·HCl is commercially available from ChemBridge Corporation and other suppliers, however, at a price of 475 USD/g. |
| 17. | de Meijere, A.; Khlebnikov, A. F.; Sünnemann, H. W.; Frank, D.; Rauch, K.; Yufit, D. S. Eur. J. Org. Chem. 2010, 3295–3301. doi:10.1002/ejoc.201000209 |
| 26. | CCDC-828827 (5) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB21EZ, UK; fax: (+44)1223-336-033; or deposit@ccdc.cam.ac.uk). |
| 3. |
Pracitto, R.; Kadow, J. F.; Bender, J. A.; Beno, B. R.; Grant-Young, K. A.; Han, Y.; Hewawasam, P.; Nickel, A.; Parcella, K. E.; Yeung, K.-S.; Chupak, L. S. Compounds for the treatment of hepatitis C. U.S. Pat. Appl. 2010/0063068 A1, March 11, 2010.
Chem. Abstr. 2010, 152, 335190. |
| 4. |
Pracitto, R.; Kadow, J. F.; Bender, J. A.; Beno, B. R.; Grant-Young, K. A.; Han, Y.; Hewawasam, P.; Nickel, A.; Parcella, K. E.; Yeung, K.-S.; Chupak, L. S. Compounds for the treatment of hepatitis C. U.S. Pat. Appl. 2010/0184800 A1, July 22, 2010.
Chem. Abstr. 2010, 153, 204354. |
| 5. |
Mita, T.; Furukawa, Y.; Iwasa, M.; Komoda, M. Isoxazoline-substituted benzamide compound and pest control agent. WO Pat. Appl. 2008/108448 A1, Sept 12, 2008.
Chem. Abstr. 2008, 149, 355887. |
| 6. |
Chen, C.-L.; Clark, R. B.; Deng, Y.; He, M.; Plamondon, L.; Sun, C.; Xiao, X.-Y. Tetracycline compounds. WO Pat. Appl. 2010/129057 A2, Nov 11, 2010.
Chem. Abstr. 2010, 153, 600600. |
| 7. |
Vettiger, T.; Stoller, A.; Jackson, D. A. Process for the preparation of anthranilamide derivatives. WO Pat. Appl. 2008/031548 A1, March 20, 2008.
Chem. Abstr. 2008, 148, 355784. |
| 8. |
Vettiger, T.; Stoller, A.; Jackson, D. A. Process for the preparation of anthranilamide derivatives. WO Pat. Appl. 2008/031549 A1, March 20, 2008.
Chem. Abstr. 2008, 148, 355782. |
| 9. |
Loiseleur, O.; Hall, R. G.; Stoller, A. D.; Graig, G. W.; Jeanguenat, A.; Edmunds, A. Condensed anthranilamide derivatives as insecticides. WO Pat. Appl 2009/024341 A2, Feb 26, 2009.
Chem. Abstr. 2009, 150, 260188. |
| 10. |
Hughes, D. J.; Peace, J. E.; Riley, S.; Russell, S.; Swanborough, J. J.; Jeanguenat, A.; Renold, P.; Hall, R. G.; Loiseleur, O.; Trah, S.; Wenger, J. Pesticidal mixtures containing bicyclic anthranilamides. WO Pat. Appl. 2007/009661 A2, Jan 25, 2007.
Chem. Abstr. 2007, 146, 178834. |
| 11. |
Loiseleur, O.; Durieux, P.; Trah, S.; Edmunds, A.; Jeanguenat, A.; Stoller, A.; Hughes, D. J. Pesticides containing a bicyclic bisamide structure. WO Pat. Appl. 2007/093402 A1, Aug 23, 2007.
Chem. Abstr. 2007, 147, 301166. |
| 12. |
Jeanguenat, A.; O'Sullivan, A. C.; Mühlebach, M.; Trah, S.; Hall, R. G. Cyano anthranilamide insecticides. WO Pat. Appl. 2006/111341 A1, Oct 26, 2006.
Chem. Abstr. 2006, 145, 455006. |
| 13. |
O'Sullivan, A. C.; Hughes, D.; Jeanguenat, A.; Mühlebach, M.; Loiseleur, O. Heterocyclic diamide insecticidal agents. WO Pat. Appl. 2006/040113 A2, April 20, 2006.
Chem. Abstr. 2006, 144, 364548. |
| 27. | Williams, D. B. G.; Lawton, M. J. Org. Chem. 2010, 75, 8351–8354. doi:10.1021/jo101589h |
| 14. | Bertus, P.; Szymoniak, J. Chem. Commun. 2001, 1792–1793. doi:10.1039/B105293B |
| 15. | Bertus, P.; Szymoniak, J. Synlett 2007, 1346–1356. doi:10.1055/s-2007-980342 |
| 19. | Weinstock, J. J. Org. Chem. 1961, 26, 3511. doi:10.1021/jo01067a604 |
| 20. | Jendralla, H. Chem. Ber. 1980, 113, 3585–3596. doi:10.1002/cber.19801131116 |
| 17. | de Meijere, A.; Khlebnikov, A. F.; Sünnemann, H. W.; Frank, D.; Rauch, K.; Yufit, D. S. Eur. J. Org. Chem. 2010, 3295–3301. doi:10.1002/ejoc.201000209 |
© 2011 Kozhushkov et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)