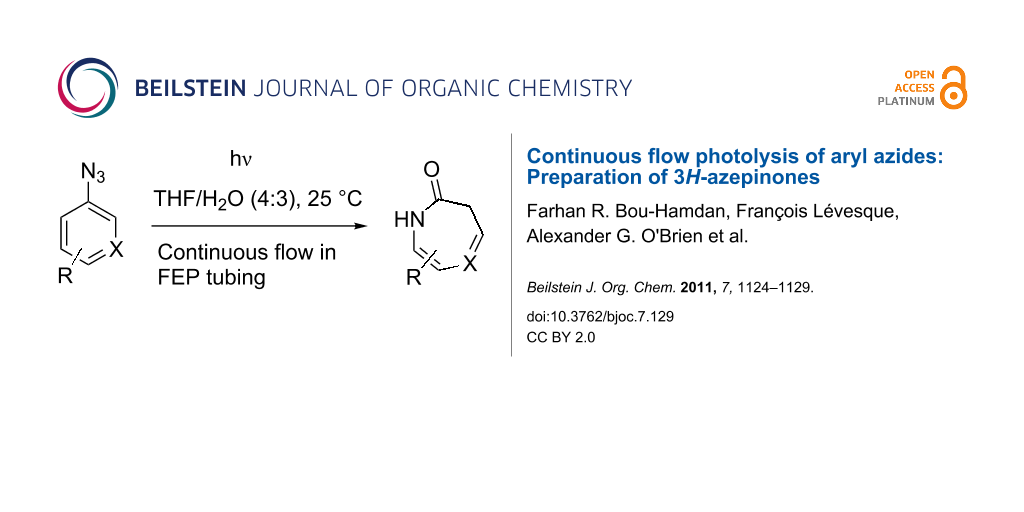
Abstract
Photolysis of aryl azides to give nitrenes, and their subsequent rearrangement in the presence of water to give 3H-azepinones, is performed in continuous flow in a photoreactor constructed of fluorinated ethylene polymer (FEP) tubing. Fine tuning of the reaction conditions using the flow reactor allowed minimization of secondary photochemical reactions.

Graphical Abstract
Findings
Although photochemical rearrangements are an important class of reactions for heterocycle synthesis [1,2], their use is often hindered by technical difficulties, both in research laboratories and in industry, particularly when large quantities of material are required [3,4]. In response, there has been a recent growth in the application of continuous flow techniques [5-7] for the deployment of photochemical reactions [8-13]. As, from the Beer–Lambert law, the intensity of light decreases exponentially with increasing distance from the light source, minimization of the path length in a continuous flow photoreactor ensures efficient and uniform irradiation of the sample [14], and scaling up is simple to achieve by running the reactor over an extended period. Additionally, the precise control of the reaction conditions and the continuous removal of products inherent in flow systems can offer improved yields and selectivities [8,15].
Nitrenes generated by aryl azide photolysis are important tools, both for preparative heterocycle synthesis [16,17] and for photoaffinity labeling of proteins [18-20]. The photolysis of aryl azide 1 [21], a well-studied and widely used reaction [22-30], generates the singlet aryl nitrene intermediate 12 (Scheme 1). Ring expansion of 12, via 2H-azirine 3, affords didehydroazepine 4, which can be trapped by variety of nucleophiles to provide the corresponding azepine 5. Alternatively, intersystem crossing (ISC) of 12 gives rise to 32, which can dimerize to form diazo compound 6. Performing the reaction in the presence of water typically affords 3H-azepinone 7 [31-34], a moiety present in natural products [35,36] and in azepinone-derived pharmaceuticals [37,38]. Despite the importance of these 7-membered nitrogen-containing heterocycles, there remain few methods for their preparation [39-44]. Overall, aryl azides, which are simple to prepare from the corresponding aniline derivative, are convenient precursors for the synthesis of azepine derivatives. However, the utility of the process is offset by the long reaction times required and by the low yields arising from poor selectivity and decomposition of the reaction mixture. We report herein the development of a continuous flow variant of the process and show how the reaction can be further optimized using a flow system [45,46].
![[1860-5397-7-129-i1]](/bjoc/content/inline/1860-5397-7-129-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Products of aryl azide photolysis.
Scheme 1: Products of aryl azide photolysis.
For this study a 14 mL photoreactor was used, constructed from fluorinated ethylene polymer (FEP) tubing, wrapped around a 450 W medium pressure Hg lamp, surrounded by a cooling jacket and Pyrex filter (see Supporting Information File 1). Continuous flow photochemical reactors based upon FEP tubing are simple to construct and are seeing increasing use in photochemical synthesis [47-49]. A recirculating cryostat maintained an apparatus temperature of 25 °C for all experiments, and a back pressure regulator (6.9 bar) was used to suppress the formation of a separate gaseous phase from the evolved nitrogen. THF and water were pumped simultaneously into the reactor at the specified rates through a commercially available Vapourtec R2 pump, [50] and mixed by a PTFE T-mixer.
Aryl azide 8a bearing a p-methyl ester substituent, prepared by treatment of the corresponding aniline with NaNO2/HCl–NaN3, was selected for initial optimization of the reaction (Scheme 2). During preliminary experiments under the conditions reported by Smalley (0.05 M in THF/H2O, 1:1) [31], blockage of the tubing was observed, which was attributed to the poor solubility of the substrate in water. Running the reaction with a reduced water content (THF/H2O, 4:3) obviated this problem and resulted in a 62% conversion of the starting azide to 3H-azepinone 9a, after a residence time of 30 min (Table 1, entry 2). Further reduction in the water content had a detrimental effect on the conversion. An inverse dependence of the conversion on substrate concentration was observed (Table 1). Although the highest conversion was observed at 0.015 M, higher productivity was obtained at 0.030 M (0.38 mmol h−1). Representing the best balance between conversion and productivity, 0.030 M was selected as the optimum concentration for further optimization. The use of other polar, aprotic, water-miscible solvents for the reaction was explored. Maintaining a residence time of 30 min, no significant change in conversion was observed upon replacing THF with DME or 1,4-dioxane; decreased conversion (22%) was observed with acetonitrile as the organic solvent.
![[1860-5397-7-129-i2]](/bjoc/content/inline/1860-5397-7-129-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Optimisation of the photolysis of aryl azide 8a.
Scheme 2: Optimisation of the photolysis of aryl azide 8a.
Table 1: Effect of aryl azide concentration on conversion.
| Entry | [8a] (M) | Conversion (%)a | Isolated Yield (%) |
|---|---|---|---|
| 1 | 0.100 | 39 | 29 |
| 2 | 0.050 | 62 | 35 |
| 3 | 0.030 | 78 | 45 |
| 4 | 0.015 | 90 | 48 |
aBy 1H NMR analysis of the crude product mixture after a residence time of 30 min.
Increasing the residence time improved the conversion of 8a (Figure 1), although this was accompanied by increased concomitant formation of the byproduct 10. Lactam 10 was obtained upon re-exposure of purified 9a to the reaction conditions and is believed to result from photochemical disrotatory electrocyclization of the 3H-azepinone diene moiety [51-54] (Scheme 3). Instead of making a direct comparison between reaction progress in batch and flow, the effects of longer residence times reported for batch photolysis were evaluated in a stopped-flow experiment by irradiating a solution of 8a for 3.5 h in the same FEP photoreactor (Figure 1). Analysis of the composition of the crude mixture showed significantly increased conversion of 9a to 10 accompanied by a slight increase in the proportion of the starting material 8a, which likely reflects decomposition of both of the reaction products. Overall, a residence time of 30 min was selected for a 0.030 M solution of 8a in THF/H2O 4:3, giving analytically pure 9a in 45% yield. Only traces of the diazo product were observed in the NMR spectrum of the crude mixture.
![[1860-5397-7-129-1]](/bjoc/content/figures/1860-5397-7-129-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Relationship between residence time and relative composition of the crude reaction mixture.
Figure 1: Relationship between residence time and relative composition of the crude reaction mixture.
![[1860-5397-7-129-i3]](/bjoc/content/inline/1860-5397-7-129-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of side product 10.
Scheme 3: Preparation of side product 10.
A variety of other 3H-azepinones was prepared in moderate to good yields under the optimized reaction conditions (Scheme 4, Table 2). Residual starting material was easily recovered by silica column chromatography. Rearrangements of aryl azides bearing electron withdrawing substituents are better represented in the literature than their electron donating congeners [31], and this was reflected in our compound selection. Methyl 2-azidobenzoate (8b), the corresponding acid 8c and dimethyl 2-azidoterephthalate (8d) each reacted favorably compared to the model substrate 8a. We were particularly interested in the preparation of 5-aryl-3H-azepinones 9e–g, examples of which have seen recent interest as γ-secretase inhibitors for the treatment of Alzheimer’s disease [37]. Photolysis of methyl 2-azido-5-phenylbenzoate 8e under the optimized conditions afforded a complex mixture of products. Upon shortening the residence time to 15 min, 3-methoxycarbonyl-5-phenyl-3H-azepinone (9e) was obtained in 35% yield. While both the corresponding acid 8f and 4-azidobiphenyl 8g decomposed upon photolysis, 8h gave the corresponding 5-chloro-3H-azepinone 9h, bearing a handle for further functionalization. To explore the preparation of bicyclic products, we studied the photolysis of 3-azidoquinoline (8i) to give the corresponding benzodiazepinone. Although the substrate decomposed under the original optimized conditions, reaction of 8i in NaOMe–MeOH [55,56] afforded 9i in good yield. Although yields were similar to those reported for batch processes [31,34,56], performing the reaction in continuous flow allows simple scaleup.
![[1860-5397-7-129-i4]](/bjoc/content/inline/1860-5397-7-129-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: General conditions for the photolysis of aryl azides in continuous flow.
Scheme 4: General conditions for the photolysis of aryl azides in continuous flow.
Table 2: 3H-azepinone derivatives prepared by photolysis of aryl azides in continuous flow.
| Entry | Substrate | Product | Yield (%)a | ||
|---|---|---|---|---|---|
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-129-i5.svg?max-width=637&scale=1.0)
|
8a |
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-129-i6.svg?max-width=637&scale=1.0)
|
9a | 45 (51) |
| 2 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-129-i7.svg?max-width=637&scale=1.0)
|
8b |
![[Graphic 4]](/bjoc/content/inline/1860-5397-7-129-i8.svg?max-width=637&scale=1.0)
|
9b | 75 (77) |
| 3 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-7-129-i9.svg?max-width=637&scale=1.0)
|
8c |
![[Graphic 6]](/bjoc/content/inline/1860-5397-7-129-i10.svg?max-width=637&scale=1.0)
|
9c | 50 (62) |
| 4 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-7-129-i11.svg?max-width=637&scale=1.0)
|
8d |
![[Graphic 8]](/bjoc/content/inline/1860-5397-7-129-i12.svg?max-width=637&scale=1.0)
|
9d | 74 (80) |
| 5b |
![[Graphic 9]](/bjoc/content/inline/1860-5397-7-129-i13.svg?max-width=637&scale=1.0)
|
8e |
![[Graphic 10]](/bjoc/content/inline/1860-5397-7-129-i14.svg?max-width=637&scale=1.0)
|
9e | 35 (47) |
| 6 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-7-129-i15.svg?max-width=637&scale=1.0)
|
8f |
![[Graphic 12]](/bjoc/content/inline/1860-5397-7-129-i16.svg?max-width=637&scale=1.0)
|
9f | 0 |
| 7 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-7-129-i17.svg?max-width=637&scale=1.0)
|
8g |
![[Graphic 14]](/bjoc/content/inline/1860-5397-7-129-i18.svg?max-width=637&scale=1.0)
|
9g | 0 |
| 8 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-7-129-i19.svg?max-width=637&scale=1.0)
|
8h |
![[Graphic 16]](/bjoc/content/inline/1860-5397-7-129-i20.svg?max-width=637&scale=1.0)
|
9h | 70 |
| 9c |
![[Graphic 17]](/bjoc/content/inline/1860-5397-7-129-i21.svg?max-width=637&scale=1.0)
|
8i |
![[Graphic 18]](/bjoc/content/inline/1860-5397-7-129-i22.svg?max-width=637&scale=1.0)
|
9i | 44d |
aYields in parentheses are based on recovered starting material. bThe residence time was reduced to 15 min. cThe photolysis was performed with a 0.030 M solution of 8i in 1.0 M NaOMe–MeOH. dDetermined by 1H NMR of the crude mixture.
In summary, the photochemical syntheses of 3H-azepinones and related azepines were performed and optimized in continuous flow through a photoreactor made from FEP tubing. Secondary photochemical reactions were minimized through careful control of the residence time. Although isolated yields were variable, a variety of aryl azides bearing electron withdrawing substituents underwent photolysis in good yield. Modification of the procedure allowed the preparation of benzodiazepine 9i.
Supporting Information
| Supporting Information File 1: Description of the flow reactor setup, experimental procedures and spectroscopic data of all compounds. | ||
| Format: PDF | Size: 4.2 MB | Download |
References
-
Hoffmann, N. Chem. Rev. 2008, 108, 1052. doi:10.1021/cr0680336
Return to citation in text: [1] -
Bach, T.; Hehn, J. P. Angew. Chem., Int. Ed. 2011, 50, 1000. doi:10.1002/anie.201002845
Return to citation in text: [1] -
André, J.-C.; Viriot, M.-L.; Villermaux, J. Pure Appl. Chem. 1986, 58, 907. doi:10.1351/pac198658060907
Return to citation in text: [1] -
Pfoertner, K. H. J. Photochem. Photobiol., A: Chem. 1990, 51, 81.
Return to citation in text: [1] -
Wirth, T., Ed. Microreactors in Organic Synthesis and Catalysis; Wiley-VCH: Weinheim, Germany, 2008.
Return to citation in text: [1] -
Ehrfeld, W.; Hessel, V.; Löwe, H. Microreactors: New Technology for Modern Chemistry; Wiley-VCH: Weinheim, Germany, 2000.
Return to citation in text: [1] -
Yoshida, J.-i., Ed. Flash Chemistry: Fast Organic Synthesis in Microsystems; John Wiley & Sons: Chichester, U.K., 2008.
Return to citation in text: [1] -
Coyle, E. E.; Oelgemöller, M. Photochem. Photobiol. Sci. 2008, 7, 1313. doi:10.1039/b808778d
Return to citation in text: [1] [2] -
Bourne, R. A.; Han, X.; Poliakoff, M.; George, M. W. Angew. Chem., Int. Ed. 2009, 48, 5322. doi:10.1002/anie.200901731
Return to citation in text: [1] -
Vasudevan, A.; Villamil, C.; Trumbull, J.; Olson, J.; Sutherland, D.; Pan, J.; Djuric, S. Tetrahedron Lett. 2010, 51, 4007. doi:10.1016/j.tetlet.2010.05.119
Return to citation in text: [1] -
Fuse, S.; Tanabe, N.; Yoshida, M.; Yoshida, H.; Doi, T.; Takahashi, T. Chem. Commun. 2010, 46, 8722. doi:10.1039/c0cc02239j
Return to citation in text: [1] -
Mimieux Vaske, Y. S.; Mahoney, M. E.; Konopelski, J. P.; Rogow, D. L.; McDonald, W. J. J. Am. Chem. Soc. 2010, 132, 11379. doi:10.1021/ja1050023
Return to citation in text: [1] -
Wegner, J.; Ceylan, S.; Kirschning, A. Chem. Commun. 2011, 47, 4583. doi:10.1039/c0cc05060a
Return to citation in text: [1] -
Shvydkiv, O.; Gallagher, S.; Nolan, K.; Oelgemöller, M. Org. Lett. 2010, 12, 5170. doi:10.1021/ol102184u
Return to citation in text: [1] -
Maeda, H.; Mukae, H.; Mizuno, K. Chem. Lett. 2005, 34, 66. doi:10.1246/cl.2005.66
Return to citation in text: [1] -
Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Angew. Chem., Int. Ed. 2005, 44, 5188. doi:10.1002/anie.200400657
Return to citation in text: [1] -
Iddon, B.; Meth-Cohn, O.; Scriven, E. F. V.; Suschitzky, H.; Gallagher, P. T. Angew. Chem., Int. Ed. Engl. 1979, 18, 900. doi:10.1002/anie.197909001
Return to citation in text: [1] -
Kotzyba-Hibert, F.; Kapfer, I.; Goeldner, M. Angew. Chem., Int. Ed. Engl. 1995, 34, 1296. doi:10.1002/anie.199512961
Return to citation in text: [1] -
Sydnes, M. O.; Doi, I.; Ohishi, A.; Kuse, M.; Isobe, M. Chem.–Asian J. 2008, 3, 102. doi:10.1002/asia.200700211
Return to citation in text: [1] -
Nielsen, P. E.; Buchardt, O. Photochem. Photobiol. 1982, 35, 317. doi:10.1111/j.1751-1097.1982.tb02568.x
Return to citation in text: [1] -
Reiser, A.; Bowes, G.; Horne, R. J. Trans. Faraday Soc. 1966, 62, 3162. doi:10.1039/tf9666203162
Return to citation in text: [1] -
von E. Doering, W.; Odum, R. A. Tetrahedron 1966, 22, 81. doi:10.1016/0040-4020(66)80104-7
Return to citation in text: [1] -
DeGraff, B. A.; Gillespie, D. W.; Sundberg, R. J. J. Am. Chem. Soc. 1974, 96, 7491. doi:10.1021/ja00831a017
Return to citation in text: [1] -
Borden, W. T.; Gritsan, N. P.; Hadad, C. M.; Karney, W. L.; Kemnitz, C. R., II; Platz, M. S. Acc. Chem. Res. 2000, 33, 765. doi:10.1021/ar990030a
Return to citation in text: [1] -
Gritsan, N. P.; Platz, M. S. Chem. Rev. 2006, 106, 3844. doi:10.1021/cr040055+
Return to citation in text: [1] -
Tsao, M.-L.; Platz, M. S. J. Am. Chem. Soc. 2003, 125, 12014. doi:10.1021/ja035833e
Return to citation in text: [1] -
Li, Y. Z.; Kirby, J. P.; George, M. W.; Poliakoff, M.; Schuster, G. B. J. Am. Chem. Soc. 1988, 110, 8092. doi:10.1021/ja00232a022
Return to citation in text: [1] -
Sundberg, R. J.; Sloan, K. B. J. Org. Chem. 1973, 38, 2052. doi:10.1021/jo00951a018
Return to citation in text: [1] -
Budyka, M. F.; Kantor, M. M.; Alfimov, M. V. Russ. Chem. Rev. 1992, 61, 25. doi:10.1070/RC1992v061n01ABEH000929
Return to citation in text: [1] -
Gritsan, N. P.; Pritchina, E. A. Russ. Chem. Rev. 1992, 61, 500. doi:10.1070/RC1992v061n05ABEH000959
Return to citation in text: [1] -
Lamara, K.; Smalley, R. K. Tetrahedron 1991, 47, 2277. doi:10.1016/S0040-4020(01)96138-1
Return to citation in text: [1] [2] [3] [4] -
Lamara, K.; Redhouse, A. D.; Smalley, R. K.; Thompson, J. R. Tetrahedron 1994, 50, 5515. doi:10.1016/S0040-4020(01)80706-7
Return to citation in text: [1] -
Koyama, K.; Takeuchi, H. J. Chem. Soc., Perkin Trans. 1 1982, 1269.
Return to citation in text: [1] -
Purvis, R.; Smalley, R. K.; Suschitzky, H.; Alkhader, M. A. J. Chem. Soc., Perkin Trans. 1 1984, 249.
Return to citation in text: [1] [2] -
O’Hagan, D. Nat. Prod. Rep. 1997, 14, 637. doi:10.1039/np9971400637
Return to citation in text: [1] -
Evans, P. A.; Holmes, B. Tetrahedron 1991, 47, 9131. doi:10.1016/S0040-4020(01)96203-9
Return to citation in text: [1] -
Miller, M.; Hope, J.; Porter, W. J.; Reel, J. K.; Rubio-Esteban, A. Azepine derivatives as gamma-secretase inhibitors. U.S. Patent Appl. 2010/0197660 Aug 5, 2010.
Return to citation in text: [1] [2] -
Robl, J. A.; Sulsky, R.; Sieber-McMaster, E.; Ryono, D. E.; Cimarusti, M. P.; Simpkins, L. M.; Karanewsky, D. S.; Chao, S.; Asaad, M. M.; Seymour, A. A.; Fox, M.; Smith, P. L.; Trippodo, N. C. J. Med. Chem. 1999, 42, 305. doi:10.1021/jm980542f
Return to citation in text: [1] -
Knobloch, K.; Eberbach, W. Org. Lett. 2000, 2, 1117. doi:10.1021/ol0056832
Return to citation in text: [1] -
Knobloch, K.; Koch, J.; Keller, M.; Eberbach, W. Eur. J. Org. Chem. 2005, 2715. doi:10.1002/ejoc.200400922
Return to citation in text: [1] -
Masaki, M.; Fukui, K.; Kita, J. Bull. Chem. Soc. Jpn. 1977, 50, 2013. doi:10.1246/bcsj.50.2013
Return to citation in text: [1] -
Atherton, F. R.; Lambert, R. W. J. Chem. Soc., Perkin Trans. 1 1973, 1079.
Return to citation in text: [1] -
Endo, Y.; Kataoko, K.-i.; Haga, N.; Shudo, K. Tetrahedron Lett. 1992, 33, 3339. doi:10.1016/S0040-4039(00)92083-5
Return to citation in text: [1] -
Reissig, H.-U.; Böttcher, G.; Zimmer, R. Can. J. Chem. 2004, 82, 166. doi:10.1139/v03-186
Return to citation in text: [1] -
O'Brien, A. G.; Lévesque, F.; Seeberger, P. H. Chem. Commun. 2011, 47, 2688. doi:10.1039/c0cc04481d
Return to citation in text: [1] -
O’Brien, A. G.; Lévesque, F.; Suzuki, Y.; Seeberger, P. H. Chim. Oggi 2011, 29, 57.
Return to citation in text: [1] -
Hook, B. D. A.; Dohle, W.; Hirst, P. R.; Pickworth, M.; Berry, M. B.; Booker-Milburn, K. I. J. Org. Chem. 2005, 70, 7558. doi:10.1021/jo050705p
Return to citation in text: [1] -
Lainchbury, M. D.; Medley, M. I.; Taylor, P. M.; Hirst, P.; Dohle, W.; Booker-Milburn, K. I. J. Org. Chem. 2008, 73, 6497. doi:10.1021/jo801108h
Return to citation in text: [1] -
Aggarwal, V. K.; Fang, G.; Kokotos, C. G.; Richardson, J.; Unthank, M. G. Tetrahedron 2006, 62, 11297. doi:10.1016/j.tet.2006.06.044
Return to citation in text: [1] -
http://www.vapourtec.co.uk. (accessed July 15, 2011).
Return to citation in text: [1] -
Odum, R. A.; Schmall, B. Chem. Commun. 1969, 1299.
Return to citation in text: [1] -
Odum, R. A.; Schmall, B. J. Chem. Res., Synop. 1997, 276. doi:10.1039/a702465g
Return to citation in text: [1] -
Chapman, O. L.; Hoganson, E. D. J. Am. Chem. Soc. 1964, 86, 498. doi:10.1021/ja01057a042
Return to citation in text: [1] -
Paquette, L. A. 2-azabicyclo[3.2.0]hep-6-en-3-ones. U.S. Patent 3,285,934 Nov 15, 1966.
Return to citation in text: [1] -
Sashida, H.; Fujii, A.; Tsuchiya, T. Chem. Pharm. Bull. 1987, 35, 4110.
Return to citation in text: [1] -
Hollywood, F.; Khan, Z. U.; Scriven, E. V. F.; Smalley, R. K.; Suschitzky, H.; Thomas, D. R.; Hull, R. J. Chem. Soc., Perkin Trans. 1 1982, 431.
Return to citation in text: [1] [2]
| 51. | Odum, R. A.; Schmall, B. Chem. Commun. 1969, 1299. |
| 52. | Odum, R. A.; Schmall, B. J. Chem. Res., Synop. 1997, 276. doi:10.1039/a702465g |
| 53. | Chapman, O. L.; Hoganson, E. D. J. Am. Chem. Soc. 1964, 86, 498. doi:10.1021/ja01057a042 |
| 54. | Paquette, L. A. 2-azabicyclo[3.2.0]hep-6-en-3-ones. U.S. Patent 3,285,934 Nov 15, 1966. |
| 31. | Lamara, K.; Smalley, R. K. Tetrahedron 1991, 47, 2277. doi:10.1016/S0040-4020(01)96138-1 |
| 1. | Hoffmann, N. Chem. Rev. 2008, 108, 1052. doi:10.1021/cr0680336 |
| 2. | Bach, T.; Hehn, J. P. Angew. Chem., Int. Ed. 2011, 50, 1000. doi:10.1002/anie.201002845 |
| 14. | Shvydkiv, O.; Gallagher, S.; Nolan, K.; Oelgemöller, M. Org. Lett. 2010, 12, 5170. doi:10.1021/ol102184u |
| 45. | O'Brien, A. G.; Lévesque, F.; Seeberger, P. H. Chem. Commun. 2011, 47, 2688. doi:10.1039/c0cc04481d |
| 46. | O’Brien, A. G.; Lévesque, F.; Suzuki, Y.; Seeberger, P. H. Chim. Oggi 2011, 29, 57. |
| 8. | Coyle, E. E.; Oelgemöller, M. Photochem. Photobiol. Sci. 2008, 7, 1313. doi:10.1039/b808778d |
| 9. | Bourne, R. A.; Han, X.; Poliakoff, M.; George, M. W. Angew. Chem., Int. Ed. 2009, 48, 5322. doi:10.1002/anie.200901731 |
| 10. | Vasudevan, A.; Villamil, C.; Trumbull, J.; Olson, J.; Sutherland, D.; Pan, J.; Djuric, S. Tetrahedron Lett. 2010, 51, 4007. doi:10.1016/j.tetlet.2010.05.119 |
| 11. | Fuse, S.; Tanabe, N.; Yoshida, M.; Yoshida, H.; Doi, T.; Takahashi, T. Chem. Commun. 2010, 46, 8722. doi:10.1039/c0cc02239j |
| 12. | Mimieux Vaske, Y. S.; Mahoney, M. E.; Konopelski, J. P.; Rogow, D. L.; McDonald, W. J. J. Am. Chem. Soc. 2010, 132, 11379. doi:10.1021/ja1050023 |
| 13. | Wegner, J.; Ceylan, S.; Kirschning, A. Chem. Commun. 2011, 47, 4583. doi:10.1039/c0cc05060a |
| 47. | Hook, B. D. A.; Dohle, W.; Hirst, P. R.; Pickworth, M.; Berry, M. B.; Booker-Milburn, K. I. J. Org. Chem. 2005, 70, 7558. doi:10.1021/jo050705p |
| 48. | Lainchbury, M. D.; Medley, M. I.; Taylor, P. M.; Hirst, P.; Dohle, W.; Booker-Milburn, K. I. J. Org. Chem. 2008, 73, 6497. doi:10.1021/jo801108h |
| 49. | Aggarwal, V. K.; Fang, G.; Kokotos, C. G.; Richardson, J.; Unthank, M. G. Tetrahedron 2006, 62, 11297. doi:10.1016/j.tet.2006.06.044 |
| 5. | Wirth, T., Ed. Microreactors in Organic Synthesis and Catalysis; Wiley-VCH: Weinheim, Germany, 2008. |
| 6. | Ehrfeld, W.; Hessel, V.; Löwe, H. Microreactors: New Technology for Modern Chemistry; Wiley-VCH: Weinheim, Germany, 2000. |
| 7. | Yoshida, J.-i., Ed. Flash Chemistry: Fast Organic Synthesis in Microsystems; John Wiley & Sons: Chichester, U.K., 2008. |
| 37. | Miller, M.; Hope, J.; Porter, W. J.; Reel, J. K.; Rubio-Esteban, A. Azepine derivatives as gamma-secretase inhibitors. U.S. Patent Appl. 2010/0197660 Aug 5, 2010. |
| 38. | Robl, J. A.; Sulsky, R.; Sieber-McMaster, E.; Ryono, D. E.; Cimarusti, M. P.; Simpkins, L. M.; Karanewsky, D. S.; Chao, S.; Asaad, M. M.; Seymour, A. A.; Fox, M.; Smith, P. L.; Trippodo, N. C. J. Med. Chem. 1999, 42, 305. doi:10.1021/jm980542f |
| 3. | André, J.-C.; Viriot, M.-L.; Villermaux, J. Pure Appl. Chem. 1986, 58, 907. doi:10.1351/pac198658060907 |
| 4. | Pfoertner, K. H. J. Photochem. Photobiol., A: Chem. 1990, 51, 81. |
| 39. | Knobloch, K.; Eberbach, W. Org. Lett. 2000, 2, 1117. doi:10.1021/ol0056832 |
| 40. | Knobloch, K.; Koch, J.; Keller, M.; Eberbach, W. Eur. J. Org. Chem. 2005, 2715. doi:10.1002/ejoc.200400922 |
| 41. | Masaki, M.; Fukui, K.; Kita, J. Bull. Chem. Soc. Jpn. 1977, 50, 2013. doi:10.1246/bcsj.50.2013 |
| 42. | Atherton, F. R.; Lambert, R. W. J. Chem. Soc., Perkin Trans. 1 1973, 1079. |
| 43. | Endo, Y.; Kataoko, K.-i.; Haga, N.; Shudo, K. Tetrahedron Lett. 1992, 33, 3339. doi:10.1016/S0040-4039(00)92083-5 |
| 44. | Reissig, H.-U.; Böttcher, G.; Zimmer, R. Can. J. Chem. 2004, 82, 166. doi:10.1139/v03-186 |
| 21. | Reiser, A.; Bowes, G.; Horne, R. J. Trans. Faraday Soc. 1966, 62, 3162. doi:10.1039/tf9666203162 |
| 31. | Lamara, K.; Smalley, R. K. Tetrahedron 1991, 47, 2277. doi:10.1016/S0040-4020(01)96138-1 |
| 32. | Lamara, K.; Redhouse, A. D.; Smalley, R. K.; Thompson, J. R. Tetrahedron 1994, 50, 5515. doi:10.1016/S0040-4020(01)80706-7 |
| 33. | Koyama, K.; Takeuchi, H. J. Chem. Soc., Perkin Trans. 1 1982, 1269. |
| 34. | Purvis, R.; Smalley, R. K.; Suschitzky, H.; Alkhader, M. A. J. Chem. Soc., Perkin Trans. 1 1984, 249. |
| 55. | Sashida, H.; Fujii, A.; Tsuchiya, T. Chem. Pharm. Bull. 1987, 35, 4110. |
| 56. | Hollywood, F.; Khan, Z. U.; Scriven, E. V. F.; Smalley, R. K.; Suschitzky, H.; Thomas, D. R.; Hull, R. J. Chem. Soc., Perkin Trans. 1 1982, 431. |
| 18. | Kotzyba-Hibert, F.; Kapfer, I.; Goeldner, M. Angew. Chem., Int. Ed. Engl. 1995, 34, 1296. doi:10.1002/anie.199512961 |
| 19. | Sydnes, M. O.; Doi, I.; Ohishi, A.; Kuse, M.; Isobe, M. Chem.–Asian J. 2008, 3, 102. doi:10.1002/asia.200700211 |
| 20. | Nielsen, P. E.; Buchardt, O. Photochem. Photobiol. 1982, 35, 317. doi:10.1111/j.1751-1097.1982.tb02568.x |
| 35. | O’Hagan, D. Nat. Prod. Rep. 1997, 14, 637. doi:10.1039/np9971400637 |
| 36. | Evans, P. A.; Holmes, B. Tetrahedron 1991, 47, 9131. doi:10.1016/S0040-4020(01)96203-9 |
| 31. | Lamara, K.; Smalley, R. K. Tetrahedron 1991, 47, 2277. doi:10.1016/S0040-4020(01)96138-1 |
| 34. | Purvis, R.; Smalley, R. K.; Suschitzky, H.; Alkhader, M. A. J. Chem. Soc., Perkin Trans. 1 1984, 249. |
| 56. | Hollywood, F.; Khan, Z. U.; Scriven, E. V. F.; Smalley, R. K.; Suschitzky, H.; Thomas, D. R.; Hull, R. J. Chem. Soc., Perkin Trans. 1 1982, 431. |
| 16. | Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Angew. Chem., Int. Ed. 2005, 44, 5188. doi:10.1002/anie.200400657 |
| 17. | Iddon, B.; Meth-Cohn, O.; Scriven, E. F. V.; Suschitzky, H.; Gallagher, P. T. Angew. Chem., Int. Ed. Engl. 1979, 18, 900. doi:10.1002/anie.197909001 |
| 31. | Lamara, K.; Smalley, R. K. Tetrahedron 1991, 47, 2277. doi:10.1016/S0040-4020(01)96138-1 |
| 8. | Coyle, E. E.; Oelgemöller, M. Photochem. Photobiol. Sci. 2008, 7, 1313. doi:10.1039/b808778d |
| 15. | Maeda, H.; Mukae, H.; Mizuno, K. Chem. Lett. 2005, 34, 66. doi:10.1246/cl.2005.66 |
| 22. | von E. Doering, W.; Odum, R. A. Tetrahedron 1966, 22, 81. doi:10.1016/0040-4020(66)80104-7 |
| 23. | DeGraff, B. A.; Gillespie, D. W.; Sundberg, R. J. J. Am. Chem. Soc. 1974, 96, 7491. doi:10.1021/ja00831a017 |
| 24. | Borden, W. T.; Gritsan, N. P.; Hadad, C. M.; Karney, W. L.; Kemnitz, C. R., II; Platz, M. S. Acc. Chem. Res. 2000, 33, 765. doi:10.1021/ar990030a |
| 25. | Gritsan, N. P.; Platz, M. S. Chem. Rev. 2006, 106, 3844. doi:10.1021/cr040055+ |
| 26. | Tsao, M.-L.; Platz, M. S. J. Am. Chem. Soc. 2003, 125, 12014. doi:10.1021/ja035833e |
| 27. | Li, Y. Z.; Kirby, J. P.; George, M. W.; Poliakoff, M.; Schuster, G. B. J. Am. Chem. Soc. 1988, 110, 8092. doi:10.1021/ja00232a022 |
| 28. | Sundberg, R. J.; Sloan, K. B. J. Org. Chem. 1973, 38, 2052. doi:10.1021/jo00951a018 |
| 29. | Budyka, M. F.; Kantor, M. M.; Alfimov, M. V. Russ. Chem. Rev. 1992, 61, 25. doi:10.1070/RC1992v061n01ABEH000929 |
| 30. | Gritsan, N. P.; Pritchina, E. A. Russ. Chem. Rev. 1992, 61, 500. doi:10.1070/RC1992v061n05ABEH000959 |
| 37. | Miller, M.; Hope, J.; Porter, W. J.; Reel, J. K.; Rubio-Esteban, A. Azepine derivatives as gamma-secretase inhibitors. U.S. Patent Appl. 2010/0197660 Aug 5, 2010. |
© 2011 Bou-Hamdan et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)