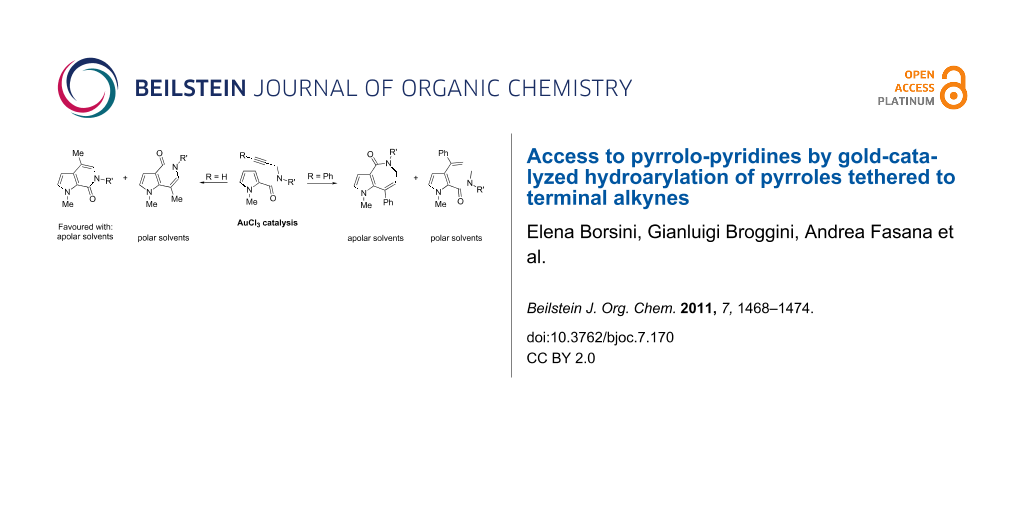
Abstract
In a simple procedure, the intramolecular hydroarylation of N-propargyl-pyrrole-2-carboxamides was accomplished with the aid of gold(III) catalysis. The reaction led to differently substituted pyrrolo[2,3-c]pyridine and pyrrolo[3,2-c]pyridine derivatives arising either from direct cyclization or from a formal rearrangement of the carboxamide group. Terminal alkynes are essential to achieve bicyclic pyrrolo-fused pyridinones by a 6-exo-dig process, while the presence of a phenyl group at the C–C triple bond promotes the 7-endo-dig cyclization giving pyrrolo-azepines.

Graphical Abstract
Introduction
Intramolecular transition-metal-catalyzed reactions represent one of the most challenging routes for the preparation of heterocyclic compounds [1-5]. Methodologies providing heterocycles, starting from readily available substrates, by different catalytic systems, as well as selective procedures for the synthesis of different heterocycles from the same starting materials by subtle modifications of the catalytic conditions, are useful tools in the hands of organic chemists. Gold catalysis has recently emerged as a suitable way to achieve such a goal, mainly thanks to the chemoselective alkynophilic properties of this attractive metal in different types of reactions [6-15]. Among the hydroarylation reactions, various kinds of heteroaryl-substituted alkynes have been demonstrated as efficient substrates for the construction of heteropolycyclic compounds. In particular, alkynyl indoles and pyrroles were successfully used to afford β-carbolines [16,17], pyrrolo-azepines [18], azepino-indoles and azocino-indoles [19] under mild conditions.
During our studies aimed at the synthesis of complex heterocyclic systems by intramolecular transition-metal-catalyzed protocols [20-32], we reported an arylative Pd-catalyzed cyclization of N-allyl-pyrrole-2-carboxamides, affording pyrrolo[1,2-a]pyrazines or pyrrolo[2,3-c]pyridines (Scheme 1) [33]. The reaction provided also the isomeric pyrrolo[3,2-c]pyridines arising from 1,2-migration of the amide moiety. Having recently broadened our studies toward gold catalysis [34,35], we have been intrigued by the investigation of a gold-catalyzed intramolecular hydroarylation of alkynyl-tethered pyrrole-2-carboxamides in order to have an alternative protocol to access pyrrolo-fused pyridine skeletons. The importance of pyrrolo[2,3-c]pyridine and pyrrolo[3,2-c]pyridine derivatives from the biological point of view [36-50] justifies new efforts to obtain them. This study on the Au-catalyzed cyclization of N-alkynyl-pyrrole-2-carboxamides is also focused on the effect of the alkyne substituent, as well as on the role of the solvent on the product distribution.
![[1860-5397-7-170-i1]](/bjoc/content/inline/1860-5397-7-170-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Pd-catalyzed cyclization of N-allyl-pyrrole-2-carboxamides.
Scheme 1: Pd-catalyzed cyclization of N-allyl-pyrrole-2-carboxamides.
Results and Discussion
On the basis of these considerations, the N-propargyl amides of 1H-pyrrole-2-carboxylic acids 1a,b, easily prepared in high yields from the corresponding 1H-pyrrole-2-carboxylic acids and N-methyl-N-propargylamine with DCC as coupling reagent, were submitted to gold-catalyzed reactions under different conditions (Table 1).
Table 1: Optimization of reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-170-i3.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Substrate | Gold catalysta | Solvent | 2b | 3b |
|---|---|---|---|---|---|
| 1 | 1a | AuCl3 | MeCN | – | – |
| 2 | 1a | NaAuCl4·2H2O | MeCN | – | – |
| 3 | 1a | AuCl | DCM | – | – |
| 4 | 1a | PPh3AuCl/AgBF4 | DCM | – | – |
| 5 | 1a | AuCl3 | DCM | – | – |
| 6 | 1b | AuCl3 | MeCN | 63 | 37 |
| 7c | 1b | NaAuCl4·2H2O | MeCN | 32 | 18 |
| 8 | 1b | AuCl | DCM | – | – |
| 9 | 1b | PPh3AuCl/AgBF4 | DCM | – | – |
aCatalyst loading: 5 mol %. bDetermined by HPLC. cIn this case, 50% of 1b was recovered.
First, it should be noted that the 1-unsubstituted pyrrole derivative 1a did not undergo cyclization, under various conditions, based on both Au(III) and Au(I) catalysts (Table 1, entries 1–5). Otherwise, AuCl3 was able to promote the intramolecular reaction of the methyl-substituted substrate 1b giving two isomeric bicyclic products. Their structures were unequivocally determined by NOESY1D, gHSQC and 1H/13C long-range correlations (gHMBC) experiments and were found to be the 6-exo-dig cyclization product 2b and the rearranged structure 3b (Figure 1). The best result was obtained when the reaction was performed in MeCN at reflux for 4 h (Table 1, entry 6); in this case, the ratio between 2b and 3b, determined by HPLC, was 1.7:1. The cyclization of substrate 1b was also accomplished by working with NaAuCl4·2H2O as catalyst to give a mixture of 2b and 3b in the same ratio, although the conversion of the substrate was only 50% (Table 1, entry 7). Conversely, gold(I) species were unable to promote the cyclization of substrate 1b (Table 1, entries 8 and 9).
![[1860-5397-7-170-1]](/bjoc/content/figures/1860-5397-7-170-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Significant relationships among hydrogen and carbon atoms arising from 2D-NMR studies to determine the pyrrolo-pyridinones.
Figure 1: Significant relationships among hydrogen and carbon atoms arising from 2D-NMR studies to determine ...
In the presence of AuCl3, the ratio between the cyclization products 2b and 3b was significantly influenced by the solvent, as shown for the investigated conditions in Table 2. In fact, nonpolar solvents enhanced the ratio of direct to rearranged products (Table 2, entries 1 and 2), while switching to the polar ones increased the yield of 3b up to 50% (Table 2, entry 4). In particular, toluene and DMF were proven to be the best solvents to gain the most selective conditions (7:1 versus 1:1.6, respectively).
Table 2: Effect of the solvent on the ratio of the isomeric products.
| Entrya | Solvent | T (°C) | t (h) | Ratiob | 2bc | 3bc |
|---|---|---|---|---|---|---|
| 1 | Toluene | 50 | 3 | 7:1 | 70 | 8 |
| 2 | DCM | reflux | 3 | 3:1 | 41 | 16 |
| 3 | MeCN | reflux | 4 | 1.7:1 | 45 | 30 |
| 4 | DMF | 90 | 4 | 1:1.6 | 32 | 50 |
aAuCl3 at 5 mol %. bHPLC–MS ratio between peak areas. cIsolated yields after column chromatography.
Other variously N-substituted propargylamides (4a–d) were submitted to the AuCl3-catalyzed cyclization in different solvents. Both the isolated yields and the HPLC–MS isomeric ratios of direct and rearranged products are summarized in Table 3. These reactions gave rise to a varied range of products, all having pyrrolo[2,3-c]pyridinone and pyrrolo[3,2-c]pyridinone structures. Analogously to what was observed for the substrate 1b, benzyl- and tosyl-substituted amides 4a and 4b afforded the expected products 5a,b and 6a,b. Also in these cases, the ratio between the different products depends on the reaction solvent (Table 3, entries 1–7). In the case of the tert-butoxycarbonyl nitrogen-substituted substrate 4c the reaction gave the two exo-methylene products 7c and 8c, with the product ratio being in the opposite sense for toluene compared to DCM (Table 3, entries 8 and 9). Unexpectedly, when working in acetonitrile, the sole product was compound 10 lacking the tert-butoxycarbonyl group (Table 3, entry 10). Finally, the cyclization of the benzoyl-substituted substrate 4d furnished only products having an internal C–C double bond, with some products lacking the benzoyl group (Table 3, entries 11–14). The highest selectivity was obtained with toluene as solvent, where only pyrrolo[2,3-c]pyridinones, i.e., 5d and 9 (Table 3, entry 11), were formed.
Table 3: Scope of the reaction on differently N-substituted N-propargyl pyrrole-carboxamides.
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-170-i4.svg?max-width=637&scale=1.0)
|
||||||||||||
| Entry | Substrate | R | Solvent | T (°C) | t (h) | Yield (%)a | Ratiob | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 6 | 7 | 8 | 9 | 10 | |||||||
| 1 | 4a | Bn | Toluene | 50 | 0.5 | 84 | 7 | – | – | – | – | 7.5:1 |
| 2 | 4a | Bn | DCM | reflux | 0.25 | 61 | 12 | – | – | – | – | 3:1 |
| 3 | 4a | Bn | MeCN | reflux | 0.25 | 50 | 25 | – | – | – | – | 1.6:1 |
| 4 | 4b | Ts | DCM | reflux | 0.5 | 75 | 9 | – | – | – | – | 6:1 |
| 5 | 4b | Ts | Toluene | 50 | 0.5 | 65 | 14 | – | – | – | – | 4:1 |
| 6 | 4b | Ts | MeCN | reflux | 0.5 | 70 | 21 | – | – | – | – | 3:1 |
| 7 | 4b | Ts | DMF | 90 | 7 | 60 | 25 | – | – | – | – | 2:1 |
| 8 | 4c | Boc | Toluene | 50 | 1 | – | – | 56 | 14 | – | – | 2.5:1 |
| 9 | 4c | Boc | DCM | reflux | 1 | – | – | 20 | 30 | – | – | 1:1.5 |
| 10 | 4c | Boc | MeCN | reflux | 3 | – | – | – | – | – | 65c | 1 |
| 11 | 4d | Bz | Toluene | 50 | 1 | 51 | – | – | – | 6 | – | 1 |
| 12 | 4d | Bz | DCM | reflux | 1.5 | 61 | 11 | – | – | – | – | 7:1 |
| 13 | 4d | Bz | MeCN | reflux | 1 | 50 | 13 | – | – | – | – | 5:1 |
| 14d | 4d | Bz | DMF | 90 | 24 | – | – | – | – | 40 | 11 | 2:1 |
aIsolated yield. bHPLC–MS ratio between peak areas. cAfter column chromatography 33% of 4c was recovered. dAt the end of the reaction 22% of 4d was still present.
Although the picture of the products generated from the treatment of the propargyl pyrrole-2-carboxamides with AuCl3 as catalyst is not completely homogeneous, it should be stressed that all the substrates cyclize following a 6-exo-dig process. This contrasts with the outcome of the cyclization of N-methyl-N-(3-arylprop-2-ynyl)-substituted 1-methyl-pyrrole-2-carboxamides in the presence of AuCl3 leading to pyrrolo-azepine derivatives, as already described in the literature by Beller and coworkers [18]. In order to shed light on the structural features determining the size of the newly formed ring, we thought it appropriate to extend this study further to N-propargyl-1-methylpyrrole-2-carboxamides bearing a phenyl group on the acetylenic carbon. Therefore, we treated amides 11a–c with AuCl3 as catalyst in different solvents. As given in Table 4, the reactions afforded only seven-membered cyclized products 12a–c and 13a–c, according to Beller’s work. This outcome confirmed the pivotal role of the substituent on the C–C triple bond to direct the cyclization following 6-exo-dig or 7-endo-dig processes. Once again, the trend in terms of direct and rearranged product formation was dependent on the solvent, the 1,2-migration products being favoured in less polar solvents. The 7-endo-dig cyclizations of amides 11a–c were highly selective with respect to the isomerization of the newly formed carbon–carbon double bond, since it keeps its initial position in both final structures, probably due to the conjugation with the benzene ring.
Table 4: Cyclization reactions on differently N-substituted N-(3-phenyl-prop-2-ynyl) pyrrole-2-carboxamides.
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-170-i5.svg?max-width=637&scale=1.0)
|
||||||||
| Entry | Substrate | R | Solvent | T (°C) | t (h) | Ratioa | Yieldb(%) | |
|---|---|---|---|---|---|---|---|---|
| 12:13 | 12 | 13 | ||||||
| 1 | 11a | Me | DCM | reflux | 8 | 1:3 | 20 | 63 |
| 2 | 11a | Me | MeCN | reflux | 2 | 1.5:1 | 43 | 31 |
| 3 | 11a | Me | DMF | 90 | 24 | 5:1 | 66 | 12 |
| 4 | 11b | Ts | DCM | reflux | 0.5 | 1:2.8 | 20 | 60 |
| 5 | 11b | Ts | MeCN | reflux | 0.25 | 3:1 | 55 | 17 |
| 6 | 11b | Ts | DMF | 90 | 24 | 10:1 | 68 | 7 |
| 7 | 11c | Bn | DCM | reflux | 5 | 1:6 | 12 | 74 |
| 8 | 11c | Bn | MeCN | reflux | 0.5 | 1:2.5 | 23 | 67 |
| 9 | 11c | Bn | DMF | 90 | 24 | 5:1 | 66 | 14 |
aHPLC–MS ratio between peak areas. bIsolated yields after column chromatography.
A plausible although speculative mechanism for the hydroarylation of terminal alkynes with pyrroles is shown in Scheme 2. It is highly probable that the products arise from the vinyl gold species B and F, in turn generated from alkynes 4 after coordination to AuCl3. The formation of F can result by a migration of the acyl group from the spiro center of the transient cationic spiro gold complex D, in turn generated by the attack of the more nucleophilic C-2 of the pyrrole nucleus on the intermediate A. On the other hand, complex B can arise from the direct attack of the pyrrolic C-3 on the activated species A or from the migration of the alkyl group on the spiro intermediate D. The complexes B and F evolve by protodeaurylation generating the exomethylenic compounds 7 and 8, which are eventually susceptible to alkene isomerization. The hypothesis that the alkene isomerization occurs prior to protodeaurylation, giving intermediates C and E, is unlikely because the protodemetallation is generally accepted to proceed much more rapidly on vinyl gold than on alkyl gold intermediates. In all cases, the product distribution plausibly reflects their thermodynamic stability. The endocyclic alkene is favoured presumably due to the higher degree of conjugation (compounds 5a,b,d and 6a,b,d), while it is reasonable to assume that the isomerization to the endocyclic alkene with the bulky N-BOC group (compounds 7c and 8c) is less favourable due to the resulting strain in the planar ring.
![[1860-5397-7-170-i2]](/bjoc/content/inline/1860-5397-7-170-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed mechanism for the formation of the six-membered products.
Scheme 2: Proposed mechanism for the formation of the six-membered products.
To explain the cyclization of alkynes 11a–c, a similar mechanism could be proposed. The formation of the seven-membered cyclized products could be due to a stabilizing effect of the phenyl group in the transition state of the reaction.
Conclusion
In summary, the cyclization of N-alkynyl pyrrole-2-carboxamides under very mild gold-catalyzed conditions has been described. The outcome of the intramolecular hydroarylation gave 6-exo-dig or 6-endo-dig processes, depending on the substitution of the alkyne. In both cases, products arising from a direct reaction as well as products involving a 1,2-shift of the amide group were obtained, their ratio being influenced by the polarity of the solvent. The gold-catalyzed procedure to achieve pyrrolo[2,3-c]pyridines and pyrrolo[3,2-c]pyridines represents a valuable alternative to the Pd-catalyzed cyclization of the analogue allylamides.
Experimental
General
Melting points were determined by capillary method with a Büchi B-540 apparatus and are uncorrected. 1H and 13C NMR spectra were recorded on an INOVA AS600 Variant spectrometer. 13C NMR spectra are 1H-decoupled and the multiplicities were determined by APT pulse sequence. Chemical shifts are given as δ values in ppm relative to residual solvent peaks (CHCl3) as the internal reference. IR spectra were measured with a Jasco FT/IR 5300 spectrometer. MS spectra were recorded on a HPLC–MS Agilent Technologies 6140 (ESI). Elemental analyses were executed on Perkin-Elmer CHN Analyzer Series II 2400. Preparative separations were performed by Biotage flash chromatography with 40M silica cartridges.
Cyclization reactions of pyrrole-carboxamides
A solution of the appropriate pyrrole-carboxamide (2 mmol) was stirred, under an argon atmosphere, with AuCl3 (0.01 mmol) in 30 mL of an appropriate solvent (see Table 3 and Table 4 for solvents, temperatures and times). At the end of the reaction, the solvent was either removed under reduced pressure (MeCN, DCM, toluene) or extracted with brine (DMF). The crude residue was purified by flash column chromatography.
1,4,6-Trimethyl-1,6-dihydro-pyrrolo[2,3-c]pyridin-7-one (2b)
Yield: 70% (Table 2, entry 1). White solid; mp 76 °C; IR (nujol) ν: 1670 cm−1; 1H NMR (599 MHz, CDCl3) δ 2.13 (s, 3H), 3.49 (s, 3H), 4.13 (s, 3H), 6.17 (d, J = 2.9 Hz, 1H), 6.57 (s, 1H), 6.91 (d, J = 2.9 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 14.7 (q), 35.5 (q), 35.5 (q), 100.1 (d), 110.3 (s), 122.3 (s), 126.2 (d), 130.7 (d), 132.4 (s), 155.6 (s); MS m/z: 177.21 [M]+; Anal. calcd for C10H12N2O: C, 68.16; H, 6.86; N, 15.90; found: C, 68.22; H, 6.80; N, 15.95.
1,5,7-Trimethyl-1,6-dihydro-pyrrolo[3,2-c]pyridin-4-one (3b)
Yield: 50% (Table 2, entry 4). Yellow solid; mp 104 °C; IR (nujol) ν: 1672 cm−1; 1H NMR (599 MHz, CDCl3) δ 2.37 (s, 3H), 3.51 (s, 3H), 3.89 (s, 3H), 6.65 (s, 1H), 6.69 (d, J = 3.1 Hz, 1H), 6.73 (d, J = 3.1 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 16.1 (q), 35.8 (q), 35.8 (q), 104.4 (d), 104.5 (s), 116.9 (s), 127.4 (d), 129.8 (d), 137.9 (s), 159.6 (s); MS m/z: 177 [M]+; Anal. calcd for C10H12N2O: C, 68.16; H, 6.86; N, 15.90; found: C, 68.13; H, 6.81; N, 15.94.
1,7-Dimethyl-4-phenyl-6,7-dihydropyrrolo[2,3-c]azepin-8(1H)-one (12a)
Yield: 66% (Table 4, entry 3). White solid; mp 118 °C; IR (nujol) ν: 1673 cm−1; 1H NMR (599 MHz, CDCl3) δ 3.17 (s, 3H), 3.78 (d, J = 6.9 Hz, 2H), 4.00 (s, 3H), 5.95 (d, J = 2.8 Hz, 1H), 6.07 (t, J = 6.9 Hz, 1H), 6.73 (d, J = 2.8 Hz, 1H), 7.33–7.37 (m, 3H), 7.38–7.40 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 34.6 (q), 36.6 (q), 47.5 (t), 107.7 (d), 119.6 (d), 126.6 (s), 126.7 (d), 127.1 (s), 127.7 (d), 128.1 (d), 128.5 (d), 140.5 (s), 142.6 (s), 162.2 (s); MS m/z: 253 [M]+; Anal. calcd for C16H16N2O: C, 76.16; H, 6.39; N, 11.10; found: C, 76.24; H, 6.35; N, 11.04.
1,5-Dimethyl-8-phenyl-5,6-dihydropyrrolo[3,2-c]azepin-4(1H)-one (13a)
Yield: 63% (Table 4, entry 1). White solid; mp 130 °C; IR (nujol) ν: 1676 cm−1; 1H NMR (599 MHz, CDCl3) δ 3.07 (s, 3H), 3.17 (s, 3H), 3.74 (d, J = 7.2 Hz, 2H), 6.13 (t, J = 7.2 Hz, 1H), 6.63 (d, J = 2.8 Hz, 1H), 6.77 (d, J = 2.8 Hz, 1H), 7.22 (d, J = 6.4 Hz, 2H), 7.32–7.35 (m, 3H); 13C NMR (150 MHz, CDCl3) δ 35.3 (q), 36.4 (q), 47.5 (t), 109.6 (d), 123.2 (s), 124.3 (d), 124.7 (d), 127.5 (d), 128.0 (d), 128.6 (d), 131.5 (s), 137.8 (s), 139.3 (s), 166.0 (s); MS m/z: 253 [M]+; Anal. calcd for C16H16N2O: C, 76.16; H, 6.39; N, 11.10; found: C, 76.23; H, 6.30; N, 11.04.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data. | ||
| Format: PDF | Size: 126.0 KB | Download |
References
-
Nakamura, I.; Yamamoto, Y. Chem. Rev. 2004, 104, 2127–2198. doi:10.1021/cr020095i
Return to citation in text: [1] -
Li, J. J.; Gribble, G. W. Palladium in Heterocyclic Chemistry. A Guide for the Synthetic Chemist; Pergamon: New York, 2000.
Return to citation in text: [1] -
Godoi, B.; Schumacher, R. F.; Zeni, G. Chem. Rev. 2011, 111, 2937–2980. doi:10.1021/cr100214d
Return to citation in text: [1] -
Beccalli, E. M.; Broggini, G.; Fasana, A.; Rigamonti, M. J. Organomet. Chem. 2011, 696, 277–295. doi:10.1016/j.jorganchem.2010.09.078
Return to citation in text: [1] -
Patil, N. T.; Yamamoto, Y. Chem. Rev. 2011, 111, 3395–3442. doi:10.1021/cr050041j
Return to citation in text: [1] -
Shen, H. C. Tetrahedron 2008, 64, 3885–3903. doi:10.1016/j.tet.2008.01.081
Return to citation in text: [1] -
Shen, H. C. Tetrahedron 2008, 64, 7847–7870. doi:10.1016/j.tet.2008.05.082
Return to citation in text: [1] -
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319
Return to citation in text: [1] -
Arcadi, A. Chem. Rev. 2008, 108, 3266–3325. doi:10.1021/cr068435d
Return to citation in text: [1] -
Marion, N.; Nolan, S. P. Chem. Soc. Rev. 2008, 37, 1776–1782. doi:10.1039/b711132k
Return to citation in text: [1] -
Fürstner, A. Chem. Soc. Rev. 2009, 38, 3208–3221. doi:10.1039/b816696j
Return to citation in text: [1] -
Shapiro, N. D.; Toste, F. D. Synlett 2010, 675–691. doi:10.1055/s-0029-1219369
Return to citation in text: [1] -
Corma, A.; Leyva-Pérez, A.; Sabater, M. J. Chem. Rev. 2011, 111, 1657–1712. doi:10.1021/cr100414u
Return to citation in text: [1] -
Krause, N.; Winter, C. Chem. Rev. 2011, 111, 1994–2009. doi:10.1021/cr1004088
Return to citation in text: [1] -
Bandini, M. Chem. Soc. Rev. 2011, 40, 1358–1367. doi:10.1039/c0cs00041h
Return to citation in text: [1] -
England, D. B.; Padwa, A. Org. Lett. 2008, 10, 3631–3634. doi:10.1021/ol801385h
Return to citation in text: [1] -
Verniest, G.; England, D.; De Kimpe, N.; Padwa, A. Tetrahedron 2010, 66, 1496–1502. doi:10.1016/j.tet.2009.10.033
Return to citation in text: [1] -
Gruit, M. M.; Michalik, D.; Krüger, K.; Spannenberg, A.; Tillack, A.; Pews-Davtyan, A.; Beller, M. Tetrahedron 2010, 66, 3341–3352. doi:10.1016/j.tet.2010.02.091
Return to citation in text: [1] [2] -
Ferrer, C.; Echavarren, A. M. Angew. Chem., Int. Ed. 2006, 45, 1105–1109. doi:10.1002/anie.200503484
Return to citation in text: [1] -
Broggini, G.; Molteni, G.; Terraneo, A.; Zecchi, G. Heterocycles 2003, 59, 823–858. doi:10.3987/REV-02-SR5
Return to citation in text: [1] -
Beccalli, E. M.; Broggini, G.; Martinelli, M.; Paladino, G.; Zoni, C. Eur. J. Org. Chem. 2005, 2091–2096. doi:10.1002/ejoc.200400817
Return to citation in text: [1] -
Abbiati, G.; Beccalli, E. M.; Broggini, G.; Paladino, G.; Rossi, E. Synthesis 2005, 2881–2886. doi:10.1055/s-2005-916033
Return to citation in text: [1] -
Abbiati, G.; Beccalli, E. M.; Broggini, G.; Martinelli, M.; Paladino, G. Synlett 2006, 73–76. doi:10.1055/s-2005-922788
Return to citation in text: [1] -
Beccalli, E. M.; Broggini, G.; Martinelli, M.; Masciocchi, N.; Sottocornola, S. Org. Lett. 2006, 8, 4521–4524. doi:10.1021/ol061693c
Return to citation in text: [1] -
Beccalli, E. M.; Borsini, E.; Broggini, G.; Rigamonti, M.; Sottocornola, S. Synlett 2008, 1053–1057. doi:10.1055/s-2008-1072583
Return to citation in text: [1] -
Beccalli, E. M.; Broggini, G.; Clerici, F.; Galli, S.; Kammerer, C.; Rigamonti, M.; Sottocornola, S. Org. Lett. 2009, 11, 1563–1566. doi:10.1021/ol900171g
Return to citation in text: [1] -
Basolo, L.; Beccalli, E. M.; Borsini, E.; Broggini, G. Tetrahedron 2009, 65, 3486–3491. doi:10.1016/j.tet.2009.02.025
Return to citation in text: [1] -
Beccalli, E. M.; Borsini, E.; Brenna, S.; Galli, S.; Rigamonti, M.; Broggini, G. Chem.–Eur. J. 2010, 16, 1670–1678. doi:10.1002/chem.200902071
Return to citation in text: [1] -
Beccalli, E. M.; Bernasconi, A.; Borsini, E.; Broggini, G.; Rigamonti, M.; Zecchi, G. J. Org. Chem. 2010, 75, 6923–6932. doi:10.1021/jo101501u
Return to citation in text: [1] -
Broggini, G.; Beccalli, E. M.; Borsini, E.; Fasana, A.; Zecchi, G. Synlett 2011, 227–230. doi:10.1055/s-0030-1259295
Return to citation in text: [1] -
Borsini, E.; Broggini, G.; Colombo, F.; Khansaa, M.; Fasana, A.; Galli, S.; Passarella, D.; Riva, E.; Riva, S. Tetrahedron: Asymmetry 2011, 22, 264–269. doi:10.1016/j.tetasy.2011.01.008
Return to citation in text: [1] -
Borsini, E.; Broggini, G.; Fasana, A.; Galli, S.; Khansaa, M.; Piarulli, U.; Rigamonti, M. Adv. Synth. Catal. 2011, 353, 985–994. doi:10.1002/adsc.201000889
Return to citation in text: [1] -
Beccalli, E. M.; Broggini, G.; Martinelli, M.; Paladino, G. Tetrahedron 2005, 61, 1077–1082. doi:10.1016/j.tet.2004.11.066
Return to citation in text: [1] -
Manzo, A. M.; Perboni, A. D.; Broggini, G.; Rigamonti, M. Tetrahedron Lett. 2009, 50, 4696–4699. doi:10.1016/j.tetlet.2009.06.011
Return to citation in text: [1] -
Manzo, A. M.; Perboni, A. D.; Broggini, G.; Rigamonti, M. Synthesis 2011, 127–132. doi:10.1055/s-0030-1258346
Return to citation in text: [1] -
Chezal, J.-M.; Paeshuyse, J.; Gaumet, V.; Canitron, D.; Maisonial, A.; Lartigue, C.; Gueiffier, A.; Moreau, E.; Teulade, J.-C.; Chavignon, O.; Neyts, J. Eur. J. Med. Chem. 2010, 45, 2044–2047. doi:10.1016/j.ejmech.2010.01.023
Return to citation in text: [1] -
Sahoo, S. P.; Chen, M.-H.; Dykstra, K. D.; Koyama, H.; Meinke, P. T.; O'Keefe, S. J.; Yang, G. X.-Q. Pyrrolo [2,3-c] pyridine derivatives as P38 kinase inhibiting agents. PCT Patent WO2009/152072 A1, Dec 17, 2009.
Return to citation in text: [1] -
Giblin, G. M. P.; Billinton, A.; Briggs, M.; Brown, A. J.; Chessell, I. P.; Clayton, N. M.; Eatherton, A. J.; Goldsmith, P.; Haslam, C.; Johnson, M. R.; Mitchell, W. L.; Naylor, A.; Perboni, A.; Slingsby, B. P.; Wilson, A. W. J. Med. Chem. 2009, 52, 5785–5788. doi:10.1021/jm9009857
Return to citation in text: [1] -
Vanotti, E.; Amici, R. R.; Bargiotti, A.; Berthelsen, J.; Bosotti, R.; Ciavolella, A.; Cirla, A.; Cristiani, C.; D’Alessio, R.; Forte, B.; Isacchi, A.; Martina, K.; Menichincheri, M.; Molinari, A.; Montagnoli, A.; Orsini, P.; Pillan, A.; Roletto, F.; Scolaro, A.; Tibolla, M.; Valsasina, B.; Varasi, M.; Volpi, D.; Santocanale, C. J. Med. Chem. 2008, 51, 487–501. doi:10.1021/jm700956r
Return to citation in text: [1] -
Cheng, L.; Helgesson, S. A. Therapeutic Agents. PCT Patent WO2008/119999 A1, Oct 9, 2008.
Return to citation in text: [1] -
Ni, F.; Kota, S.; Takahashi, V.; Strosberg, A. D.; Snyder, J. K. Bioorg. Med. Chem. Lett. 2011, 21, 2198–2202. doi:10.1016/j.bmcl.2011.03.014
Return to citation in text: [1] -
Vintonyak, V. V.; Waldmann, H.; Rauh, D. Bioorg. Med. Chem. 2011, 19, 2145–2155. doi:10.1016/j.bmc.2011.02.047
Return to citation in text: [1] -
Li, G.-B.; Yang, L.-L.; Feng, S.; Zhou, J.-P.; Huang, Q.; Xie, H.-Z.; Li, L.-L.; Yang, S.-Y. Bioorg. Med. Chem. Lett. 2011, 21, 1736–1740. doi:10.1016/j.bmcl.2011.01.087
Return to citation in text: [1] -
Chen, J.; Liu, T.; Wu, R.; Lou, J.; Dong, X.; He, Q.; Yang, B.; Hu, Y. Eur. J. Med. Chem. 2011, 46, 1343–1347. doi:10.1016/j.ejmech.2011.01.057
Return to citation in text: [1] -
Martini, E.; Mannelli, L. C.; Bartolucci, G.; Bertucci, C.; Dei, S.; Ghelardini, C.; Guandalini, L.; Manetti, D.; Scapecchi, S.; Teodori, E.; Romanelli, M. N. J. Med. Chem. 2011, 54, 2512–2516. doi:10.1021/jm101376k
Return to citation in text: [1] -
Simard, D.; Leblanc, Y.; Berthelette, C.; Zaghdane, M. H.; Molinaro, C.; Wang, Z.; Gallant, M.; Lau, S.; Thao, T.; Hamel, M.; Stocco, R.; Sawyer, N.; Sillaots, S.; Gervais, F.; Houle, R.; Lévesque, J.-F. Bioorg. Med. Chem. Lett. 2011, 21, 841–845. doi:10.1016/j.bmcl.2010.11.084
Return to citation in text: [1] -
Johnson, T. W.; Tanis, S. P.; Butler, S. L.; Dalvie, D.; DeLisle, D. M.; Dress, K. R.; Flahive, E. J.; Hu, Q.; Kuehler, J. E.; Kuki, A.; Liu, W.; McClellan, G. A.; Peng, Q.; Plewe, M. B.; Richardson, P. F.; Smith, G. L.; Solowiej, J.; Tran, K. T.; Wang, H.; Yu, X.; Zhang, J.; Zhu, H. J. Med. Chem. 2011, 54, 3393–3417. doi:10.1021/jm200208d
Return to citation in text: [1] -
Bi, W.; Bi, Y.; Xue, P.; Zhang, Y.; Gao, X.; Wang, Z.; Li, M.; Baudy-Floc'h, M.; Ngerebara, N.; Gibson, M. K.; Bi, L. Eur. J. Med. Chem. 2011, 46, 1453–1462. doi:10.1016/j.ejmech.2011.01.021
Return to citation in text: [1] -
Yang, M.-L.; Kuo, P.-C.; Hwang, T.-L.; Chiou, W.-F.; Qian, K.; Lai, C.-Y.; Lee, K.-H.; Wu, T.-S. Bioorg. Med. Chem. 2011, 19, 1674–1682. doi:10.1016/j.bmc.2011.01.034
Return to citation in text: [1] -
Duan, J.-X.; Cai, X.; Meng, F.; Sun, J. D.; Liu, Q.; Jung, D.; Jiao, H.; Matteucci, J.; Jung, B.; Bhupathi, D.; Ahluwalia, D.; Huang, H.; Hart, C. P.; Matteucci, M. J. Med. Chem. 2011, 54, 1715–1723. doi:10.1021/jm101354u
Return to citation in text: [1]
| 1. | Nakamura, I.; Yamamoto, Y. Chem. Rev. 2004, 104, 2127–2198. doi:10.1021/cr020095i |
| 2. | Li, J. J.; Gribble, G. W. Palladium in Heterocyclic Chemistry. A Guide for the Synthetic Chemist; Pergamon: New York, 2000. |
| 3. | Godoi, B.; Schumacher, R. F.; Zeni, G. Chem. Rev. 2011, 111, 2937–2980. doi:10.1021/cr100214d |
| 4. | Beccalli, E. M.; Broggini, G.; Fasana, A.; Rigamonti, M. J. Organomet. Chem. 2011, 696, 277–295. doi:10.1016/j.jorganchem.2010.09.078 |
| 5. | Patil, N. T.; Yamamoto, Y. Chem. Rev. 2011, 111, 3395–3442. doi:10.1021/cr050041j |
| 19. | Ferrer, C.; Echavarren, A. M. Angew. Chem., Int. Ed. 2006, 45, 1105–1109. doi:10.1002/anie.200503484 |
| 18. | Gruit, M. M.; Michalik, D.; Krüger, K.; Spannenberg, A.; Tillack, A.; Pews-Davtyan, A.; Beller, M. Tetrahedron 2010, 66, 3341–3352. doi:10.1016/j.tet.2010.02.091 |
| 16. | England, D. B.; Padwa, A. Org. Lett. 2008, 10, 3631–3634. doi:10.1021/ol801385h |
| 17. | Verniest, G.; England, D.; De Kimpe, N.; Padwa, A. Tetrahedron 2010, 66, 1496–1502. doi:10.1016/j.tet.2009.10.033 |
| 6. | Shen, H. C. Tetrahedron 2008, 64, 3885–3903. doi:10.1016/j.tet.2008.01.081 |
| 7. | Shen, H. C. Tetrahedron 2008, 64, 7847–7870. doi:10.1016/j.tet.2008.05.082 |
| 8. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319 |
| 9. | Arcadi, A. Chem. Rev. 2008, 108, 3266–3325. doi:10.1021/cr068435d |
| 10. | Marion, N.; Nolan, S. P. Chem. Soc. Rev. 2008, 37, 1776–1782. doi:10.1039/b711132k |
| 11. | Fürstner, A. Chem. Soc. Rev. 2009, 38, 3208–3221. doi:10.1039/b816696j |
| 12. | Shapiro, N. D.; Toste, F. D. Synlett 2010, 675–691. doi:10.1055/s-0029-1219369 |
| 13. | Corma, A.; Leyva-Pérez, A.; Sabater, M. J. Chem. Rev. 2011, 111, 1657–1712. doi:10.1021/cr100414u |
| 14. | Krause, N.; Winter, C. Chem. Rev. 2011, 111, 1994–2009. doi:10.1021/cr1004088 |
| 15. | Bandini, M. Chem. Soc. Rev. 2011, 40, 1358–1367. doi:10.1039/c0cs00041h |
| 36. | Chezal, J.-M.; Paeshuyse, J.; Gaumet, V.; Canitron, D.; Maisonial, A.; Lartigue, C.; Gueiffier, A.; Moreau, E.; Teulade, J.-C.; Chavignon, O.; Neyts, J. Eur. J. Med. Chem. 2010, 45, 2044–2047. doi:10.1016/j.ejmech.2010.01.023 |
| 37. | Sahoo, S. P.; Chen, M.-H.; Dykstra, K. D.; Koyama, H.; Meinke, P. T.; O'Keefe, S. J.; Yang, G. X.-Q. Pyrrolo [2,3-c] pyridine derivatives as P38 kinase inhibiting agents. PCT Patent WO2009/152072 A1, Dec 17, 2009. |
| 38. | Giblin, G. M. P.; Billinton, A.; Briggs, M.; Brown, A. J.; Chessell, I. P.; Clayton, N. M.; Eatherton, A. J.; Goldsmith, P.; Haslam, C.; Johnson, M. R.; Mitchell, W. L.; Naylor, A.; Perboni, A.; Slingsby, B. P.; Wilson, A. W. J. Med. Chem. 2009, 52, 5785–5788. doi:10.1021/jm9009857 |
| 39. | Vanotti, E.; Amici, R. R.; Bargiotti, A.; Berthelsen, J.; Bosotti, R.; Ciavolella, A.; Cirla, A.; Cristiani, C.; D’Alessio, R.; Forte, B.; Isacchi, A.; Martina, K.; Menichincheri, M.; Molinari, A.; Montagnoli, A.; Orsini, P.; Pillan, A.; Roletto, F.; Scolaro, A.; Tibolla, M.; Valsasina, B.; Varasi, M.; Volpi, D.; Santocanale, C. J. Med. Chem. 2008, 51, 487–501. doi:10.1021/jm700956r |
| 40. | Cheng, L.; Helgesson, S. A. Therapeutic Agents. PCT Patent WO2008/119999 A1, Oct 9, 2008. |
| 41. | Ni, F.; Kota, S.; Takahashi, V.; Strosberg, A. D.; Snyder, J. K. Bioorg. Med. Chem. Lett. 2011, 21, 2198–2202. doi:10.1016/j.bmcl.2011.03.014 |
| 42. | Vintonyak, V. V.; Waldmann, H.; Rauh, D. Bioorg. Med. Chem. 2011, 19, 2145–2155. doi:10.1016/j.bmc.2011.02.047 |
| 43. | Li, G.-B.; Yang, L.-L.; Feng, S.; Zhou, J.-P.; Huang, Q.; Xie, H.-Z.; Li, L.-L.; Yang, S.-Y. Bioorg. Med. Chem. Lett. 2011, 21, 1736–1740. doi:10.1016/j.bmcl.2011.01.087 |
| 44. | Chen, J.; Liu, T.; Wu, R.; Lou, J.; Dong, X.; He, Q.; Yang, B.; Hu, Y. Eur. J. Med. Chem. 2011, 46, 1343–1347. doi:10.1016/j.ejmech.2011.01.057 |
| 45. | Martini, E.; Mannelli, L. C.; Bartolucci, G.; Bertucci, C.; Dei, S.; Ghelardini, C.; Guandalini, L.; Manetti, D.; Scapecchi, S.; Teodori, E.; Romanelli, M. N. J. Med. Chem. 2011, 54, 2512–2516. doi:10.1021/jm101376k |
| 46. | Simard, D.; Leblanc, Y.; Berthelette, C.; Zaghdane, M. H.; Molinaro, C.; Wang, Z.; Gallant, M.; Lau, S.; Thao, T.; Hamel, M.; Stocco, R.; Sawyer, N.; Sillaots, S.; Gervais, F.; Houle, R.; Lévesque, J.-F. Bioorg. Med. Chem. Lett. 2011, 21, 841–845. doi:10.1016/j.bmcl.2010.11.084 |
| 47. | Johnson, T. W.; Tanis, S. P.; Butler, S. L.; Dalvie, D.; DeLisle, D. M.; Dress, K. R.; Flahive, E. J.; Hu, Q.; Kuehler, J. E.; Kuki, A.; Liu, W.; McClellan, G. A.; Peng, Q.; Plewe, M. B.; Richardson, P. F.; Smith, G. L.; Solowiej, J.; Tran, K. T.; Wang, H.; Yu, X.; Zhang, J.; Zhu, H. J. Med. Chem. 2011, 54, 3393–3417. doi:10.1021/jm200208d |
| 48. | Bi, W.; Bi, Y.; Xue, P.; Zhang, Y.; Gao, X.; Wang, Z.; Li, M.; Baudy-Floc'h, M.; Ngerebara, N.; Gibson, M. K.; Bi, L. Eur. J. Med. Chem. 2011, 46, 1453–1462. doi:10.1016/j.ejmech.2011.01.021 |
| 49. | Yang, M.-L.; Kuo, P.-C.; Hwang, T.-L.; Chiou, W.-F.; Qian, K.; Lai, C.-Y.; Lee, K.-H.; Wu, T.-S. Bioorg. Med. Chem. 2011, 19, 1674–1682. doi:10.1016/j.bmc.2011.01.034 |
| 50. | Duan, J.-X.; Cai, X.; Meng, F.; Sun, J. D.; Liu, Q.; Jung, D.; Jiao, H.; Matteucci, J.; Jung, B.; Bhupathi, D.; Ahluwalia, D.; Huang, H.; Hart, C. P.; Matteucci, M. J. Med. Chem. 2011, 54, 1715–1723. doi:10.1021/jm101354u |
| 34. | Manzo, A. M.; Perboni, A. D.; Broggini, G.; Rigamonti, M. Tetrahedron Lett. 2009, 50, 4696–4699. doi:10.1016/j.tetlet.2009.06.011 |
| 35. | Manzo, A. M.; Perboni, A. D.; Broggini, G.; Rigamonti, M. Synthesis 2011, 127–132. doi:10.1055/s-0030-1258346 |
| 33. | Beccalli, E. M.; Broggini, G.; Martinelli, M.; Paladino, G. Tetrahedron 2005, 61, 1077–1082. doi:10.1016/j.tet.2004.11.066 |
| 20. | Broggini, G.; Molteni, G.; Terraneo, A.; Zecchi, G. Heterocycles 2003, 59, 823–858. doi:10.3987/REV-02-SR5 |
| 21. | Beccalli, E. M.; Broggini, G.; Martinelli, M.; Paladino, G.; Zoni, C. Eur. J. Org. Chem. 2005, 2091–2096. doi:10.1002/ejoc.200400817 |
| 22. | Abbiati, G.; Beccalli, E. M.; Broggini, G.; Paladino, G.; Rossi, E. Synthesis 2005, 2881–2886. doi:10.1055/s-2005-916033 |
| 23. | Abbiati, G.; Beccalli, E. M.; Broggini, G.; Martinelli, M.; Paladino, G. Synlett 2006, 73–76. doi:10.1055/s-2005-922788 |
| 24. | Beccalli, E. M.; Broggini, G.; Martinelli, M.; Masciocchi, N.; Sottocornola, S. Org. Lett. 2006, 8, 4521–4524. doi:10.1021/ol061693c |
| 25. | Beccalli, E. M.; Borsini, E.; Broggini, G.; Rigamonti, M.; Sottocornola, S. Synlett 2008, 1053–1057. doi:10.1055/s-2008-1072583 |
| 26. | Beccalli, E. M.; Broggini, G.; Clerici, F.; Galli, S.; Kammerer, C.; Rigamonti, M.; Sottocornola, S. Org. Lett. 2009, 11, 1563–1566. doi:10.1021/ol900171g |
| 27. | Basolo, L.; Beccalli, E. M.; Borsini, E.; Broggini, G. Tetrahedron 2009, 65, 3486–3491. doi:10.1016/j.tet.2009.02.025 |
| 28. | Beccalli, E. M.; Borsini, E.; Brenna, S.; Galli, S.; Rigamonti, M.; Broggini, G. Chem.–Eur. J. 2010, 16, 1670–1678. doi:10.1002/chem.200902071 |
| 29. | Beccalli, E. M.; Bernasconi, A.; Borsini, E.; Broggini, G.; Rigamonti, M.; Zecchi, G. J. Org. Chem. 2010, 75, 6923–6932. doi:10.1021/jo101501u |
| 30. | Broggini, G.; Beccalli, E. M.; Borsini, E.; Fasana, A.; Zecchi, G. Synlett 2011, 227–230. doi:10.1055/s-0030-1259295 |
| 31. | Borsini, E.; Broggini, G.; Colombo, F.; Khansaa, M.; Fasana, A.; Galli, S.; Passarella, D.; Riva, E.; Riva, S. Tetrahedron: Asymmetry 2011, 22, 264–269. doi:10.1016/j.tetasy.2011.01.008 |
| 32. | Borsini, E.; Broggini, G.; Fasana, A.; Galli, S.; Khansaa, M.; Piarulli, U.; Rigamonti, M. Adv. Synth. Catal. 2011, 353, 985–994. doi:10.1002/adsc.201000889 |
| 18. | Gruit, M. M.; Michalik, D.; Krüger, K.; Spannenberg, A.; Tillack, A.; Pews-Davtyan, A.; Beller, M. Tetrahedron 2010, 66, 3341–3352. doi:10.1016/j.tet.2010.02.091 |
© 2011 Borsini et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)