Abstract
New trans- and cis-o-stilbene-methylene-sydnones 3a,b were synthesized by transforming the trans- and cis-o-aminomethylstilbene derivative, obtained by reduction of corresponding o-cyano derivatives, into glycine ester derivatives (43 and 31% yield) followed by hydrolysis (90 and 96% yield), nitrosation and ring closure with acetic acid anhydride (30 and 40% yield). The products were submitted to photochemical and thermal intramolecular [3 + 2] cycloadditions to afford diverse heteropolycyclic compounds. Photochemical reactions afforded cis-3-(4-methylphenyl)-3a,8-dihydro-3H-pyrazolo[5,1-a]isoindole (11, 12.5% yield) and trans-3-(4-methylphenyl)-3a,8-dihydro-3H-pyrazolo[5,1-a]isoindole (12, 5% yield). Thermal reactions afforded 3-(4-methylphenyl)-3,3a,8,8a-tetrahydroindeno[2,1-c]pyrazole (14, 50% yield) and 11-(4-methylphenyl)-9,10-diazatricyclo[7.2.1.02,7]dodeca-2,4,6,10-tetraene (15, 22% yield).

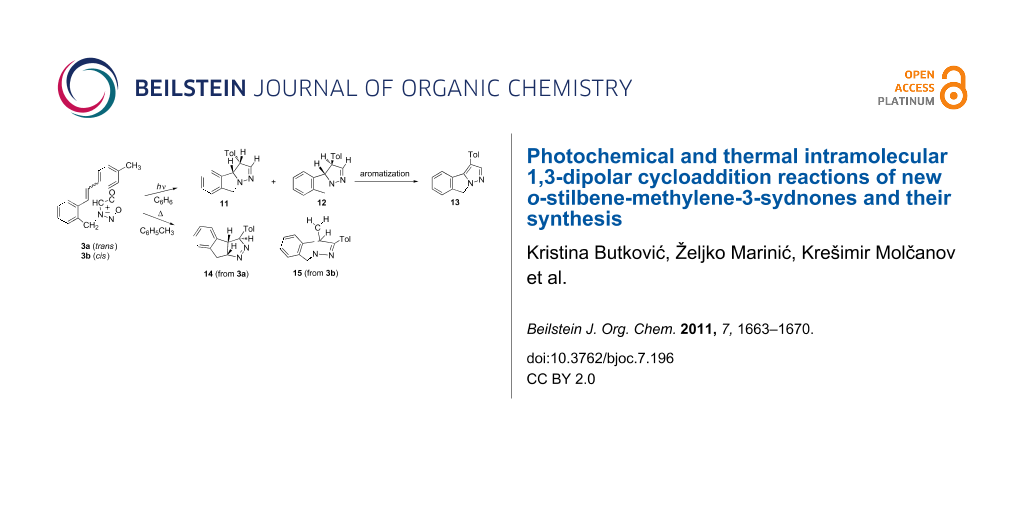
Graphical Abstract
Introduction
Sydnones belong to the group of five-membered heterocyclic compounds referred to as being "mesoionic" and have been widely studied since their discovery [1-5]. They can be represented as hybrids of a number of mesomeric ionic structures (Figure 1).
![[1860-5397-7-196-1]](/bjoc/content/figures/1860-5397-7-196-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Resonance structures of the sydnone ring.
Figure 1: Resonance structures of the sydnone ring.
One of the most characteristic reactions of sydnones is the intermolecular 1,3-dipolar cycloaddition. In the presence of acetylenic or ethylenic dipolarophiles, sydnones undergo cycloaddition reactions, which can be induced thermally [4,6,7] or photochemically [8-17], giving different pyrazole and/or pyrazoline derivatives, depending on the applied dipolarophile (Scheme 1). Namely, sydnones are masked 1,3-dipoles that by photolysis give nitrile imine intermediates, or in thermal reactions react as cyclic azomethine imines.
![[1860-5397-7-196-i1]](/bjoc/content/inline/1860-5397-7-196-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Thermal and photochemical intermolecular [3 + 2] cycloadditions.
Scheme 1: Thermal and photochemical intermolecular [3 + 2] cycloadditions.
Intramolecular 1,3-dipolar cycloadditions of sydnone derivatives have not been as thoroughly investigated, and so far only a few examples are known [18-20]. Photochemically induced intramolecular 1,3-dipolar cycloadditions have been studied on 3,4-disubstituted sydnone derivatives [18,19] (Figure 2, A and B), wherein indolopyrazole and pyrazolobenzoxazine structures are formed (Figure 2, C and D).
![[1860-5397-7-196-2]](/bjoc/content/figures/1860-5397-7-196-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Illustration of intramolecular [3 + 2] cycloadditions.
Figure 2: Illustration of intramolecular [3 + 2] cycloadditions.
Heimgartner and coworkers also carried out the thermally induced reaction of 3-(o-allylphenyl)-4-phenylsydnone (A) and obtained the cycloadduct E (Figure 2) with the oxycarbonyl group remaining in the structure [18].
We have been studying photochemical reactions of conjugated heterostilbene derivatives in which the sydnone moiety is part of a heterostilbene [17] (1, Figure 3) or is directly attached at the ortho position to the stilbene 2 [21-23]. Upon photolysis of compound 1, where the sydnone moiety is part of a heterostilbene system, cis–trans isomerization was the main process, and no intramolecular cycloadducts were found owing to the unfavourable conformation of the formed intermediate in the trans configuration. The existence of the nitrile imine intermediate as a result of competitive photolysis of the sydnone moiety was confirmed on irradiation of 1 in the presence of acrolein and isolation of the pyrazoline derivative (F, Figure 3) [17]. In the case of stilbenylsydnones 2, where the sydnone moiety is directly connected to the ortho position of the stilbene, the cyclization of the formed nitrile imine intermediate leading to benzodiazepine ring closure (G, Figure 3) was the main intramolecular process [22]. In a continuation of our interest in the synthesis of heteropolycyclic compounds we extended our research to new stilbene-sydnone derivatives 3 (Figure 3). In such a system, where two chromophores, stilbene and sydnone, are divided by a methylene bridge, an intramolecular 1,3-dipolar cycloaddition and the formation of diverse polycyclic compounds could be expected. Herein we describe, for the first time, the synthesis of cis- and trans-3-(stilbenylmethyl)sydnones and their photochemical and thermal intramolecular transformations to heteropolycyclic structures.
![[1860-5397-7-196-3]](/bjoc/content/figures/1860-5397-7-196-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Styryl-sydnone 1 and stilbenyl sydnone 2 and their photoproducts F and G, respectively; target molecules 3 in this work.
Figure 3: Styryl-sydnone 1 and stilbenyl sydnone 2 and their photoproducts F and G, respectively; target mole...
Results and Discussion
In the investigation of 3-{2-[2-(4-tolyl)ethenyl]phenyl}methylsydnones, 3a (trans) and 3b (cis), were prepared by a sequence of reactions (Scheme 2) starting from o-cyanotoluene (see Supporting Information File 1 for full experimental data). Bromination of o-cyanotoluene afforded 2-(bromomethyl)benzonitrile (4) [24], which was transformed to triphenylphosphonium salt 5 [25] followed by Wittig reaction with 4-methylbenzaldehyde to 2-(4-methylstyryl)benzonitrile (6a,b) [26]. The product was obtained as a mixture of 6a (trans isomer, 40%) and 6b (cis isomer, 60%).
![[1860-5397-7-196-i2]](/bjoc/content/inline/1860-5397-7-196-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the target molecules 3a and 3b.
Scheme 2: Synthesis of the target molecules 3a and 3b.
The isomers were separated by column chromatography and further treated separately to achieve the final products in cis and trans configurations. Reduction of 6a (trans) or 6b (cis) with LiAlH4 in anhydrous ether afforded amino derivative 7a (trans, 94%) or 7b (cis, 93%). In the 1H NMR spectra new signals from the methylene protons appeared at 3.88 ppm (7a) and 3.62 ppm (7b) confirming the reduction. By further nucleophilic substitution, from 7a (trans) or 7b (cis) and ethyl bromoacetate, the ester 8a (trans) or 8b (cis) was prepared. On purification by column chromatography, the byproducts, obtained by disubstitution reaction of the amino compound, were separated, and the pure 8a (43%) or 8b (31%) was isolated. The obtained esters showed the presence of the carbonyl group at ~1740 cm−1 in the IR spectra and the carbonyl carbon at ~172 ppm in the 13C NMR spectra. The esters 8a or 8b were hydrolysed to the amino acid 9a (trans, 90%) or 9b (cis, 96%). The obtained amino acids were transformed to N-nitroso glycine 10a or 10b and, without isolation or further purification, were submitted to dehydration with acetic acid anhydride to give sydnone 3a (trans) or 3b (cis). After column chromatography the pure 3a (30%) or 3b (42%) was isolated. The best indication that the sydnone structures were formed was given by the singlet at ~6 ppm in the 1H NMR spectrum, characteristic for the proton H-4 in the sydnone ring, as well as those in the 13C NMR spectrum, namely the CH and CO sydnone carbons at ~94 and ~169 ppm, respectively.
The irradiation experiments with the trans isomer (3a) or cis isomer (3b) were performed in ~10−3 M benzene solution in a Rayonet reactor at 300 nm under anaerobic conditions (purged with argon). The absorption maximum of trans isomer (3a) is at 300 nm (ε 37453) and of cis isomer (3b) at 291 nm (ε 16228), thus upon irradiation both isomers were excited. The irradiation of the isomers was performed until full conversion. The irradiation of either the trans or cis isomer, or of the mixture of isomers, resulted in the formation of two products in the same mutual ratio, along with large amounts of unidentified high-molecular-weight products. Separation by column chromatography in combination with thin layer chromatography gave dihydropyrazolo-isoindoles, 11 (12.5%) and 12 (5%) (Scheme 3).
![[1860-5397-7-196-i3]](/bjoc/content/inline/1860-5397-7-196-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
The structure of the photoproducts was determined by spectroscopic methods. The molecular ions of 11 and 12, m/z 248 in the mass spectra, indicate that the structures have lost CO2 relative to the starting compound. In the 1H and 13C NMR spectra, the signals are found in an area which is characteristic for saturated cyclic compounds. The major product is assigned to compound 11 based on the following data: In 1H NMR spectrum the signals at 4.98 ppm and 4.43 ppm are doublets with coupling constants of 15.6 Hz and are assigned to geminal protons G-1 and G-2. The other two signals at 4.92 and 4.24 ppm are also doublets but with coupling constants of 10.8 Hz. Based on the interaction of the proton at 5.88 with the proton at 4.92 ppm in the NOESY spectrum, the doublet at 5.88 is assigned to proton H-5, the doublet at 4.92 ppm to proton B and the doublet at 4.24 to proton C. Interaction of proton H-5 with proton B is also visible in HMBC spectrum. The signal at 6.53 ppm was assigned to proton A based on COSY interaction with proton C. The rather large high-field shift of the aromatic proton H-5 can be explained by an anisotropic effect of the tolyl group and thus confirms the cis orientation of protons B and C.
The structure of the minor photoproduct 12, different from the structure 11 only in trans orientation of the protons B and C, was also evident from NMR spectra by using 2D NMR techniques. The doublets at 4.90 ppm and 4.37 ppm with a coupling constant of 15.6 Hz are assigned to geminal protons G. The singlet at 6.57 ppm is assigned to proton A. The chemical shift of proton A is the same as in structure 11. The other two signals at 3.97 ppm and 4.70 ppm appear as singlets, and they are assigned to protons C and B, respectively, based on weak interactions in the COSY and NOESY spectra. Nevertheless, the proton H-5 of the fused benzene ring in structure 12 is in the multiplet together with other aromatic protons, which is in accordance with the proposed structure.
The structure of the photoproducts was confirmed by an additional experiment on the crude reaction mixture with DDQ (Scheme 4) in which, as expected for the predicted structures, the aromatization reaction took place forming the pyrazolo-isoindole 13. Compound 13 arose also on silica gel during the purification of either 11 or 12.
![[1860-5397-7-196-i4]](/bjoc/content/inline/1860-5397-7-196-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
The irradiation of 3a or 3b until full conversion, as previously mentioned, produced a mixture of 11 and 12 along with decomposition and high-molecular-weight products. On shorter irradiation time (10 min, in benzene or acetonitrile) 3a (trans isomer) afforded, according to 1H NMR, the photomixture of predominantly 3b (cis isomer) with only traces of starting 3a and tricyclic photoproducts 11 and 12. Under the same irradiation conditions, 3b (cis isomer) as starting compound gave a photomixture of cis isomer and the newly formed product 11 in 1:1 ratio with only traces of 3a (trans isomer), along with some unidentified side products. The experimental results show that the trans-(3a) and cis-(3b) isomerize and react with different efficiency, and that the isomerization, as in the case of stilbene itself [27], is shifted toward the cis isomer. It follows that the reaction is stereospecific and that photoproduct 11 is formed from the cis configuration of the stilbene moiety and the photoproduct 12 from the trans configuration, although the formation of 12 via epimerization of 11 could not be eliminated. It is also evident that there are several competitive processes, which are summarized in Scheme 5.
![[1860-5397-7-196-i5]](/bjoc/content/inline/1860-5397-7-196-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Possible mechanism for the formation of the photoproducts.
Scheme 5: Possible mechanism for the formation of the photoproducts.
On irradiation of 3a (trans) or 3b (cis) parallel competitive processes are in operation, namely, trans–cis and cis–trans isomerization of the stilbene moiety, and photolysis of the sydnone ring resulting in the formation of the nitrile imine intermediate. The nitrile imine species is, in intramolecular dipolar [3 + 2] cycloaddition, trapped by the cis- or trans-double bond of the stilbene, giving cycloadducts 11 or 12, respectively.
We also performed the thermal intramolecular reactions with the starting compounds 3a and 3b. Theoretically the intramolecular 1,3-dipolar cycloaddition of the sydnone moiety, acting as a masked azomethine dipole, and the double bond of the stilbene moiety could proceed in different ways. The orientation of the sydnone ring toward the π bond of the stilbene in combination with the double bond configuration can give various formal [3 + 2] intramolecular cycloadducts.
On heating of 3a (trans) in toluene until full conversion (4 h) one product in 50% yield was isolated from the reaction mixture after column chromatography. From the molecular ion (m/z 248) of the product and its 13C NMR spectrum it was obvious that in the cycloaddition CO2 elimination took place. Fragmentation of the product and the presence of an ion at m/z 220 suggests a structure in which the expulsion of nitrogen is possible. The structure 14 (Scheme 6) was determined by additional NMR techniques, NOE and HMBC interactions, and by single crystal X-ray structure analysis (Figure 4) of the crystal formed in an NMR tube by slow evaporation of the solvent.
![[1860-5397-7-196-i6]](/bjoc/content/inline/1860-5397-7-196-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-7-196-4]](/bjoc/content/figures/1860-5397-7-196-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Also, when compound 3b (cis) was refluxed in xylene (9 h) or toluene (19 h) only one cycloadduct 15 was isolated (Scheme 7), besides decomposition products, in 22% yield. The structure of 15 was determined by spectroscopic methods.
![[1860-5397-7-196-i7]](/bjoc/content/inline/1860-5397-7-196-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
In the 1H NMR spectrum two pairs of geminal protons were found at 4.51 and 4.21 ppm (A, B) and at 3.71 and 3.35 ppm (E, D). The doublet at 3.95 ppm, coupled with one geminal proton (D), was assigned to proton C. In the 13C NMR spectrum, one of the five quaternary carbons is shifted to 176 ppm, which corresponds to an sp2-carbon in the vicinity of nitrogen. In the NOESY spectrum the interaction of protons A and B with an aromatic proton (H-2) at 7.00 ppm is seen, as well as the interaction of proton C with protons E and D. Since the NOE interaction is seen between protons A and E we concluded that protons A and E must lie on the same side of the six-membered ring. In addition, the interaction of proton C with tolyl (H-10) and H-5 protons was seen.
In order to explain the diverse structures (14 and 15) and their formation mechanism, we analysed the possible ways of intramolecular [3 + 2] cycloaddition relating to the arrangement of the sydnone ring towards the cis and trans double bond (Figure 5 and Figure 6).
![[1860-5397-7-196-5]](/bjoc/content/figures/1860-5397-7-196-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Proposed stereochemical pathway of sydnone ring (CH–N) and trans- and cis-stilbene (α–β).
Figure 5: Proposed stereochemical pathway of sydnone ring (CH–N) and trans- and cis-stilbene (α–β).
![[1860-5397-7-196-6]](/bjoc/content/figures/1860-5397-7-196-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Proposed stereochemical pathway of sydnone ring (N–CH) and trans- and cis-stilbene (α–β).
Figure 6: Proposed stereochemical pathway of sydnone ring (N–CH) and trans- and cis-stilbene (α–β).
As presented in Figure 5, the sydnone ring could be oriented to the double bond in such a way that the bonds are formed at the C(Sy)–α(St) and N(Sy)–β(St) positions, or, as presented in Figure 6, at the N(Sy)–α(St) and C(Sy)–β(St) positions. The favoured arrangement of the sydnone ring toward the cis and trans double bond, leading to the products, is the pathway presented in Figure 5. The regiospecific and stereospecific formation of the products 14 and 15 could be explained by this approach of the sydnone ring (Scheme 8). The cycloadducts, cA from trans isomer and cB from cis isomer, lose CO2 under the reaction conditions to afford intermediates 14A and 14 B, respectively. Owing to the favourable conformation in the case of biradical 15A, the 1,3-H abstraction and formation of the C–N double bond in product 15 is possible. In the biradical 14A the intramolecular hydrogen abstraction is not favourable, but 1,2-alkyl shift takes place followed by formation of the N–N double bond in product 14.
![[1860-5397-7-196-i8]](/bjoc/content/inline/1860-5397-7-196-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Possible formation of thermal products 14 (from trans-3) and 15 (from cis-3).
Scheme 8: Possible formation of thermal products 14 (from trans-3) and 15 (from cis-3).
Monitoring the reaction by thin layer chromatography revealed that the [3 + 2] cycloaddition is much faster in the case of the trans isomer (3a). After the 4 h reflux of the toluene solution of the trans isomer, the 1H NMR spectrum of the crude reaction mixture showed complete conversion, while the cis isomer (3b) under the same conditions showed complete conversion only after 19 h. This evidence led us to believe that the formation of the "C–α/Ν–β" adduct cA proceeds via an energetically favoured transition state due to a possible secondary π–π interaction of the tolyl and carbonyl groups.
Conclusion
In photochemical and thermal intramolecular reactions the investigated compounds 3a and 3b, in which the stilbene and sydnone ring are bridged by a methylene group, show the characteristic reaction for stilbene and sydnone moieties. The stilbene moiety photochemically isomerizes and the process of trans–cis isomerization is in competition with the photolysis of the sydnone ring. Photolysis of the sydnone moiety leads to a nitrile imine, followed by its intramolecular trapping by the cis or trans double bond of stilbene moiety, affording polycyclic compounds 11 and 12, respectively. The same starting compounds also react thermally: The sydnone moiety in 3a reacts as a masked azomethine dipole with trans configuration on the stilbene moiety by intramolecular [3 + 2] cycloaddition, giving polycyclic compound 14, while the sydnone moiety in the cis isomer 3b gives polycyclic compound 15. Stilbene-methylene-sydnones are useful substrates for photochemical and thermal intramolecular [3 + 2] cycloaddition reactions to heteropolycyclic compounds.
Supporting Information
| Supporting Information File 1: Experimental details and characterization data for all compounds. | ||
| Format: PDF | Size: 198.3 KB | Download |
| Supporting Information File 2: 1H NMR and APT spectra of 3a, 3b, 11–15, NOESY spectra of 11, 12, 14 and 15 and X-ray data for 14. | ||
| Format: PDF | Size: 1.2 MB | Download |
References
-
Stewart, F. H. C. Chem. Rev. 1964, 64, 129–147. doi:10.1021/cr60228a004
Return to citation in text: [1] -
Newton, C. G.; Ramsden, C. A. Tetrahedron 1982, 38, 2965–3011. doi:10.1016/0040-4020(82)80186-5
Return to citation in text: [1] -
Clapp, L. B. 1,2,3- and 1,2,4-Oxadiazoles. In Comprehensive Heterocyclic Chemistry; Katritzky, A. R.; Rees, C. W., Eds.; Pergamon Press: Oxford, 1984; Vol. 6, pp 365–378. doi:10.1016/B978-008096519-2.00089-8
Return to citation in text: [1] -
Gribble, G. W. Mesoionic Ring Systems. In Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; Padwa, A.; Pearson, W. H., Eds.; Wiley & Sons: New Jersey, 2003; pp 681–753.
Return to citation in text: [1] [2] -
Browne, D. L.; Harrity, J. P. A. Tetrahedron 2010, 66, 553–568. doi:10.1016/j.tet.2009.10.085
Return to citation in text: [1] -
Chang, E.-M.; Chen, T.-H.; Wong, F. F.; Chang, E.-C.; Yeh, M.-Y. Synlett 2006, 901–904. doi:10.1055/s-2006-939041
Return to citation in text: [1] -
Rai, G.; Puranik, V. G.; Kalluraya, B.; Hedge, J. C. Synth. Commun. 2006, 36, 1285–1290. doi:10.1080/00397910500518874
Return to citation in text: [1] -
Krauch, C. H.; Kuhls, J.; Piek, H.-J. Tetrahedron Lett. 1966, 7, 4043–4048. doi:10.1016/S0040-4039(00)90284-3
Return to citation in text: [1] -
Chinone, A.; Huseya, Y.; Ohta, M. Bull. Chem. Soc. Jpn. 1970, 43, 2650. doi:10.1246/bcsj.43.2650
Return to citation in text: [1] -
Huseya, Y.; Chinone, A.; Ohta, M. Bull. Chem. Soc. Jpn. 1971, 44, 1667–1668. doi:10.1246/bcsj.44.1667
Return to citation in text: [1] -
Märky, M.; Hansen, H.-J.; Schmid, H. Helv. Chim. Acta 1971, 54, 1275–1278. doi:10.1002/hlca.19710540506
Return to citation in text: [1] -
Gotthardt, H.; Reiter, F. Tetrahedron Lett. 1971, 12, 2749–2752. doi:10.1016/S0040-4039(01)96970-9
Return to citation in text: [1] -
George, M. V.; Angadiyavar, C. S. J. Org. Chem. 1971, 36, 1589–1594. doi:10.1021/jo00811a004
Return to citation in text: [1] -
Gotthardt, H.; Reiter, F. Chem. Ber. 1979, 112, 1206–1225. doi:10.1002/cber.19791120415
Return to citation in text: [1] -
Gotthardt, H.; Reiter, F. Chem. Ber. 1979, 112, 1635–1649. doi:10.1002/cber.19791120514
Return to citation in text: [1] -
Pfoertner, K.-H.; Foricher, J. Helv. Chim. Acta 1980, 63, 653–657. doi:10.1002/hlca.19800630312
Return to citation in text: [1] -
Butković, K.; Basarić, N.; Lovreković, K.; Marinić, Ž.; Višnjevac, A.; Kojić-Prodić, B.; Šindler-Kulyk, M. Tetrahedron Lett. 2004, 45, 9057–9060. doi:10.1016/j.tetlet.2004.10.036
Return to citation in text: [1] [2] [3] -
Meier, H.; Heimgartner, H.; Schmid, H. Helv. Chim. Acta 1977, 60, 1087–1090. doi:10.1002/hlca.19770600333
Return to citation in text: [1] [2] [3] -
Meier, H.; Heimgartner, H. Helv. Chim. Acta 1986, 69, 927–940. doi:10.1002/hlca.19860690421
Return to citation in text: [1] [2] -
Yeu, J.-P.; Yeh, J.-T.; Chen, T.-Y.; Uang, B.-J. Synthesis 2001, 1775–1777. doi:10.1055/s-2001-17517
Return to citation in text: [1] -
Butković, K.; Marinić, Ž.; Šindler-Kulyk, M. Magn. Reson. Chem. 2004, 42, 1053–1055. doi:10.1002/mrc.1488
Return to citation in text: [1] -
Butković, K.; Vuk, D.; Marinić, Ž.; Penić, J.; Šindler-Kulyk, M. Tetrahedron 2010, 66, 9356–9362. doi:10.1016/j.tet.2010.10.013
Return to citation in text: [1] [2] -
Butković, K.; Marinić, Ž.; Šindler-Kulyk, M. ARKIVOC 2011, 10, 1–15.
Return to citation in text: [1] -
Gorsane, M.; Defay, N.; Martin, R. H. Bull. Soc. Chim. Belg. 1985, 94, 215–231. doi:10.1002/bscb.19850940309
Return to citation in text: [1] -
Palmer, B. D.; Thompson, A. M.; Booth, R. J.; Dobrusin, E. M.; Kraker, A. J.; Lee, H. H.; Lunney, E. A.; Mitchell, L. H.; Ortwine, D. F.; Smaill, J. B.; Swan, L. M.; Denny, W. A. J. Med. Chem. 2006, 49, 4896–4911. doi:10.1021/jm0512591
Return to citation in text: [1] -
Husemoen, G.; Olsson, R.; Andersson, C.-M.; Harvey, S. C.; Hansen, H. C. J. Comb. Chem. 2003, 5, 606–609.
(The isomers are not separarted.)
Return to citation in text: [1] -
Saltiel, J.; Sears, D. F., Jr.; Ko, D.-H.; Park, K.-M. Cis-Trans Isomerization of Alkenes. In CRC Handbook of Photochemistry and Photobiology; Horspool, W., Ed.; CRC Press: Boca Raton, 1995; pp 3–15.
Return to citation in text: [1]
| 1. | Stewart, F. H. C. Chem. Rev. 1964, 64, 129–147. doi:10.1021/cr60228a004 |
| 2. | Newton, C. G.; Ramsden, C. A. Tetrahedron 1982, 38, 2965–3011. doi:10.1016/0040-4020(82)80186-5 |
| 3. | Clapp, L. B. 1,2,3- and 1,2,4-Oxadiazoles. In Comprehensive Heterocyclic Chemistry; Katritzky, A. R.; Rees, C. W., Eds.; Pergamon Press: Oxford, 1984; Vol. 6, pp 365–378. doi:10.1016/B978-008096519-2.00089-8 |
| 4. | Gribble, G. W. Mesoionic Ring Systems. In Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; Padwa, A.; Pearson, W. H., Eds.; Wiley & Sons: New Jersey, 2003; pp 681–753. |
| 5. | Browne, D. L.; Harrity, J. P. A. Tetrahedron 2010, 66, 553–568. doi:10.1016/j.tet.2009.10.085 |
| 18. | Meier, H.; Heimgartner, H.; Schmid, H. Helv. Chim. Acta 1977, 60, 1087–1090. doi:10.1002/hlca.19770600333 |
| 19. | Meier, H.; Heimgartner, H. Helv. Chim. Acta 1986, 69, 927–940. doi:10.1002/hlca.19860690421 |
| 18. | Meier, H.; Heimgartner, H.; Schmid, H. Helv. Chim. Acta 1977, 60, 1087–1090. doi:10.1002/hlca.19770600333 |
| 19. | Meier, H.; Heimgartner, H. Helv. Chim. Acta 1986, 69, 927–940. doi:10.1002/hlca.19860690421 |
| 20. | Yeu, J.-P.; Yeh, J.-T.; Chen, T.-Y.; Uang, B.-J. Synthesis 2001, 1775–1777. doi:10.1055/s-2001-17517 |
| 8. | Krauch, C. H.; Kuhls, J.; Piek, H.-J. Tetrahedron Lett. 1966, 7, 4043–4048. doi:10.1016/S0040-4039(00)90284-3 |
| 9. | Chinone, A.; Huseya, Y.; Ohta, M. Bull. Chem. Soc. Jpn. 1970, 43, 2650. doi:10.1246/bcsj.43.2650 |
| 10. | Huseya, Y.; Chinone, A.; Ohta, M. Bull. Chem. Soc. Jpn. 1971, 44, 1667–1668. doi:10.1246/bcsj.44.1667 |
| 11. | Märky, M.; Hansen, H.-J.; Schmid, H. Helv. Chim. Acta 1971, 54, 1275–1278. doi:10.1002/hlca.19710540506 |
| 12. | Gotthardt, H.; Reiter, F. Tetrahedron Lett. 1971, 12, 2749–2752. doi:10.1016/S0040-4039(01)96970-9 |
| 13. | George, M. V.; Angadiyavar, C. S. J. Org. Chem. 1971, 36, 1589–1594. doi:10.1021/jo00811a004 |
| 14. | Gotthardt, H.; Reiter, F. Chem. Ber. 1979, 112, 1206–1225. doi:10.1002/cber.19791120415 |
| 15. | Gotthardt, H.; Reiter, F. Chem. Ber. 1979, 112, 1635–1649. doi:10.1002/cber.19791120514 |
| 16. | Pfoertner, K.-H.; Foricher, J. Helv. Chim. Acta 1980, 63, 653–657. doi:10.1002/hlca.19800630312 |
| 17. | Butković, K.; Basarić, N.; Lovreković, K.; Marinić, Ž.; Višnjevac, A.; Kojić-Prodić, B.; Šindler-Kulyk, M. Tetrahedron Lett. 2004, 45, 9057–9060. doi:10.1016/j.tetlet.2004.10.036 |
| 26. |
Husemoen, G.; Olsson, R.; Andersson, C.-M.; Harvey, S. C.; Hansen, H. C. J. Comb. Chem. 2003, 5, 606–609.
(The isomers are not separarted.) |
| 4. | Gribble, G. W. Mesoionic Ring Systems. In Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; Padwa, A.; Pearson, W. H., Eds.; Wiley & Sons: New Jersey, 2003; pp 681–753. |
| 6. | Chang, E.-M.; Chen, T.-H.; Wong, F. F.; Chang, E.-C.; Yeh, M.-Y. Synlett 2006, 901–904. doi:10.1055/s-2006-939041 |
| 7. | Rai, G.; Puranik, V. G.; Kalluraya, B.; Hedge, J. C. Synth. Commun. 2006, 36, 1285–1290. doi:10.1080/00397910500518874 |
| 27. | Saltiel, J.; Sears, D. F., Jr.; Ko, D.-H.; Park, K.-M. Cis-Trans Isomerization of Alkenes. In CRC Handbook of Photochemistry and Photobiology; Horspool, W., Ed.; CRC Press: Boca Raton, 1995; pp 3–15. |
| 17. | Butković, K.; Basarić, N.; Lovreković, K.; Marinić, Ž.; Višnjevac, A.; Kojić-Prodić, B.; Šindler-Kulyk, M. Tetrahedron Lett. 2004, 45, 9057–9060. doi:10.1016/j.tetlet.2004.10.036 |
| 24. | Gorsane, M.; Defay, N.; Martin, R. H. Bull. Soc. Chim. Belg. 1985, 94, 215–231. doi:10.1002/bscb.19850940309 |
| 21. | Butković, K.; Marinić, Ž.; Šindler-Kulyk, M. Magn. Reson. Chem. 2004, 42, 1053–1055. doi:10.1002/mrc.1488 |
| 22. | Butković, K.; Vuk, D.; Marinić, Ž.; Penić, J.; Šindler-Kulyk, M. Tetrahedron 2010, 66, 9356–9362. doi:10.1016/j.tet.2010.10.013 |
| 23. | Butković, K.; Marinić, Ž.; Šindler-Kulyk, M. ARKIVOC 2011, 10, 1–15. |
| 25. | Palmer, B. D.; Thompson, A. M.; Booth, R. J.; Dobrusin, E. M.; Kraker, A. J.; Lee, H. H.; Lunney, E. A.; Mitchell, L. H.; Ortwine, D. F.; Smaill, J. B.; Swan, L. M.; Denny, W. A. J. Med. Chem. 2006, 49, 4896–4911. doi:10.1021/jm0512591 |
| 17. | Butković, K.; Basarić, N.; Lovreković, K.; Marinić, Ž.; Višnjevac, A.; Kojić-Prodić, B.; Šindler-Kulyk, M. Tetrahedron Lett. 2004, 45, 9057–9060. doi:10.1016/j.tetlet.2004.10.036 |
| 18. | Meier, H.; Heimgartner, H.; Schmid, H. Helv. Chim. Acta 1977, 60, 1087–1090. doi:10.1002/hlca.19770600333 |
| 22. | Butković, K.; Vuk, D.; Marinić, Ž.; Penić, J.; Šindler-Kulyk, M. Tetrahedron 2010, 66, 9356–9362. doi:10.1016/j.tet.2010.10.013 |
© 2011 Butković et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)