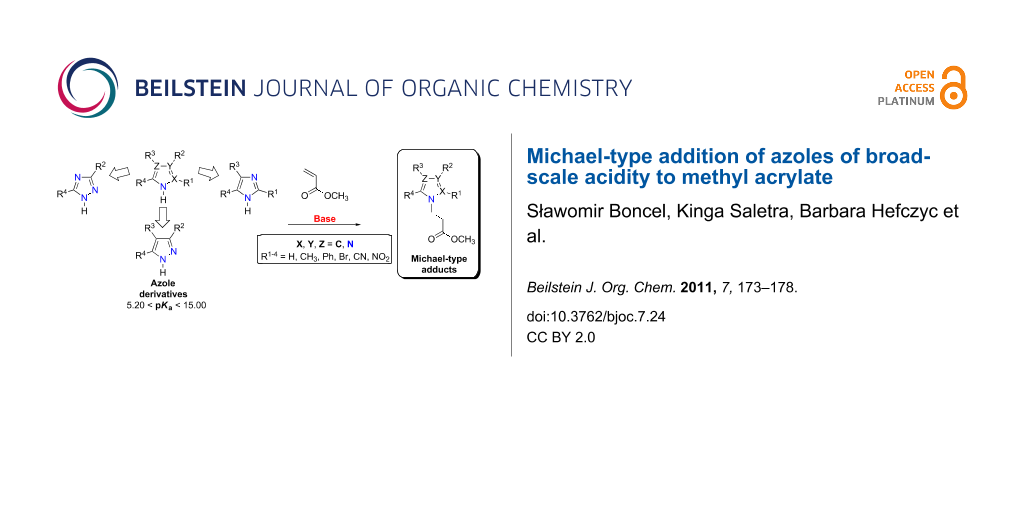
Abstract
An optimisation of Michael-type addition of azole derivatives of broad-scale acidity – ranging from 5.20 to 15.00 pKa units – namely 4-nitropyrazole, 3,5-dimethyl-4-nitropyrazole, 4(5)-nitroimidazole, 4,5-diphenylimidazole, 4,5-dicyanoimidazole, 2-methyl-4(5)-nitroimidazole, 5(4)-bromo-2-methyl-4(5)-nitroimidazole and 3-nitro-1,2,4-triazole to methyl acrylate as an acceptor was carried out. The optimisation process involved the use of an appropriate basic catalyst (DBU, DIPEA, NaOH, NaH, TEDA), a donor/base/acceptor ratio and the reaction temperature. The reactions were performed in DMF as solvent. Target Michael adducts were obtained in medium to excellent yields. Importantly, for imidazole and 1,2,4-triazole derivatives, no corresponding regioisomers were obtained.

Graphical Abstract
Introduction
Derivatives of azoles are important biologically active compounds. Many, especially those containing imidazole, pyrazole and 1,2,4-triazole moieties, constitute building blocks for a variety of therapeutic agents (Figure 1).
![[1860-5397-7-24-1]](/bjoc/content/figures/1860-5397-7-24-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of azole derivatives as important therapeutic agents.
Figure 1: Examples of azole derivatives as important therapeutic agents.
1,2-Arylimidazole derivatives, e.g., 2-(3-chloro-4-methylphenyl)-1-phenyl-4-(trifluoro-methyl)imidazole (I) are selective cyclooxygenase (COX-2) inhibitors [1]. Metronidazole (2-(2-methyl-5-nitroimidazol-1-yl)ethanol) is an antibiotic and antiprotozoal agent whose uses include, among others, the treatment of anaerobic bacterial infections [2]. Complexes of Co(II) and Cu(II) with 3-(2-nitroimidazol-1-yl)propanoic acid (III) are radiosensitisers in cancer therapy [3]. 4,4'-(4-Ethyl-1-phenylpyrazole-3,5-diyl)diphenol (IV) and its derivatives are known modulators of receptors regulating the secretion of estrogens crucial in the pathogenesis of breast cancer [4]. 4-(Benzo[d][1,3]dioxol-5-ylmethyl)-1-(3-methoxybenzyl)-3-phenyl-pyrazole-5-carboxylic acid (V) is an antagonist of endothelial receptors and is used as a clinical regulator of the cardiac cycle [5]. Fluconazole (VI) 2-(2,4-difluorophenyl)-1,3-bis(1,2,4-triazol-1-yl)propan-2-ol is an antifungal drug used in the treatment and prevention of superficial and systemic fungal infections [6].
The base-catalysed Michael-type addition reaction of various azoles to α,β-unsaturated carbonyl and nitro derivatives has been frequently exploited in the synthesis of precursors of biologically active compounds [3,7-15]. This reaction, although structurally restricted to α,β-unsaturated carbonyl compounds and their analogues, is advantageous due to its higher regioselectivity compared to alkylation reactions using alkyl halides [16-19], alkyl sulfates [20] or oxiranes [18] under basic conditions, or alcohols over zeolites [21]. Moreover, it is usually described as a green-chemistry with an effective synthetic protocol and a simple work-up. In the Michael-type additions of azole N–H acids, numerous catalysts such as KF/Al2O3 [8], CeCl3∙7 H2O/NaI [9], NaH or ZnCl2 [10], K2CO3 [11], anhydrous K3PO4 [12], (NH4)2Ce(NO3)6 (CAN) [22] or ionic liquids, e.g., Cu(acac)2 immobilised in [bmim][BF4] [15] have been used. Other reaction conditions including enzymatic catalysis (Bacillus subtilis) [13], zinc-active-site acylases from Escherichia coli and Aspergillus oryzae [23] as well as ultrasonic irradiation in the presence of montmorillonite [14] have also been reported. In addition, there has been several recent reports on the synthesis of Michael-type adducts of azoles by the use of microwave irradiation [7,24]. The optimisation of aza-Michael reactions with N-methylimidazole as the base catalyst was published in 2007 by Liu et al [25]. However, when this catalyst was used with azoles of narrower-scale acidity (pKa range from 8.93 to 15.1) including imidazole and its 2- and 4(5)-methyl, 4(5)-nitro-, 2-methyl-4(5)-nitro- derivatives as well as with 1,2,4-triazole, there was no evidence for a regioselective course of the reactions. Furthermore, extremely high acidic azoles, which are substrates for many important drugs, have seldomly been considered as aza-Michael donors due to their poor nucleophilicity and the possible tendency of adducts to undergo retro-Michael reactions [26].
Here, we present Michael-type addition of azoles, inter alia imidazole and 1,2,4-triazole derivatives of high acidity (pKa ≤ 6.05), to methyl acrylate under optimised conditions (temperature, type of base and donor/base/acceptor ratio) as a regioselective, efficient and useful reaction for the formation of adducts that can be utilised in the synthesis of more complex systems including intermediates for the pharmaceutical industry[27].
Results and Discussion
4-Nitropyrazole (1a), 3,5-dimethyl-4-nitropyrazole (1b), 4(5)-nitroimidazole (1c), 4,5-diphenylimidazole (1d), 4,5-dicyanoimidazole (1e), 2-methyl-4(5)-nitroimidazole (1f), 4(5)-bromo-2-methyl-5(4)-nitroimidazole (1g) and 3-nitro-1,2,4-triazole (1h) were used as Michael donors. The azoles derivatives (1a–h) were subjected to the reaction with methyl acrylate (2) (Michael acceptor) in the presence of an appropriate base, namely DBU (pKa = 12 [28]), diisopropylethylamine (DIPEA, Hünig’s base, pKa = 10.75 [29]), NaOH (pKa = 15.7 [30]), NaH (pKa = 37 [29]) or TEDA (pKa = 8.82 [31]) in polar aprotic (DMF) or protic (MeOH) solvent (Scheme 1, Table 1). Although the above pKa values were determined in water, their relative basicity is usually in good agreement in other polar solvents, e.g., there is a linear relationship between pKa values in water and DMF for the set of N-centred organic bases expressed by the equation: pKa (DMF) = 0.9463 pKa (water) + 1.4154 [32,33]. This semi-empirical relationship enables an initial selection of a base required for a sufficient level of deprotonation of N–H azole acids.
![[1860-5397-7-24-i1]](/bjoc/content/inline/1860-5397-7-24-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Michael-type addition of azoles of broad-scale acidity 1a–h to methyl acrylate (2) under basic conditions.
Scheme 1: Michael-type addition of azoles of broad-scale acidity 1a–h to methyl acrylate (2) under basic cond...
Table 1: Optimised conditions of Michael-type addition of azoles of broad-scale acidity 1a–h to methyl acrylate (2), physical properties and yields of the products 3a–h.
| 1 | pKa (H2O) | Conditions | 3 | ||||
|---|---|---|---|---|---|---|---|
| A/D | Base | Base / A | T [°C] | t [h] | Yield [%] | ||
| a | 9.67 [34] | 1:1 | DBU | 1.0 | 20 | 1 | 31 |
| 2:1 | DIPEA | 1.0 | 20 | 5 | 97 | ||
| b | 15.00 [35] | 1:1 | DBU | 1.0 | 20 | 1 | 54 |
| 2:1 | DIPEA | 1.0 | 20 | 24 | 98 | ||
| c | 8.93 [36] | 1:1 | DBU | 1.0 | 20 | 2 | 30 |
| 1.1:1 | DIPEA | 1.0 | 20 | 12 | 98 | ||
| d | 5.70a [37] | 4:1 | DIPEA | 1.0 | 20 | 192 | 0 |
| 4:1 | DIPEA | 1.0 | 80 | 48 | 0 | ||
| 1:1 | w/ob | — | 60 | 168 | 0 | ||
| 4:1 | NaOHb,c | 0.02 | 65 | 24 | 8 | ||
| 2:1 | NaH | 1.0 | 60 | 72 | 60 | ||
| e | 5.20 [38] | 2:1 | DIPEA | 1.0 | 20 | 192 | 0 |
| 1:1 | DIPEA | 1.0 | 60 | 120 | 0 | ||
| 1:1 | DBU | 1.0 | 60 | 120 | 0 | ||
| 2:1 | NaOHc | 1.0 | 65 | 24 | 25 | ||
| 1:1 | NaHb,d | 2.5 | 60 | 144 | 0 | ||
| 1.5:1 | NaHb,d | 1.2 | 60 | 144 | 0 | ||
| 1:2 | NaHb,d | 1.2 | 60 | 144 | 14 | ||
| 1:2 | NaHb,d | 1.2 | 60 | 168 | 62 | ||
| f | 9.64 [39] | 1:1 | DIPEA | 1.0 | 20 | 120 | 97 |
| g | — | 3:1 | DIPEA | 1.0 | 60 | 96 | 22e |
| 2:1 | DBU | 1.0 | 20 | 72 | 0 | ||
| 1:1 | TEDA | 1.0 | 20 | 120 | 0 | ||
| 2:1 | NaOHc | 0.02 | 65 | 48 | 13 | ||
| 1:2 | NaHb,d | 1.2 | 60 | 72 | 55 | ||
| h | 6.05 [18] | 2:1 | DIPEA | 1.0 | 20 | 114 | 80 |
a30 mol % DMSO aqueous solution at 30 °C, bReaction performed under an inert (N2) atmosphere, creaction performed in MeOH, d80% solution of NaH in mineral oil was used; etotal yield, a regioisomer 4g was obtained in 10% yield (1H NMR); A/D = acceptor to donor molar ratio.
1a and 1b as weak N–H acids required an equimolar amount of a strong base (DBU) and reacted with 2 in stoichiometric proportions to give the appropriate products 3a and 3b in high purity but in low/medium yields. The presence of the unreacted substrates 1a, 1b in the post-reaction mixtures points to the tendency of the adducts to undergo the reverse reaction – a retro-Michael reaction – in equilibrium with Michael-type addition. By contrast, DIPEA, with a lower pKa gave the same adducts 3a and 3b in excellent yields. For 1b (the azole with the lowest acidity) a similar yield to 1a was achieved only after a significantly prolonged reaction time due to its lower concentration of the anionic form in the reaction mixture. For 1c, analogous behaviour was observed when treated with 2 in the presence of DBU. Here again, a use of DIPEA, instead of DBU, in a slight molar excess with respect to the azole furnished 3c in excellent yield. These conditions were also adequate in the reactions of 1f and 1h with 2 and gave the corresponding products 3f and 3h respectively, in excellent yields. No regioisomers were obtained for these C-nitro azole derivatives. The structure of 3c was established by an independent synthesis from 1,4-dinitroimidazole (4) and β-alanine methyl ester hydrochloride (5). This latter reaction proceeds via an ANRORC mechanism (Addition of Nucleophile, Ring Opening, Ring Closure) [40,41] (Scheme 2). Product 3c obtained by the two independent routes, possessed identical physicochemical and spectral properties. The observed difference in yields, 98% for Michael-type addition and 60% for ANRORC, arises from some unidentified side reactions.
![[1860-5397-7-24-i2]](/bjoc/content/inline/1860-5397-7-24-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Chemical evidence for a regioselective Michael-type addition of 4(5)-nitroimidazole (1c) to methyl acrylate (2) based on the ANRORC reaction of 1,4-dinitroimidazole (4) with β-alanine methyl ester hydrochloride (5).
Scheme 2: Chemical evidence for a regioselective Michael-type addition of 4(5)-nitroimidazole (1c) to methyl ...
In the case of the reaction of 1d, an imidazole derivative of acidity three orders of magnitude higher compared to 1c, no reaction was observed in the presence of an equimolar amount of DIPEA and even with a four-fold molar excess of the acceptor at ambient or elevated temperature. We then carried out an additional trial in the absence of a deprotonating agent at elevated temperature on the assumption that due to the increased acidity of 1d, a small concentration of the anionic form would be sufficient for completion of the reaction. However, gain no reaction was observed. On the other hand, the use of a catalytic amount of NaOH at elevated temperature furnished only a small amount of the expected product 3d. However, when 1d was treated with a double molar excess of the acceptor in the presence of NaH at elevated temperature, the expected product was isolated in good yield (60%). The reaction of the most acidic imidazole derivative within the group 1e was more complicated to optimise. DBU, DIPEA and NaOH all appeared as ineffective basic catalysts and only when a double molar excess of the donor was used in the presence of NaH was a satisfactory yield of 3e obtained. Similarly, for 1g only the former conditions gave 3g in good yield (62%). Interestingly, a regioisomeric product of the addition, i.e., methyl 3-(4-bromo-2-methyl-5-nitroimidazol-1-yl)propanoate (4g) was not obtained when NaH was used as a catalyst. However, when DIPEA was used as a base, this regioisomer was obtained in ca. 10% yield, as determined by 1H NMR spectroscopy. The structures of all the products were established by means of 1H and 13C NMR spectroscopy.
It is worth noting that in none of the reactions polymerisation or hydrolysis of methyl acrylate was observed. Nevertheless, decreased yields for azole adducts when DBU or NaH were used as catalysts at elevated temperature could originate from a reversibility of the Michael reaction since unreacted azoles were isolated. This observation is in a good agreement with our recent findings for Michael-type addition of uracils to acrylic acceptors [27,42], where a a strongly basic catalyst (DBU) enabled control of regioselectivity via a retro-Michael reaction.
Conclusion
We have developed and optimised a simple and effective synthetic protocol for Michael-type addition of azoles of broad-scale acidity including pyrazole, imidazole and 1,2,4-triazole derivatives. Importantly, for 4(5)-nitroimidazole, 2-methyl-4(5)-nitroimidazole, 4(5)-bromo-2-methyl-5(4)-nitroimidazole and 3-nitro-1,2,4-triazole no regioisomers were obtained. The adducts constitute biologically active model compounds themselves and which, after acidic hydrolysis [43], can be anchored to drug delivery systems, including, e.g., chemically modified carbon nanotubes, via amide bonds.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 339.7 KB | Download |
References
-
Desiraju, G. R.; Gopalakrishnan, B.; Jetti, R. K. R.; Nagaraju, A.; Raveendra, D.; Sarma, J. A. R. P.; Sobhia, M. E.; Thilagavathi, R. J. Med. Chem. 2002, 45, 4847–4857. doi:10.1021/jm020198t
Return to citation in text: [1] -
Katritzky, A. R.; Rees, C. W. Comprehensive Heterocyclic Chemistry 1984, 5, 469–498.
Return to citation in text: [1] -
Goodgame, D. M. L.; Page, C. J.; Williams, D. J. Polyhedron 1992, 11, 2507–2515. doi:10.1016/S0277-5387(00)83571-8
Return to citation in text: [1] [2] -
Stauffer, S. R.; Katzenellenbogen, J. A. J. Comb. Chem. 2000, 2, 318–329. doi:10.1021/cc0000040
Return to citation in text: [1] -
Zhang, J.; Didierlaurent, S.; Fortin, M.; Lefrançois, D.; Uridat, E.; Vevert, J. P. Bioorg. Med. Chem. Lett. 2000, 10, 1351–1355. doi:10.1016/S0960-894X(00)00232-8
Return to citation in text: [1] -
Potts, K. T. Chem. Rev. 1961, 61, 87–127. doi:10.1021/cr60210a001
Return to citation in text: [1] -
Xu, J.-M.; Qian, C.; Liu, B.-K.; Wu, Q.; Lin, X.-F. Tetrahedron 2007, 63, 986–990. doi:10.1016/j.tet.2006.11.013
Return to citation in text: [1] [2] -
Yang, L.; Xu, L.-W.; Xia, C.-G. Tetrahedron Lett. 2005, 46, 3279–3282. doi:10.1016/j.tetlet.2005.03.112
Return to citation in text: [1] [2] -
Bartoli, G.; Bartolacci, M.; Giuliani, A.; Marcantoni, E.; Massaccesi, M.; Torregiani, E. J. Org. Chem. 2005, 70, 169–174. doi:10.1021/jo048329g
Return to citation in text: [1] [2] -
Díez-Barra, E.; Guerra, J.; Hornillos, V.; Merino, S.; Tejada, J. Tetrahedron Lett. 2004, 45, 6937–6939. doi:10.1016/j.tetlet.2004.07.083
Return to citation in text: [1] [2] -
Ferreira, P. M. T.; Maia, H. L. S.; Monteiro, L. S. Tetrahedron Lett. 1999, 40, 4099–4102. doi:10.1016/S0040-4039(99)00691-7
Return to citation in text: [1] [2] -
Hou, X.; Hemit, H.; Yong, J.; Nie, L.; Aisa, H. A. Synth. Commun. 2010, 40, 973–979. doi:10.1080/00397910903029867
Return to citation in text: [1] [2] -
Cai, Y.; Yao, S.-P.; Wu, Q.; Lin, X.-F. Biotechnol. Lett. 2004, 26, 525–528. doi:10.1023/B:BILE.0000019562.21256.39
Return to citation in text: [1] [2] -
Martín-Aranda, R. M.; Ortega-Cantero, E.; Rojas-Cervantes, M. L.; Vicente-Rodríguez, M. A.; Bañares-Muñoz, M. A. Catal. Lett. 2002, 84, 201–204. doi:10.1023/A:1021480020425
Return to citation in text: [1] [2] -
Kantam, M. L.; Neelima, B.; Reddy, C. V.; Chakravarti, R. Ind. Eng. Chem. Res. 2007, 46, 8614–8619. doi:10.1021/ie070080g
Return to citation in text: [1] [2] -
Khabnadideh, S.; Rezaei, Z.; Khalafi-Nezhad, A.; Bahrinajafi, R.; Mohamadi, R.; Farrokhroz, A. A. Bioorg. Med. Chem. Lett. 2003, 13, 2863–2865. doi:10.1016/S0960-894X(03)00591-2
Return to citation in text: [1] -
Begtrup, M.; Larsen, P. Acta Chem. Scand. 1990, 44, 1050–1057. doi:10.3891/acta.chem.scand.44-1050
Return to citation in text: [1] -
Kofman, T. P.; Kartseva, G. Y. Russ. J. Org. Chem. 2001, 37, 707–716. doi:10.1023/A:1012460103654
Return to citation in text: [1] [2] [3] -
Chu, T.; Hu, S.; Wei, B.; Wang, Y.; Liu, X.; Wang, X. Bioorg. Med. Chem. Lett. 2004, 14, 747–749. doi:10.1016/j.bmcl.2003.11.017
Return to citation in text: [1] -
Sukhanov, G. T.; Sukhanova, A. G.; Ilyasova, Y. V. Chem. Heterocycl. Compd. 2006, 42, 1197–1199. doi:10.1007/s10593-006-0225-9
Return to citation in text: [1] -
Ono, Y.; Izawa, Y.; Fu, Z.-H. Catal. Lett. 1997, 47, 251–253. doi:10.1023/A:1019065323538
Return to citation in text: [1] -
Duan, Z.; Xuan, X.; Li, T.; Yang, C.; Wu, Y. Tetrahedron Lett. 2006, 47, 5433–5436. doi:10.1016/j.tetlet.2006.05.182
Return to citation in text: [1] -
Qian, C.; Xu, J.-M.; Wu, Q.; Lv, D.-S.; Lin, X.-F. Tetrahedron Lett. 2007, 48, 6100–6104. doi:10.1016/j.tetlet.2007.06.164
Return to citation in text: [1] -
Martín-Aranda, R. M.; Vicente-Rodríguez, M. A.; López-Pestaña, J. M.; López-Peinado, A. J.; Jerez, A.; López-González, J. de D.; Bañares-Muñoz, M. A. J. Mol. Catal. A: Chem. 1997, 124, 115–121. doi:10.1016/S1381-1169(97)00070-8
Return to citation in text: [1] -
Liu, B. K.; Wu, Q.; Qian, X. Q.; Lv, D. S.; Lin, X. F. Synthesis 2007, 17, 2653–2659. doi:10.1055/s-2007-983816
Return to citation in text: [1] -
Boncel, S.; Mączka, M.; Walczak, K. Z. Tetrahedron 2010, 66, 8450–8457. doi:10.1016/j.tet.2010.08.059
Return to citation in text: [1] -
Boncel, S.; Osyda, D.; Walczak, K. Z. Beilstein J. Org. Chem. 2007, 3, No. 40. doi:10.1186/1860-5397-3-40
Return to citation in text: [1] [2] -
Granitza, D.; Beyermann, M.; Wenschuh, H.; Haber, H.; Carpino, L. A.; Truranb, G. A.; Bienerta, M. J. Chem. Soc., Chem. Commun. 1995, 21, 2223–2224.
Return to citation in text: [1] -
Perrin, D. D. Dissociation constans of organic acids and bases; Butterworths: London, 1965.
Supplement 1972.
Return to citation in text: [1] [2] -
Smith, M. B.; March, J. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5th ed.; Wiley-Interscience, 2001.
Return to citation in text: [1] -
Benoit, R. L.; Lefebvre, D.; Fréchette, M. Can. J. Chem. 1987, 65, 996–1001. doi:10.1139/v87-170
Return to citation in text: [1] -
Kolthoff, I. M.; Chantooni, M. K., Jr.; Smagowski, H. Anal. Chem. 1970, 42, 1662.
Return to citation in text: [1] -
Granitza, D.; Beyermann, M.; Wenschuh, H.; Haber, H.; Carpino, L. A.; Truranb, G. A.; Bienerta, M. J. Chem. Soc., Chem. Commun. 1995, 21, 2223–2224.
Return to citation in text: [1] -
Vokin, A. I.; Lopyrev, V. A.; Shulunova, A. M.; Komarova, T. N.; Turchaninov, V. K. Russ. Chem. Bull. 1997, 46, 834–836. doi:10.1007/BF02495226
Return to citation in text: [1] -
Elguero, J.; Yranzo, G. I.; Laynez, J.; Jimenez, P.; Menendez, M.; Catalan, J.; De Paz, J. L. G.; Anvia, F.; Taft, R. W. J. Org. Chem. 1991, 56, 3942–3947. doi:10.1021/jo00012a030
Return to citation in text: [1] -
Li, W.; Norris, B. C.; Snodgrass, P.; Prasad, K.; Stockett, A. S.; Pryamitsyn, V.; Ganesan, V.; Bielawski, C. W.; Manthiram, A. J. Phys. Chem. B 2009, 113, 10063–10067. doi:10.1021/jp904192t
Return to citation in text: [1] -
Bowden, K.; Brownhill, A. J. Chem. Soc., Perkin Trans. 2 1997, 219–221. doi:10.1039/a605649k
Return to citation in text: [1] -
Vargeese, C.; Carter, J.; Yegge, J.; Krivjansky, S.; Settle, A.; Kropp, E.; Peterson, K.; Pieken, W. Nucleic Acids Res. 1998, 26, 1046–1050. doi:10.1093/nar/26.4.1046
Return to citation in text: [1] -
Yang, S. J. West China Univ. Med. Sci. 1999, 30, 345–346.
Return to citation in text: [1] -
Gondela, A.; Walczak, K. Carbohydr. Res. 2005, 340, 1379–1385. doi:10.1016/j.carres.2005.03.005
Return to citation in text: [1] -
Gondela, A.; Walczak, K. Tetrahedron: Asymmetry 2005, 16, 2107–2112. doi:10.1016/j.tetasy.2005.05.009
Return to citation in text: [1] -
Boncel, S.; Gondela, A.; Walczak, K. Synthesis 2010, 10, 1573–1589. doi:10.1055/s-0029-1218757
Return to citation in text: [1] -
Boncel, S.; Walczak, K. Nucleosides, Nucleotides Nucleic Acids 2009, 28, 103–117. doi:10.1080/15257770902736467
Return to citation in text: [1]
| 30. | Smith, M. B.; March, J. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5th ed.; Wiley-Interscience, 2001. |
| 29. |
Perrin, D. D. Dissociation constans of organic acids and bases; Butterworths: London, 1965.
Supplement 1972. |
| 31. | Benoit, R. L.; Lefebvre, D.; Fréchette, M. Can. J. Chem. 1987, 65, 996–1001. doi:10.1139/v87-170 |
| 1. | Desiraju, G. R.; Gopalakrishnan, B.; Jetti, R. K. R.; Nagaraju, A.; Raveendra, D.; Sarma, J. A. R. P.; Sobhia, M. E.; Thilagavathi, R. J. Med. Chem. 2002, 45, 4847–4857. doi:10.1021/jm020198t |
| 5. | Zhang, J.; Didierlaurent, S.; Fortin, M.; Lefrançois, D.; Uridat, E.; Vevert, J. P. Bioorg. Med. Chem. Lett. 2000, 10, 1351–1355. doi:10.1016/S0960-894X(00)00232-8 |
| 11. | Ferreira, P. M. T.; Maia, H. L. S.; Monteiro, L. S. Tetrahedron Lett. 1999, 40, 4099–4102. doi:10.1016/S0040-4039(99)00691-7 |
| 4. | Stauffer, S. R.; Katzenellenbogen, J. A. J. Comb. Chem. 2000, 2, 318–329. doi:10.1021/cc0000040 |
| 12. | Hou, X.; Hemit, H.; Yong, J.; Nie, L.; Aisa, H. A. Synth. Commun. 2010, 40, 973–979. doi:10.1080/00397910903029867 |
| 18. | Kofman, T. P.; Kartseva, G. Y. Russ. J. Org. Chem. 2001, 37, 707–716. doi:10.1023/A:1012460103654 |
| 3. | Goodgame, D. M. L.; Page, C. J.; Williams, D. J. Polyhedron 1992, 11, 2507–2515. doi:10.1016/S0277-5387(00)83571-8 |
| 9. | Bartoli, G.; Bartolacci, M.; Giuliani, A.; Marcantoni, E.; Massaccesi, M.; Torregiani, E. J. Org. Chem. 2005, 70, 169–174. doi:10.1021/jo048329g |
| 37. | Bowden, K.; Brownhill, A. J. Chem. Soc., Perkin Trans. 2 1997, 219–221. doi:10.1039/a605649k |
| 2. | Katritzky, A. R.; Rees, C. W. Comprehensive Heterocyclic Chemistry 1984, 5, 469–498. |
| 10. | Díez-Barra, E.; Guerra, J.; Hornillos, V.; Merino, S.; Tejada, J. Tetrahedron Lett. 2004, 45, 6937–6939. doi:10.1016/j.tetlet.2004.07.083 |
| 38. | Vargeese, C.; Carter, J.; Yegge, J.; Krivjansky, S.; Settle, A.; Kropp, E.; Peterson, K.; Pieken, W. Nucleic Acids Res. 1998, 26, 1046–1050. doi:10.1093/nar/26.4.1046 |
| 20. | Sukhanov, G. T.; Sukhanova, A. G.; Ilyasova, Y. V. Chem. Heterocycl. Compd. 2006, 42, 1197–1199. doi:10.1007/s10593-006-0225-9 |
| 21. | Ono, Y.; Izawa, Y.; Fu, Z.-H. Catal. Lett. 1997, 47, 251–253. doi:10.1023/A:1019065323538 |
| 35. | Elguero, J.; Yranzo, G. I.; Laynez, J.; Jimenez, P.; Menendez, M.; Catalan, J.; De Paz, J. L. G.; Anvia, F.; Taft, R. W. J. Org. Chem. 1991, 56, 3942–3947. doi:10.1021/jo00012a030 |
| 16. | Khabnadideh, S.; Rezaei, Z.; Khalafi-Nezhad, A.; Bahrinajafi, R.; Mohamadi, R.; Farrokhroz, A. A. Bioorg. Med. Chem. Lett. 2003, 13, 2863–2865. doi:10.1016/S0960-894X(03)00591-2 |
| 17. | Begtrup, M.; Larsen, P. Acta Chem. Scand. 1990, 44, 1050–1057. doi:10.3891/acta.chem.scand.44-1050 |
| 18. | Kofman, T. P.; Kartseva, G. Y. Russ. J. Org. Chem. 2001, 37, 707–716. doi:10.1023/A:1012460103654 |
| 19. | Chu, T.; Hu, S.; Wei, B.; Wang, Y.; Liu, X.; Wang, X. Bioorg. Med. Chem. Lett. 2004, 14, 747–749. doi:10.1016/j.bmcl.2003.11.017 |
| 8. | Yang, L.; Xu, L.-W.; Xia, C.-G. Tetrahedron Lett. 2005, 46, 3279–3282. doi:10.1016/j.tetlet.2005.03.112 |
| 36. | Li, W.; Norris, B. C.; Snodgrass, P.; Prasad, K.; Stockett, A. S.; Pryamitsyn, V.; Ganesan, V.; Bielawski, C. W.; Manthiram, A. J. Phys. Chem. B 2009, 113, 10063–10067. doi:10.1021/jp904192t |
| 3. | Goodgame, D. M. L.; Page, C. J.; Williams, D. J. Polyhedron 1992, 11, 2507–2515. doi:10.1016/S0277-5387(00)83571-8 |
| 7. | Xu, J.-M.; Qian, C.; Liu, B.-K.; Wu, Q.; Lin, X.-F. Tetrahedron 2007, 63, 986–990. doi:10.1016/j.tet.2006.11.013 |
| 8. | Yang, L.; Xu, L.-W.; Xia, C.-G. Tetrahedron Lett. 2005, 46, 3279–3282. doi:10.1016/j.tetlet.2005.03.112 |
| 9. | Bartoli, G.; Bartolacci, M.; Giuliani, A.; Marcantoni, E.; Massaccesi, M.; Torregiani, E. J. Org. Chem. 2005, 70, 169–174. doi:10.1021/jo048329g |
| 10. | Díez-Barra, E.; Guerra, J.; Hornillos, V.; Merino, S.; Tejada, J. Tetrahedron Lett. 2004, 45, 6937–6939. doi:10.1016/j.tetlet.2004.07.083 |
| 11. | Ferreira, P. M. T.; Maia, H. L. S.; Monteiro, L. S. Tetrahedron Lett. 1999, 40, 4099–4102. doi:10.1016/S0040-4039(99)00691-7 |
| 12. | Hou, X.; Hemit, H.; Yong, J.; Nie, L.; Aisa, H. A. Synth. Commun. 2010, 40, 973–979. doi:10.1080/00397910903029867 |
| 13. | Cai, Y.; Yao, S.-P.; Wu, Q.; Lin, X.-F. Biotechnol. Lett. 2004, 26, 525–528. doi:10.1023/B:BILE.0000019562.21256.39 |
| 14. | Martín-Aranda, R. M.; Ortega-Cantero, E.; Rojas-Cervantes, M. L.; Vicente-Rodríguez, M. A.; Bañares-Muñoz, M. A. Catal. Lett. 2002, 84, 201–204. doi:10.1023/A:1021480020425 |
| 15. | Kantam, M. L.; Neelima, B.; Reddy, C. V.; Chakravarti, R. Ind. Eng. Chem. Res. 2007, 46, 8614–8619. doi:10.1021/ie070080g |
| 32. | Kolthoff, I. M.; Chantooni, M. K., Jr.; Smagowski, H. Anal. Chem. 1970, 42, 1662. |
| 33. | Granitza, D.; Beyermann, M.; Wenschuh, H.; Haber, H.; Carpino, L. A.; Truranb, G. A.; Bienerta, M. J. Chem. Soc., Chem. Commun. 1995, 21, 2223–2224. |
| 18. | Kofman, T. P.; Kartseva, G. Y. Russ. J. Org. Chem. 2001, 37, 707–716. doi:10.1023/A:1012460103654 |
| 34. | Vokin, A. I.; Lopyrev, V. A.; Shulunova, A. M.; Komarova, T. N.; Turchaninov, V. K. Russ. Chem. Bull. 1997, 46, 834–836. doi:10.1007/BF02495226 |
| 13. | Cai, Y.; Yao, S.-P.; Wu, Q.; Lin, X.-F. Biotechnol. Lett. 2004, 26, 525–528. doi:10.1023/B:BILE.0000019562.21256.39 |
| 22. | Duan, Z.; Xuan, X.; Li, T.; Yang, C.; Wu, Y. Tetrahedron Lett. 2006, 47, 5433–5436. doi:10.1016/j.tetlet.2006.05.182 |
| 40. | Gondela, A.; Walczak, K. Carbohydr. Res. 2005, 340, 1379–1385. doi:10.1016/j.carres.2005.03.005 |
| 41. | Gondela, A.; Walczak, K. Tetrahedron: Asymmetry 2005, 16, 2107–2112. doi:10.1016/j.tetasy.2005.05.009 |
| 15. | Kantam, M. L.; Neelima, B.; Reddy, C. V.; Chakravarti, R. Ind. Eng. Chem. Res. 2007, 46, 8614–8619. doi:10.1021/ie070080g |
| 27. | Boncel, S.; Osyda, D.; Walczak, K. Z. Beilstein J. Org. Chem. 2007, 3, No. 40. doi:10.1186/1860-5397-3-40 |
| 42. | Boncel, S.; Gondela, A.; Walczak, K. Synthesis 2010, 10, 1573–1589. doi:10.1055/s-0029-1218757 |
| 43. | Boncel, S.; Walczak, K. Nucleosides, Nucleotides Nucleic Acids 2009, 28, 103–117. doi:10.1080/15257770902736467 |
| 28. | Granitza, D.; Beyermann, M.; Wenschuh, H.; Haber, H.; Carpino, L. A.; Truranb, G. A.; Bienerta, M. J. Chem. Soc., Chem. Commun. 1995, 21, 2223–2224. |
| 29. |
Perrin, D. D. Dissociation constans of organic acids and bases; Butterworths: London, 1965.
Supplement 1972. |
| 26. | Boncel, S.; Mączka, M.; Walczak, K. Z. Tetrahedron 2010, 66, 8450–8457. doi:10.1016/j.tet.2010.08.059 |
| 27. | Boncel, S.; Osyda, D.; Walczak, K. Z. Beilstein J. Org. Chem. 2007, 3, No. 40. doi:10.1186/1860-5397-3-40 |
| 7. | Xu, J.-M.; Qian, C.; Liu, B.-K.; Wu, Q.; Lin, X.-F. Tetrahedron 2007, 63, 986–990. doi:10.1016/j.tet.2006.11.013 |
| 24. | Martín-Aranda, R. M.; Vicente-Rodríguez, M. A.; López-Pestaña, J. M.; López-Peinado, A. J.; Jerez, A.; López-González, J. de D.; Bañares-Muñoz, M. A. J. Mol. Catal. A: Chem. 1997, 124, 115–121. doi:10.1016/S1381-1169(97)00070-8 |
| 25. | Liu, B. K.; Wu, Q.; Qian, X. Q.; Lv, D. S.; Lin, X. F. Synthesis 2007, 17, 2653–2659. doi:10.1055/s-2007-983816 |
| 23. | Qian, C.; Xu, J.-M.; Wu, Q.; Lv, D.-S.; Lin, X.-F. Tetrahedron Lett. 2007, 48, 6100–6104. doi:10.1016/j.tetlet.2007.06.164 |
| 14. | Martín-Aranda, R. M.; Ortega-Cantero, E.; Rojas-Cervantes, M. L.; Vicente-Rodríguez, M. A.; Bañares-Muñoz, M. A. Catal. Lett. 2002, 84, 201–204. doi:10.1023/A:1021480020425 |
© 2011 Boncel et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)