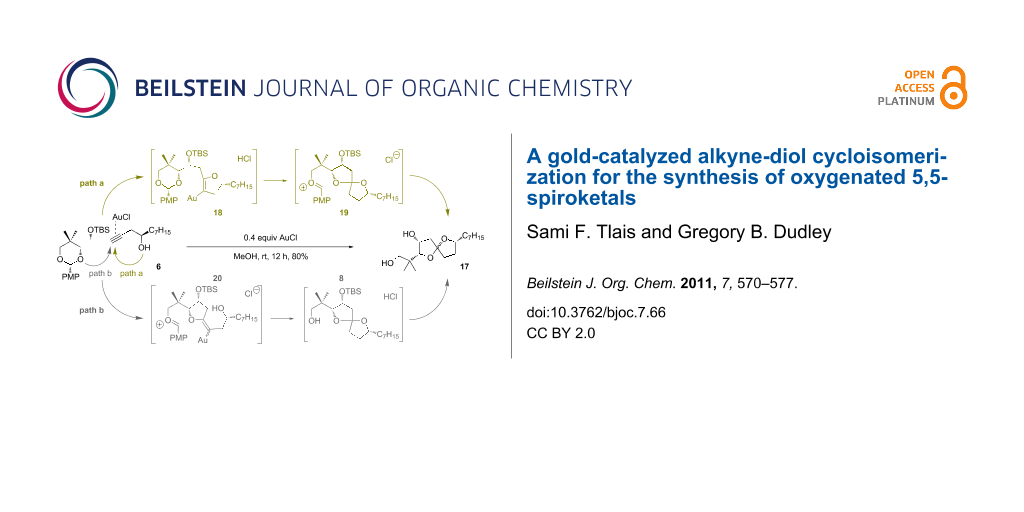
Abstract
A highly efficient synthesis of oxygenated 5,5-spiroketals was performed towards the synthesis of the cephalosporolides. Gold(I) chloride in methanol induced the cycloisomerization of a protected alkyne triol with concomitant deprotection to give a strategically hydroxylated 5,5-spiroketal, despite the potential for regiochemical complications and elimination to furan. Other late transition metal Lewis acids were less effective. The use of methanol as solvent helped suppress the formation of the undesired furan by-product. This study provides yet another example of the advantages of gold catalysis in the activation of alkyne π-systems.

Graphical Abstract
Introduction
Spiroketals, exemplified by structure shown in Figure 1, are prominent structural features of many biomedically relevant natural and non-natural target structures [1-4]. As such, the synthesis of spiroketals has received considerable attention, with most progress having been made on systems that include at least one six-membered ring [5]. 5,5-Spiroketals (m, n = 0, Figure 1), particularly oxygenated 5,5-spiroketals such as are found in the cephalosporolides (Figure 2), are the focus of this study.
![[1860-5397-7-66-1]](/bjoc/content/figures/1860-5397-7-66-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-7-66-2]](/bjoc/content/figures/1860-5397-7-66-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Spiroketal-containing cephalosporolide natural products.
Figure 2: Spiroketal-containing cephalosporolide natural products.
A variety of synthetic methods are available for the synthesis of 5,5-spiroketals, including cyclocondensation of ketone diols [6,7], the cycloisomerization of alkyne diols (Scheme 1) [8-16], oxidative spirocyclization of tetrahydrofuryl propanols [17-20], and others. Cyclocondensation of ketone diols is perhaps the most straighforward and the most used method, but the alternative procedures offer specific advantages. For example, the cycloisomerization of alkyne diols is more exothermic (Scheme 1) [21] and atom economical [22], and non-polar alkyne π-bonds are more compatible than ketones (kinetically stable) towards a number of common reaction conditions. Conversely, the use of alkynes in the synthesis of spiroketals introduces regiochemistry concerns as to which of the two alkyne carbons becomes the spiroketal carbon, and the kinetic stability of alkynes must be overcome when alkyne reactivity is desired.
![[1860-5397-7-66-i1]](/bjoc/content/inline/1860-5397-7-66-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Cyclocondensation vs. cycloisomerization for the synthesis of spiroketals.
Scheme 1: Cyclocondensation vs. cycloisomerization for the synthesis of spiroketals.
As an off-shoot of our program devoted to the synthesis of functionalized alkynes by fragmentation reactions [23-27], we became interested in the application of alkyne-diol cycloisomerization to the synthesis of the cephalosporolides and other oxygenated spiroketals. Our retrosynthetic analysis of the reported structure of cephalosporolide H (1) is outlined in Scheme 2. We recently demonstrated the use of inter-cycle chelation effects to control the spiroketal stereochemistry [28,29]. However, formation of the requisite oxygenated spiroketals (by cycloisomerization) posed significant challenges that required a focused study.
![[1860-5397-7-66-i2]](/bjoc/content/inline/1860-5397-7-66-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Retrosynthetic analysis of cephalosporolide H.
Scheme 2: Retrosynthetic analysis of cephalosporolide H.
For this thematic issue on gold catalysis in organic synthesis, we detail here the challenges and considerations involved in the cycloisomerization of alkynes to oxygenated spiroketals and outline our screening of various late transition metal catalysts and conditions that ultimately resulted in the acquisition of our target structures [28]. Gold(I) chloride emerged as the best choice for the desired transformation.
The key precedents for the desired cycloisomerization are shown in Scheme 3, although many methods are available [30-34] and no consensus option has emerged. Utimoto studied the palladium-catalyzed cycloisomerization [8] and reported that a range of spiroketals are available in excellent yield (e.g., Scheme 3, Reaction 1). However, regiochemistry is sometimes difficult to control, and De Brabander later found variability in reaction selectivity using the Utimoto conditions to prepare 6,6-spiroketals. Therefore, he suggested the preferred use of Ziese’s dimer, a platinum catalyst (Scheme 3, Reaction 2), for such cyclizations [9]. In an unrelated study that also bears on the current work, Aponick and co-workers described a gold-catalyzed cyclocondensation of alkyne diols to give substituted furans (Scheme 3, Reaction 3) [35].
![[1860-5397-7-66-i3]](/bjoc/content/inline/1860-5397-7-66-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Key precedents for the desired cycloisomerization.
Scheme 3: Key precedents for the desired cycloisomerization.
Our objective, laid out in Scheme 4, was to initiate cycloisomerization with a 5-endo-dig cyclization of the homopropargyl alcohol 6, followed by 5-exo-trig cyclization onto the resulting dihydrofuran, whilst avoiding dehydration to furan 10. The use of alkyne-diol cycloisomerization instead of ketone-diol cyclocondensation is important for the potential success of this approach, since β-alkoxy ketone 9 (Scheme 4, inset) would be more prone to undesired elimination than homopropargyl ether 6. We addressed regiochemistry by blocking one of the alcohols as an acetal (the alcohol that otherwise could undergo either 5-exo or 6-endo cyclization [9,13,36]), thus favoring the initial 5-endo cyclization of the other. In this way we aimed to ensure that the desired regioisomer could form, with the expectation of acetal hydrolysis during the course of the reaction. Indeed, attempts to induce spiroketalization after removal of the acetal resulted in complex product mixtures (not shown).
![[1860-5397-7-66-i4]](/bjoc/content/inline/1860-5397-7-66-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed cycloisomerization with acetal hydrolysis.
Scheme 4: Proposed cycloisomerization with acetal hydrolysis.
Results and Discussion
Initial studies on the cycloisomerization took advantage of chiral propargyl alcohol 12, which is readily available from pantolactone (11, Scheme 5) [37]. An alkyne zipper reaction, protection, and coupling with propylene oxide gave homopropargyl 13.
![[1860-5397-7-66-i5]](/bjoc/content/inline/1860-5397-7-66-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of model cyclization substrate 13.
Scheme 5: Synthesis of model cyclization substrate 13.
Cycloisomerization of 13 to the spiroketal (14) was investigated under a variety of conditions, some of which are featured in Table 1. Utimoto’s general conditions as reported (Table 1, entry 1) resulted in decomposition of the substrate, but at room temperature the spiroketal was obtained in modest yield (Table 1, entry 2). Reactions involving Ziese’s dimer were disappointing (Table 1, entry 3), but gold(I) chloride in methylene chloride (cf. Scheme 3, Reaction 3) gave more encouraging results. Other gold catalysts and solvents were screened, with the best results being achieved with a higher catalyst loading of gold(I) chloride in methanol (Table 1, entry 10). The need for higher catalyst loading is tentatively ascribed to some form of instability of the gold catalyst in methanol, as pre-mixing the gold(I) chloride with methanol and aging this mixture prior to adding the substrate results in a less efficient reaction. This is not the first time that we have observed the importance of the order of addition in a gold-catalyzed reaction in a protic solvent [38], but nonetheless we were satisfied with these results for our current study. Furan 16, which presumably arises by analogy to Aponick’s cyclocondensation, was observed in varying amounts in many cases and was the major product in Table 1, entry 11: The use of methanol as a solvent seems to help suppress formation of the (undesired for our purposes) furan product.
Table 1: Spiroketalization using late transition metal salt complexes.
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-66-i9.svg?max-width=637&scale=1.0)
|
|||
| Entry | Conditions | Major Product | Yield |
|---|---|---|---|
| 1 | 1% PdCl2, CH3CN, reflux, 1 h | — | —a |
| 2 | 1% PdCl2, CH3CN, rt, 1.5 h | 14 | 43%b |
| 3 | 1% [Cl2Pt(CH2=CH2)]2, Et2O, rt, then CSA | — | —a |
| 4 | 5% AuCl, CH2Cl2, rt, 6 h | 14 | 36% |
| 5 | 5% AuCl, PPTS, CH2Cl2, rt, 14 h | 14 | 37% |
| 6 | 5% AuCl(PPh3)3, CH2Cl2, rt | — | —a |
| 7 | 5% AuCl(PPh3)3, AgSbF6, CH2Cl2, rt, 12 h | — | —a |
| 8 | 5% AuCl3, CH2Cl2, rt, 12 h | — | —a |
| 9 | 5% AuCl, MeOH, rt, 12 h | 15 | 35% |
| 10c | 25% + 25% AuCl, MeOH, rt, 12 h | 15 | 68% |
| 11 | 35% AuCl, MeCN, rt, 4 h | 16 | 18% |
acomplex mixture of products was observed, bno increase in yield after a longer reaction time, ca second portion of AuCl (25 mol %) was added after 1 h to achieve full conversion.
For the synthesis of cephalosporolide H, we prepared homopropargyl alcohol 6 by a two-step inversion of 12, followed by an alkyne zipper reaction and coupling with nonene oxide (Scheme 6) [28]. Treatment of 6 with 40 mol % gold(I) chloride in methanol resulted in cycloisomerization with simultaneous hydrolysis of the PMP acetal. Meanwhile, cleavage of the silyl ether also occurred under the reaction conditions, and spiroketal diol 17 was isolated in 80% yield as a roughly 1:1 mixture of spiroketal epimers. This mixture of epimers led to a single diastereomer upon chelation with zinc chloride. TEMPO oxidation gave lactone 1, which corresponds to the reported structure of cephalosporolide H. A more detailed discussion is found in our earlier report [28].
![[1860-5397-7-66-i6]](/bjoc/content/inline/1860-5397-7-66-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of reported structure of cephalosporolide H.
Scheme 6: Synthesis of reported structure of cephalosporolide H.
Two mechanistic alternatives (Scheme 7) are proposed for the conversion of 6 → 17 in methanol. Path a, which corresponds roughly to our original experimental designs, involves initial gold-catalyzed 5-endo-dig cyclization to dihydrofuran 18. Once the regiochemistry is established, any number of condensation pathways would lead to spiroketal 17. For example, protonation of the enol ether could assist in the opening of the acetal, with simultaneous formation of spiroketal 19. Any carbenium intermediates could be intercepted reversibly by methanol. The acidity of the gold(I) chloride in methanol mixture is sufficient to hydrolyze the secondary silyl ether group in a separate event: The reaction time was intentionally extended to ensure complete desilylation.
![[1860-5397-7-66-i7]](/bjoc/content/inline/1860-5397-7-66-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
A second mechanistic alternative, path b, cannot be ruled out at this time. Path b involves gold-activation of the alkyne followed by 5-exo-dig nucleophilic attack of the acetal oxygen. Methanolysis of the acetal and spirocyclization would quickly follow. Although this pathway seems unlikely to compete effectively with path a, a control experiment suggests that path b is feasible under certain conditions (Scheme 8, Reaction 4): We subjected terminal alkyne 21 to gold(I) chloride in methylene chloride and observed the formation of furan 23 in low yield, along with other products.
![[1860-5397-7-66-i8]](/bjoc/content/inline/1860-5397-7-66-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Control experiment for gold-activation of the alkyne.
Scheme 8: Control experiment for gold-activation of the alkyne.
Conclusion
Gold(I) chloride effectively catalyzed the cycloisomerization of homopropargyl alcohol 6 to spiroketal 17 in good yield, despite the potential for regiochemical complications and elimination to give furan by-products. Other late transition metal Lewis acids were less effective. This study provides yet another example of the advantages of gold catalysis in the activation of alkyne π-systems.
Experimental
1H NMR and 13C NMR spectra were recorded in CDCl3 as the deuterated solvent. The chemical shifts (δ) are reported in parts per million (ppm) relative to the residual CHCl3 peak (7.26 ppm for 1H NMR and 77.0 ppm for 13C NMR) with TMS as internal standard. The coupling constants (J) are reported in hertz (Hz). IR spectra were recorded on a FT-IR spectrometer (100). Mass spectra were recorded either by electron ionization (EI) or fast-atom bombardment (FAB). Yields refer to isolated material judged to be ≥95% pure by 1H NMR spectroscopy following silica gel chromatography. All solvents, solutions and liquid reagents were added via syringe. Methanol (MeOH), methylene chloride (CH2Cl2) and acetonitrile (CH3CN) were used without any purification. All reactions were carried out under an inert nitrogen atmosphere unless otherwise stated. Purifications were performed by flash chromatography on silica gel F-254 (230–499 mesh particle size).
Typical procedure for gold-catalyzed spiroketalization: AuCl (8 mg, 0.036 mmol) was added to a solution of 6 (50 mg, 0.091 mmol) in MeOH (5 mL) at room temperature to give a black mixture. After 4 h, the reaction mixture was filtered, mixed with 100 mg of silica gel, and concentrated under reduced pressure. The silica gel admixed with the crude reaction mixture was transferred to a silica gel column and eluted with 15% EtOAc in hexane to afford pure product 17 (23 mg, 80%).
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-66-i10.svg?max-width=637&scale=1.18182)
Characterization data for 17a: 1H NMR (600 MHz, CDCl3) δ 4.26 (m, 1H), 3.98 (ddd, J = 13.1, 9.4, 6.1 Hz, 1H), 3.59 (d, J = 3.1 Hz, 1H), 3.51 (d, J = 10.9 Hz, 1H), 3.46 (d, J = 10.9 Hz, 1H), 2.15 (dd, J = 13.4, 4.3 Hz, 1H), 2.10–1.91 (m, 4H), 1.75–1.66 (m, 2H), 1.55–1.47 (m, 1H), 1.40–1.20 (m, 13H), 1.09 (s, 3H), 1.02 (s, 3H), 0.87 (t, J = 7.0 Hz, 4H); 13C NMR (150 MHz, CDCl3) δ 114.1, 91.0, 81.3, 73.3, 71.1, 43.9, 38.0, 37.3, 36.1, 31.8, 30.6, 29.5, 29.2, 26.3, 23.4, 22.6, 20.9, 14.1; IR (Neat): 3280, 2926, 2857, 1461, 1334, 1108; HRMS (ESI+): calcd. for C18H34O4Na 337.2354, found: 337.2354.
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-66-i11.svg?max-width=637&scale=1.18182)
Characterization data for 17b (obtained as a mixture with 17a): 1H NMR (400 MHz, CDCl3) δ 4.39–4.33 (m, 1H), 4.07–3.97 (m, 1H), 3.67 (d, J = 10.7 Hz, 1H), 3.65 (d, J = 2.9 Hz, 1H), 2.44 (dd, J = 14.3, 5.5 Hz, 1H), 2.21–1.98 (m, 5H), 1.54–1.46 (m, 1H), 1.36–1.23 (m, 12H), 1.07 (s, 3H), 1.05 (s, 3H), 0.89 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 113.2, 87.9, 78.3, 72.6, 69.7, 69.0, 46.4, 37.5, 36.9, 35.6, 31.8, 30.2, 29.7, 29.3, 25.8, 24.2, 22.7, 21.8, 14.1. IR (Neat): 3280, 2926, 2857, 1461, 1334, 1108; HRMS (ESI+): calcd. for C18H34O4Na 337.2354, found: 337.2354.
![[Graphic 4]](/bjoc/content/inline/1860-5397-7-66-i12.svg?max-width=637&scale=1.18182)
Characterization data for 14: 1H NMR (400 MHz, CDCl3) δ 4.49 (m, 1H) [Major], 4.18 (m, 1H) [ Minor], 3.76 (d, J = 6.9 Hz, 1H) [Minor], 3.60 (d, J = 6.5 Hz, 1H) [Major], 3.49 (m, 1H), 3.39–3.30 (m, 2H), 3.31–2.20 (m, 1H), 2.15–1.86 (m, 4H), 1.68 (m, 1H), 1.43 (m, 1H), 1.29 (d, J = 6.1 Hz, 3H) [Major], 1.21 (d, J = 6.2 Hz, 3H) [Minor], 0.87 (s, 9H) [Minor], 0.86 (s, 9H) [Major], 0.071 (d, J = 1.8 Hz, 6H) [Minor], 0.07(d, J = 7.4 Hz, 6H) [Major]; 13C NMR (101 MHz, CDCl3) δ 113.28, 112.79, 92.23, 89.87, 76.85, 74.43, 72.49, 71.61, 71.49, 71.43, 45.43, 45.15, 37.54, 37.18, 36.75, 36.70, 32.34, 31.77, 25.73, 25.70, 22.68, 22.39, 21.20, 21.04, 20.46, 19.73, 17.78, 17.70, −3.95, −4.01, −4.86, −4.95. HRMS (CI+): calcd. for C18H37O4Si 345.2455, found: 345.2455.
![[Graphic 5]](/bjoc/content/inline/1860-5397-7-66-i13.svg?max-width=637&scale=1.18182)
Characterization data for 15: 1H NMR (400 MHz, CDCl3) δ 4.57 (dd, J = 16.2, 7.1 Hz, 1H), 4.24 (dq, J = 6.2, 12.6 Hz, 1H), 4.17–4.06 (m, 2H), 3.86 (d, J = 2.6 Hz, 1H), 3.55–3.32 (m, 5H), 3.08 (br s, 1H), 2.95 (br d, J = 9.3 Hz, 1H), 2.65 (br s, 1H), 2.57 (br s, 1H), 2.31 (dd, J = 12.5, 7.0 Hz, 1H), 2.19–1.88 (m, 9H), 1.76–1.57 (m, 4H), 1.53–1.40 (m, 1H), 1.29 (d, J = 6.1 Hz, 3H), 1.22 (d, J = 6.2 Hz, 3H), 0.97 (s, 3H), 0.96 (s, 3H), 0.93 (s, 3H), 0.86 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 114.6, 113.0, 94.5, 91.7, 76.8, 74.8, 72.9, 72.1, 70.9, 70.8, 44.3, 44.1, 37.5, 37.3, 36.9, 34.0, 32.3, 31.6, 22.5, 21.5, 21.3, 20.99, 20.84, 18.8; IR (Neat): 3389, 3005, 2969, 2873, 1461, 1350; HRMS (ESI+): calcd. for C12H22O4SiNa 253.1416, found: 253.1413.
![[Graphic 6]](/bjoc/content/inline/1860-5397-7-66-i14.svg?max-width=637&scale=1.18182)
Characterization data for 16: 1H NMR (400 MHz, CDCl3) δ 5.97 (d, J = 3.1 Hz, 1H), 5.90 (d, J = 3.0 Hz, 1H), 3.83 (m, 2H), 3.56 (s, 2H), 2.76–2.62 (m, 2H), 1.77 (m, 2H), 1.23 (d, J = 12.8 Hz, 9H); 13C NMR (101 MHz, CDCl3) δ 114.32, 91.01, 77.14, 73.25, 70.84, 43.98, 38.01, 36.43, 32.16, 23.77, 22.66, 21.01. HRMS (ESI+): calcd. for C12H20O3Na 235.1310, found: 235.1315.
![[Graphic 7]](/bjoc/content/inline/1860-5397-7-66-i15.svg?max-width=637&scale=1.18182)
Characterization data for 23: 1H NMR (400 MHz, CDCl3) δ 7.40 (d, J = 8.9 Hz, 2H), 6.91 (d, J = 16.24 Hz, 1H), 6.88 (d, J = 8.68 Hz, 2H), 6.71 (d, J = 16.24 Hz, 1H), 6.21 (d, J = 3.24 Hz, 1H), 6.12 (d, J = 3.24 Hz, 1H), 3.82 (s, 3H), 3.64 (d, J = 6.56 Hz, 2H), 1.62 (t, J = 6.60 Hz, 1H) 1.32 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 159.9, 159.1, 152.4, 129.9, 127.4, 125.8, 114.7, 114.1, 108.4, 106.9, 71.0, 55.2, 38.5, 23.4. HRMS (CI+): calcd. for C17H20O3 272.1412, found: 272.1421.
Acknowledgements
This research was supported by a grant from the National Science Foundation (NSF-CHE 0749918). We thank Dr Tom Gedris (FSU) for assistance with NMR analysis and the FSU High Performance Computing group for assistance with the calculations. Helpful discussions with Dr. Abdulkader Baroudi (FSU) regarding computational chemistry and with Prof. Aaron Aponick (UF) regarding gold-catalyzed cyclocondensations are gratefully acknowledged.
References
-
Perron, F.; Albizati, K. F. Chem. Rev. 1989, 89, 1617–1661. doi:10.1021/cr00097a015
Return to citation in text: [1] -
Aho, J. E.; Pihko, P. M.; Rissa, T. K. Chem. Rev. 2005, 105, 4406–4440. doi:10.1021/cr050559n
Return to citation in text: [1] -
Rama Raju, B.; Saikia, A. K. Molecules 2008, 13, 1942–2038. doi:10.3390/molecules13081942
Return to citation in text: [1] -
Blunt, J. W.; Copp, B. R.; Hu, W.-P.; Munro, M. H. G.; Northcote, P. T.; Prinsep, M. R. Nat. Prod. Rep. 2009, 26, 170–244. doi:10.1039/b805113p
Return to citation in text: [1] -
Mead, K. T.; Brewer, B. N. Curr. Org. Chem. 2003, 7, 227–256. doi:10.2174/1385272033372969
Return to citation in text: [1] -
Brimble, M. A.; Bryant, C. J. Chem. Commun. 2006, 4506–4508. doi:10.1039/b612757f
Return to citation in text: [1] -
Izquierdo Cubero, I.; Plaza Lopez-Espinosa, M. T.; Kari, N. Carbohydr. Res. 1994, 261, 231–242. doi:10.1016/0008-6215(94)84020-2
Return to citation in text: [1] -
Utimoto, K. Pure Appl. Chem. 1983, 55, 1845–1852. doi:10.1351/pac198355111845
Return to citation in text: [1] [2] -
Liu, B.; De Brabander, J. K. Org. Lett. 2006, 8, 4907–4910. doi:10.1021/ol0619819
Return to citation in text: [1] [2] [3] -
Messerle, B. A.; Vuong, K. Q. Pure Appl. Chem. 2006, 78, 385–390. doi:10.1351/pac200678020385
Return to citation in text: [1] -
Fang, C.; Pang, Y.; Forsyth, C. J. Org. Lett. 2010, 12, 4528–4531. doi:10.1021/ol101833h
Return to citation in text: [1] -
Dai, L.-Z.; Shi, M. Chem.–Eur. J. 2008, 14, 7011–7018. doi:10.1002/chem.200701954
Return to citation in text: [1] -
Zhang, Y.; Xue, J.; Xin, Z.; Xie, Z.; Li, Y. Synlett 2008, 6, 940–944. doi:10.1055/s-2008-1042910
Return to citation in text: [1] [2] -
Sherry, B. D.; Maus, L.; Laforteza, B. N.; Toste, F. D. J. Am. Chem. Soc. 2006, 128, 8132–8133. doi:10.1021/ja061344d
Return to citation in text: [1] -
Belting, V.; Krause, N. Org. Lett. 2006, 8, 4489–4492. doi:10.1021/ol061751u
Return to citation in text: [1] -
Antoniotti, S.; Genin, E.; Michelet, V.; Genêt, J.-P. J. Am. Chem. Soc. 2005, 127, 9976–9977. doi:10.1021/ja0530671
Return to citation in text: [1] -
Martín, A.; Salazar, J. A.; Suárez, E. J. Org. Chem. 1996, 61, 3999–4006. doi:10.1021/jo960060g
Return to citation in text: [1] -
Majetich, G.; Wheless, K. Tetrahedron 1995, 51, 7095–7129. doi:10.1016/0040-4020(95)00406-X
Return to citation in text: [1] -
Phillips, S. T.; Shair, M. D. J. Am. Chem. Soc. 2007, 129, 6589–6598. doi:10.1021/ja0705487
Return to citation in text: [1] -
Pastine, S. J.; Sames, D. Org. Lett. 2005, 7, 5429–5431. doi:10.1021/ol0522283
Return to citation in text: [1] -
Enthalpies calculated at the B3LYP/6-31G level with the assistance of the FSU High Performance Computing group.
Return to citation in text: [1] -
Trost, B. M. Science 1991, 254, 1471–1477. doi:10.1126/science.1962206
Return to citation in text: [1] -
Tummatorn, J.; Dudley, G. B. Org. Lett. 2011, 13, 158–160. doi:10.1021/ol102760q
Return to citation in text: [1] -
Jones, D. M.; Dudley, G. B. Tetrahedron 2010, 66, 4860–4866. doi:10.1016/j.tet.2010.03.014
Return to citation in text: [1] -
Jones, D. M.; Lisboa, M. P.; Kamijo, S.; Dudley, G. B. J. Org. Chem. 2010, 75, 3260–3267. doi:10.1021/jo100249g
Return to citation in text: [1] -
Tummatorn, J.; Dudley, G. B. J. Am. Chem. Soc. 2008, 130, 5050–5051. doi:10.1021/ja801018r
Return to citation in text: [1] -
Kamijo, S.; Dudley, G. B. J. Am. Chem. Soc. 2006, 128, 6499–6507. doi:10.1021/ja0608085
Return to citation in text: [1] -
Tlais, S. F.; Dudley, G. B. Org. Lett. 2010, 12, 4698–4701. doi:10.1021/ol102201z
Return to citation in text: [1] [2] [3] [4] -
Solladie, G.; Huser, N.; Fischer, J.; Decian, A. J. Org. Chem. 1995, 60, 4988–4990. doi:10.1021/jo00121a015
Return to citation in text: [1] -
Aponick, A.; Li, C.-Y.; Palmes, J. A. Org. Lett. 2008, 11, 121–124. doi:10.1021/ol802491m
Return to citation in text: [1] -
Selvaratnam, S.; Ho, J. H. H.; Huleatt, P. B.; Messerle, B. A.; Chai, C. L. L. Tetrahedron Lett. 2009, 50, 1125–1127. doi:10.1016/j.tetlet.2008.12.075
Return to citation in text: [1] -
Trost, B. M.; Weiss, A. H. Angew. Chem., Int. Ed. 2007, 46, 7664–7666. doi:10.1002/anie.200702637
Return to citation in text: [1] -
Trost, B. M.; Horne, D. B.; Woltering, M. J. Angew. Chem., Int. Ed. 2003, 42, 5987–5990. doi:10.1002/anie.200352857
Return to citation in text: [1] -
Ramana, C. V.; Suryawanshi, S. B.; Gonnade, R. G. J. Org. Chem. 2009, 74, 2842–2845. doi:10.1021/jo802539z
Return to citation in text: [1] -
Aponick, A.; Li, C.-Y.; Malinge, J.; Marques, E. F. Org. Lett. 2009, 11, 4624–4627. doi:10.1021/ol901901m
Return to citation in text: [1] -
Trost, B. M.; Dong, G. Nature 2008, 456, 485–488. doi:10.1038/nature07543
Return to citation in text: [1] -
Tlais, S. F.; Clark, R. J.; Dudley, G. B. Molecules 2009, 14, 5216–5222. doi:10.3390/molecules14125216
Return to citation in text: [1] -
Lopez, S. S.; Engel, D. A.; Dudley, G. B. Synlett 2007, 949–953. doi:10.1055/s-2007-973885
Return to citation in text: [1]
| 28. | Tlais, S. F.; Dudley, G. B. Org. Lett. 2010, 12, 4698–4701. doi:10.1021/ol102201z |
| 38. | Lopez, S. S.; Engel, D. A.; Dudley, G. B. Synlett 2007, 949–953. doi:10.1055/s-2007-973885 |
| 28. | Tlais, S. F.; Dudley, G. B. Org. Lett. 2010, 12, 4698–4701. doi:10.1021/ol102201z |
| 1. | Perron, F.; Albizati, K. F. Chem. Rev. 1989, 89, 1617–1661. doi:10.1021/cr00097a015 |
| 2. | Aho, J. E.; Pihko, P. M.; Rissa, T. K. Chem. Rev. 2005, 105, 4406–4440. doi:10.1021/cr050559n |
| 3. | Rama Raju, B.; Saikia, A. K. Molecules 2008, 13, 1942–2038. doi:10.3390/molecules13081942 |
| 4. | Blunt, J. W.; Copp, B. R.; Hu, W.-P.; Munro, M. H. G.; Northcote, P. T.; Prinsep, M. R. Nat. Prod. Rep. 2009, 26, 170–244. doi:10.1039/b805113p |
| 17. | Martín, A.; Salazar, J. A.; Suárez, E. J. Org. Chem. 1996, 61, 3999–4006. doi:10.1021/jo960060g |
| 18. | Majetich, G.; Wheless, K. Tetrahedron 1995, 51, 7095–7129. doi:10.1016/0040-4020(95)00406-X |
| 19. | Phillips, S. T.; Shair, M. D. J. Am. Chem. Soc. 2007, 129, 6589–6598. doi:10.1021/ja0705487 |
| 20. | Pastine, S. J.; Sames, D. Org. Lett. 2005, 7, 5429–5431. doi:10.1021/ol0522283 |
| 9. | Liu, B.; De Brabander, J. K. Org. Lett. 2006, 8, 4907–4910. doi:10.1021/ol0619819 |
| 13. | Zhang, Y.; Xue, J.; Xin, Z.; Xie, Z.; Li, Y. Synlett 2008, 6, 940–944. doi:10.1055/s-2008-1042910 |
| 36. | Trost, B. M.; Dong, G. Nature 2008, 456, 485–488. doi:10.1038/nature07543 |
| 8. | Utimoto, K. Pure Appl. Chem. 1983, 55, 1845–1852. doi:10.1351/pac198355111845 |
| 9. | Liu, B.; De Brabander, J. K. Org. Lett. 2006, 8, 4907–4910. doi:10.1021/ol0619819 |
| 10. | Messerle, B. A.; Vuong, K. Q. Pure Appl. Chem. 2006, 78, 385–390. doi:10.1351/pac200678020385 |
| 11. | Fang, C.; Pang, Y.; Forsyth, C. J. Org. Lett. 2010, 12, 4528–4531. doi:10.1021/ol101833h |
| 12. | Dai, L.-Z.; Shi, M. Chem.–Eur. J. 2008, 14, 7011–7018. doi:10.1002/chem.200701954 |
| 13. | Zhang, Y.; Xue, J.; Xin, Z.; Xie, Z.; Li, Y. Synlett 2008, 6, 940–944. doi:10.1055/s-2008-1042910 |
| 14. | Sherry, B. D.; Maus, L.; Laforteza, B. N.; Toste, F. D. J. Am. Chem. Soc. 2006, 128, 8132–8133. doi:10.1021/ja061344d |
| 15. | Belting, V.; Krause, N. Org. Lett. 2006, 8, 4489–4492. doi:10.1021/ol061751u |
| 16. | Antoniotti, S.; Genin, E.; Michelet, V.; Genêt, J.-P. J. Am. Chem. Soc. 2005, 127, 9976–9977. doi:10.1021/ja0530671 |
| 37. | Tlais, S. F.; Clark, R. J.; Dudley, G. B. Molecules 2009, 14, 5216–5222. doi:10.3390/molecules14125216 |
| 6. | Brimble, M. A.; Bryant, C. J. Chem. Commun. 2006, 4506–4508. doi:10.1039/b612757f |
| 7. | Izquierdo Cubero, I.; Plaza Lopez-Espinosa, M. T.; Kari, N. Carbohydr. Res. 1994, 261, 231–242. doi:10.1016/0008-6215(94)84020-2 |
| 9. | Liu, B.; De Brabander, J. K. Org. Lett. 2006, 8, 4907–4910. doi:10.1021/ol0619819 |
| 5. | Mead, K. T.; Brewer, B. N. Curr. Org. Chem. 2003, 7, 227–256. doi:10.2174/1385272033372969 |
| 35. | Aponick, A.; Li, C.-Y.; Malinge, J.; Marques, E. F. Org. Lett. 2009, 11, 4624–4627. doi:10.1021/ol901901m |
| 28. | Tlais, S. F.; Dudley, G. B. Org. Lett. 2010, 12, 4698–4701. doi:10.1021/ol102201z |
| 29. | Solladie, G.; Huser, N.; Fischer, J.; Decian, A. J. Org. Chem. 1995, 60, 4988–4990. doi:10.1021/jo00121a015 |
| 30. | Aponick, A.; Li, C.-Y.; Palmes, J. A. Org. Lett. 2008, 11, 121–124. doi:10.1021/ol802491m |
| 31. | Selvaratnam, S.; Ho, J. H. H.; Huleatt, P. B.; Messerle, B. A.; Chai, C. L. L. Tetrahedron Lett. 2009, 50, 1125–1127. doi:10.1016/j.tetlet.2008.12.075 |
| 32. | Trost, B. M.; Weiss, A. H. Angew. Chem., Int. Ed. 2007, 46, 7664–7666. doi:10.1002/anie.200702637 |
| 33. | Trost, B. M.; Horne, D. B.; Woltering, M. J. Angew. Chem., Int. Ed. 2003, 42, 5987–5990. doi:10.1002/anie.200352857 |
| 34. | Ramana, C. V.; Suryawanshi, S. B.; Gonnade, R. G. J. Org. Chem. 2009, 74, 2842–2845. doi:10.1021/jo802539z |
| 23. | Tummatorn, J.; Dudley, G. B. Org. Lett. 2011, 13, 158–160. doi:10.1021/ol102760q |
| 24. | Jones, D. M.; Dudley, G. B. Tetrahedron 2010, 66, 4860–4866. doi:10.1016/j.tet.2010.03.014 |
| 25. | Jones, D. M.; Lisboa, M. P.; Kamijo, S.; Dudley, G. B. J. Org. Chem. 2010, 75, 3260–3267. doi:10.1021/jo100249g |
| 26. | Tummatorn, J.; Dudley, G. B. J. Am. Chem. Soc. 2008, 130, 5050–5051. doi:10.1021/ja801018r |
| 27. | Kamijo, S.; Dudley, G. B. J. Am. Chem. Soc. 2006, 128, 6499–6507. doi:10.1021/ja0608085 |
| 21. | Enthalpies calculated at the B3LYP/6-31G level with the assistance of the FSU High Performance Computing group. |
| 28. | Tlais, S. F.; Dudley, G. B. Org. Lett. 2010, 12, 4698–4701. doi:10.1021/ol102201z |
© 2011 Tlais and Dudley; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)