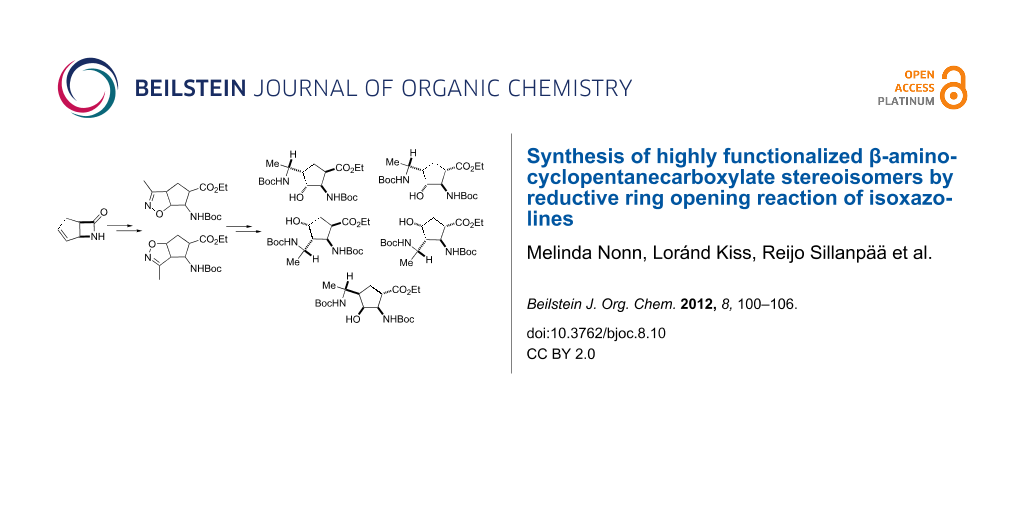
Abstract
A rapid and simple procedure was devised for the synthesis of multifunctionalized cyclic β-amino esters and γ-amino alcohols via the 1,3-dipolar cycloaddition of nitrile oxides to β-aminocyclopentenecarboxylates. The opening of the isoxazoline reductive ring to the corresponding highly functionalized 2-aminocyclopentanecarboxylates occurred stereoselectively with good yields.

Graphical Abstract
Introduction
Isoxazoline-fused amino acids are important bioactive derivatives in organic and medicinal chemistry (e.g., conformationally restricted aspartate and glutamate analogues) [1-6]. As a consequence of their ability to undergo reductive ring opening, isoxazolines are of interest as precursors for the synthesis of highly functionalized molecules such as β-hydroxyketones [7-10], amino alcohols or amino acids [11-17], etc. The multifunctionalized cyclic amino acids – e.g., the antibiotic Oryzoxymycin [18-21], the antiviral agents Tamiflu [22-33], Zanamivir and 2,3-didehydro-2-deoxy-N-acetylneuraminic acid (DANA) [34-38] – are bioactive derivatives of great significance for medicinal chemistry. A promising neuraminidase inhibitor, BCX-1812 (Peramivir), is currently under evaluation in clinical trials [39-45] (Figure 1). A series of Peramivir analogues has recently been investigated as potential antiviral agents [46,47].
![[1860-5397-8-10-1]](/bjoc/content/figures/1860-5397-8-10-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of neuraminidase inhibitors.
Figure 1: Structures of neuraminidase inhibitors.
Results and Discussion
We recently reported a regio- and stereoselective procedure for the formation of a series of isoxazoline-fused cispentacin and transpentacin regio- and stereoisomers (2–6) from bicyclic β-lactam 1 [48,49] (Scheme 1). The syntheses consisted of a dipolar cycloaddition of nitrile oxide (generated with Boc2O, Et3N and DMAP) to the olefinic bond of cis-ethyl 2-aminocyclopent-3-enecarboxylate derived from 1, during which the isoxazoline-fused amino ester regio- and stereoisomers (2 and 4) were formed, then separated and isolated. The cycloaddition of nitrile oxide to trans-ethyl 2-aminocyclopent-3-enecarboxylate under similar conditions proceeded selectively with the formation of 6. Epimerization of 2 and 4 afforded trans derivatives 3 and 5 [48,49].
![[1860-5397-8-10-i1]](/bjoc/content/inline/1860-5397-8-10-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Isoxazoline-fused β-aminocyclopentanecarboxylate regio- and stereoisomers [8].
Scheme 1: Isoxazoline-fused β-aminocyclopentanecarboxylate regio- and stereoisomers [8].
Since isoxazoline-functionalized molecules are excellent precursors for the construction of different functional groups through reductive ring cleavage, our recent aim was to synthesize highly functionalized β-aminocyclopentanecarboxylate regio- and stereoisomers from the earlier prepared isoxazoline-fused cispentacin and transpentacin derivatives.
A number of methods are known for the reductive opening of the isoxazoline ring: Catalytic hydrogenation or reduction with Fe in the presence of NH4Cl, NaBH4, LiAlH4, Raney Ni, BH3·THF, or SmI2/B(OH)3/H2O [7-17].
For the reduction, we selected model compound 2 from earlier prepared isoxazoline-fused cispentacin stereoisomers to execute the reduction under different conditions. The isoxazoline-fused derivative was treated with the above-mentioned reducing agents. Unfortunately, neither transformation nor isoxazoline opening with ester reduction was observed. When the reduction was carried out with NaBH4 in EtOH, three products were obtained: The epimerized isoxazoline-fused amino carboxylate 7 and amino alcohols 8 and 9 which were separated by chromatography and isolated (Scheme 2).
![[1860-5397-8-10-i2]](/bjoc/content/inline/1860-5397-8-10-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Treatment of isoxazoline-fused amino ester 2 with NaBH4.
Scheme 2: Treatment of isoxazoline-fused amino ester 2 with NaBH4.
Thus, this reaction did not lead to the formation of highly functionalized isoxazoline ring-opened β-amino ester either. When ammonium formate in EtOH in the presence of Pd/C was investigated for the reduction of 2, the ring opening resulted in carbonyl compound 10 in rather low yield through the corresponding hydroxyimine intermediate, followed by elimination and saturation (Scheme 3).
![[1860-5397-8-10-i3]](/bjoc/content/inline/1860-5397-8-10-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reduction with Pd/C in the presence of HCO2NH4.
Scheme 3: Reduction with Pd/C in the presence of HCO2NH4.
Combinations of NaBH4 (as a mild and selective reducing agent) with cobalt, nickel, iridium or rhodium halide have previously been employed for cleavage of the isoxazoline ring system, which is otherwise inert to NaBH4 without such metal halide additives [50]. Accordingly, we investigated the reduction of isoxazoline-fused amino ester stereoisomers 2 [48,49] with NaBH4 in the presence of NiCl2 (Scheme 4), which was found to be a suitable reducing system.
![[1860-5397-8-10-i4]](/bjoc/content/inline/1860-5397-8-10-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Transformation of isoxazoline-fused cispentacin stereoisomer 2 into multifunctionalized β-amino acid derivative 12.
Scheme 4: Transformation of isoxazoline-fused cispentacin stereoisomer 2 into multifunctionalized β-amino aci...
The reduction carried out by adding NaBH4 to a mixture of NiCl2 and isoxazoline derivative 2 in EtOH/H2O, followed by amino group protection with Boc2O, selectively afforded only isoxazoline-opened product 12 as a single diastereomer in good yield. The reaction was exothermic and deposited a black granular precipitate, reflecting the presence of metal boride. The product was purified by column chromatography and the structure of 12 was certified by X-ray analysis (Figure 2).
![[1860-5397-8-10-2]](/bjoc/content/figures/1860-5397-8-10-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ORTEP diagram of 12 showing the atomic labeling scheme. The thermal ellipsoids are drawn at the 20% probability level.
Figure 2: ORTEP diagram of 12 showing the atomic labeling scheme. The thermal ellipsoids are drawn at the 20%...
The isoxazoline opening occurred with the formation of a new stereocenter at a one-carbon distance from C-3. In accordance with earlier results [39-47], the hydrogenation of the isoxazoline proceeded through hydrogen attack from the carbamate side (cis to –NHBoc) of the cyclopentane skeleton. This was confirmed by X-ray analysis of 12.
In order to increase the number of multifunctionalized amino ester stereoisomers, we next examined the reductions of isoxazoline-fused cispentacin and transpentacin stereoisomers (3–6) [49]. Reactions were carried out similarly with NaBH4 in the presence of NiCl2 in EtOH/H2O and led selectively to the corresponding multifunctionalized amino esters 13–16 in good yields (Scheme 5) as single diastereoisomers.
![[1860-5397-8-10-i5]](/bjoc/content/inline/1860-5397-8-10-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of multifunctionalized β-amino acid derivatives 13–16. Reaction conditions: NaBH4, NiCl2, Boc2O, EtOH/H2O, rt, 6 h.
Scheme 5: Synthesis of multifunctionalized β-amino acid derivatives 13–16. Reaction conditions: NaBH4, NiCl2,...
Conclusion
The present work has furnished a facile and efficient stereoselective reduction of isoxazoline-fused cyclic β-amino esters to multifunctionalized 2-aminocyclopentanecarboxylates through the use of NaBH4/NiCl2 as reducing agent. As Peramivir related derivatives, highly functionalized cyclic amino esters may be regarded as promising bioactive compounds.
Experimental
The chemicals were purchased from Aldrich. The solvents were used as received from the supplier. Melting points were determined with a Kofler apparatus. NMR spectra were recorded on a Bruker DRX 400 MHz spectrometer in deuterated DMSO or CDCl3. Chemical shifts are expressed in ppm (δ) from the signal of internal tetramethylsilane. Mass spectra were recorded on a Finnigan MAT 95S spectrometer. Elemental analyses were recorded on a Perkin-Elmer CHNS-2400 Ser II Elemental Analyzer. FTIR spectra were recorded on a Perkin-Elmer Spectrum 100 instrument. Cycloadducts 2–6 were synthesized according to previously published procedures [8].
General procedure for the synthesis of compounds 8 and 9
To a solution of izoxazoline-fused β-aminocyclopentanecarboxylate 2 (0.96 mmol) in dry EtOH (15 mL) NaBH4 (2.88 mmol) was added and the reaction mixture was stirred under reflux for 16 h. The reaction was quenched by the addition of H2O (10 mL) and then, the mixture was concentrated under reduced pressure. The reaction mixture was diluted with H2O (20 mL), washed with EtOAc (3 × 15 mL), dried (Na2SO4) and concentrated under reduced pressure. The crude residue was purified by column chromatography on silica gel (n-hexane/EtOAc) giving 8 and 9.
General procedure for the synthesis of 10
To a stirred solution of isoxazoline-fused β-aminocyclopentanecarboxylate 2 (1.6 mmol) in dry EtOH (15 mL), HCOONH4 (3.2 mmol) and Pd/C (0.10 g) were added and the reaction mixture was stirred under reflux for 24 h. The mixture was filtered through a celite pad and the filtrate was evaporated in vacuo. The crude residue was diluted with EtOAc (30 mL), washed with H2O (3 × 15 mL), dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (n-hexane/EtOAc), giving 10.
General procedure for isoxazoline ring opening
To a stirred solution of isoxazoline-fused β-aminocyclopentanecarboxylates 2–6 (0.96 mmol) in 8 mL of EtOH/THF (v:v = 3:1), NiCl2 (1.92 mmol) and Boc2O (1.92 mmol) were added. After stirring for 10 min, NaBH4 (1.92 mmol) was added in portions. The reaction mixture was further stirred for 6 h at room temperature and the reaction was quenched by the addition of H2O (5 mL). The reaction mixture was filtered through a celite pad and the filtrate was evaporated in vacuo. The crude residue was diluted with EtOAc (30 mL), washed with H2O (3 × 15 mL), dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (n-hexane/EtOAc), giving the corresponding reduced product.
tert-Butyl (3aR*,4R*,5R*,6aR*)-[5-(hydroxymethyl)-3-methyl-4,5,6,6a-tetrahydro-3aH-cyclopenta[d]isoxazol-4-yl]carbamate (8): Light-yellow oil; yield 48% (124 mg); Rf 0.35 (n-hexane/EtOAc); IR (KBr) ν/cm–1: 3344, 3265, 2979, 1678, 1563, 1184; 1H NMR (400 MHz, CDCl3) δ 1.45 (s, 3H, CH3), 1.56 (s, 9H, CH3), 1.65–1.72 (m, 2H, CH2), 2.19–2.25 (m, 1H, H-5), 2.75–2.81 (m, 1H, H-3a), 3.19–3.25 (m, 1H, H-6a), 3.59–3.71 (m, 1H, H-4), 3.63–3.72 (m, 2H, CH2), 5.42 (br s, 1H, N-H), OH group not observed – exchanged; 13C NMR (100 MHz, CDCl3) δ 16.0, 28.6, 30.2, 32.5, 43.0, 44.4, 59.2, 63.9, 78.0, 155.2, 155.6; MS (ESI) m/z: 293 [M + Na]+; Anal. calcd for C13H22N2O4: C, 57.76; H, 8.20; N, 10.36; found: C, 57.60; H, 8.07; N, 10.23.
tert-Butyl (3S*,3aR*,4R*,5R*,6aR*)-[5-(hydroxymethyl)-3-methylhexahydro-2H-cyclopenta[d]isoxazol-4-yl]carbamate (9): Colorless oil; yield 12% (31 mg); Rf 0.29 (n-hexane/EtOAc); IR (KBr) ν/cm–1: 3460, 3331, 2978, 1683, 1531, 1174; 1H NMR (400 MHz, CDCl3) δ 0.98–1.05 (m, 3H, CH3), 1.36 (s, 9H, CH3), 1.55–1.75 (m, 2H, CH2), 2.22–2.27 (m, 1H, H-5), 2.38–2.47 (m, 1H, H-3a), 2.78–2.86 (m, 1H, H-3), 3.17–3.24 (m, 1H, H-6a), 3.59–3.69 (m, 1H, H-4), 3.36–3.68 (m, 2H, CH2), 5.32 (br s, 1H, N-H), 6.12 (br s, 1H, N-H), OH group not observed – exchanged; 13C NMR (100 MHz, CDCl3) δ 15.0, 27.1, 29.0, 35.8, 42.4, 51.7, 57.2, 62.6, 77.4, 80.4, 155.6; MS (ESI) m/z: 295 [M + Na]+; Anal. calcd for C13H24N2O4: C, 57.33; H, 8.88; N, 10.29; found: C, 57.20; H, 8.71; N, 10.42.
Ethyl (1R*,2S*,3S*)-3-acetyl-2-(tert-butoxycarbonylamino)cyclopentanecarboxylate (10): White solid; yield 32% (153 mg); mp 109–110 °C; Rf 0.62 (n-hexane/EtOAc); IR (KBr) ν/cm–1: 3354, 2978, 1716, 1684, 1531, 1171; 1H NMR (400 MHz, CDCl3) δ 1.29 (t, J = 7.54 Hz, 3H, CH3), 1.41 (s, 9H, CH3), 1.59–1.71 (m, 2H, CH2), 1.74–1.95 (m, 2H, CH2), 2.05 (s, 3H, CH3), 2.83–2.97 (m, 1H, H-1), 3.01–3.15 (m, 1H, H-3), 4.18–4.29 (m, 2H, OCH2), 4.31–4.44 (m, 1H, H-2), 5.76 (br s, 1H, N-H); 13C NMR (100 MHz, CDCl3) δ 13.98, 20.05, 25.76, 29.31, 31.21, 43.97, 46.01, 52.70, 82.01, 155.67, 176.01, 206.52; MS (ESI) m/z: 322 [M + Na]+; Anal. calcd for C15H25NO5: C, 60.18; H, 8.42; N, 4.68; found: C, 60.05; H, 8.35; N, 4.54.
Ethyl (1R*,2S*,3S*,4R*)-2-(tert-butoxycarbonyl)-3-((S*)-1-(tert-butoxycarbonyl)ethyl)-4-hydroxycyclopentanecarboxylate (12): White solid; yield 80% (320 mg); mp 120–121 °C; Rf 0.22 (n-hexane/EtOAc 1:1); IR (KBr) ν/cm–1: 3457, 3348, 2982, 1720, 1698, 1531, 1160; 1H NMR (400 MHz, DMSO) δ 0.96 (t, J = 7.34 Hz, 3H, CH3), 1.27–1.33 (m, 3H, CH3), 1.45–1.50 (m, 18H, CH3), 1.94–2.02 (m, 2H, CH2), 2.07–2.16 (m, 1H, H-4), 3.30–3.39 (m, 1H, H-1), 3.80–3.89 (m, 1H, CH), 4.13–4.23 (m, 2H, OCH2), 4.24–4.30 (m, 1H, H-2), 4.44–4.56 (m, 1H, H-3), 5.28–5.35 (m, 1H, NH), 5.61–5.72 (m, 1H, NH), OH group not observed – exchanged; 13C NMR (100 MHz, CDCl3) δ 11.7, 14.6, 28.8, 28.9, 29.3, 30.1, 31.8, 37.3, 44.6, 51.1, 54.6, 61.2, 73.7, 80.1, 80.4, 155.0, 156.5, 172.0; MS (ESI) m/z: 418 [M + 2H]+; Anal. calcd for C20H36N2O7: C, 57.67; H, 8.71; N, 6.73; found: C, 57,44; H, 8.86; N, 6.58.
Ethyl (1R*,2R*,3R*,4S*)-2-(tert-butoxycarbonyl)-4-((R*)-1-(tert-butoxycarbonyl)ethyl)-3-hydroxycyclopentanecarboxylate (13): White solid; yield 72% (288 mg); mp 129–130 °C; Rf 0.59 (n-hexane/EtOAc 1:1); IR (KBr) ν/cm–1: 3479, 3347, 3353, 1725, 1685, 1662, 1531, 1163; 1H NMR (400 MHz, CDCl3) δ 1.17–1.29 (m, 6H, CH3), 1.40–1.46 (m, 18H, CH3), 1.79–1.91 (m, 1H, CH2), 2.05–2.19 (m, 2H, CH2, H-1), 3.26–3.34 (m, 1H, H-4), 3.86–4.01 (m, 2H, H-2, CH), 4.08–4.19 (m, 3H, OCH2, H-3), 4.53 (br s, 1H, N-H), 5.05 (br s, 1H, N-H), OH group not observed – exchanged; 13C NMR (100 MHz, CDCl3) δ 14.6, 21.1, 28.4, 28.7, 28.8, 44.0, 46.6, 60.5, 61.2, 67.5, 77.6, 80.2, 86.4, 156.1, 156.4, 174.8; MS (ESI) m/z: 418 [M + 2H]+; Anal. calcd for C20H36N2O7: C, 57.67; H, 8.71; N, 6.73; found: C, 57.50; H, 8.98; N, 6.39.
Ethyl (1S*,2S*,3S*,4R*)-2-(tert-butoxycarbonyl)-3-((S*)-1-(tert-butoxycarbonyl)ethyl)-4-hydroxycyclopentanecarboxylate (14): White solid; yield 75% (300 mg); mp 144–145 °C; Rf 0.3 (n-hexane/EtOAc 1:1); IR (KBr) ν/cm–1: 3420, 3363, 2980, 1692, 1537, 1185; 1H NMR (400 MHz, CDCl3) δ 1.26–1.33 (m, 6H, CH3), 1.43–1.48 (m, 18H, CH3), 1.82–1.93 (m, 1H, CH2), 1.98–2.15 (m, 1H, H-1), 2.24–2.36 (m, 1H, CH2), 2.76–2.89 (m, 1H, H-3), 3.58–3.72 (m, 1H, H-4), 3.93–4.05 (m, 1H, H-2), 4.15–4.25 (m, 3H, OCH2, CH), 4.87 (br s, 1H, N-H), 5.09 (br s, 1H, N-H), OH group not observed – exchanged; 13C NMR (100 MHz, CDCl3) δ 14.5, 21.4, 28.8, 28.9, 35.9, 45.7, 49.1, 52.3, 54.5, 58.3, 73.4, 80.1, 152.5, 156.8, 172.6; MS (ESI) m/z: 418 [M + 2H]+; Anal. calcd for C20H36N2O7: C, 57.67; H, 8.71; N, 6.73; found: C, 57.41; H, 8.37; N, 6.59.
Ethyl (1S*,2R*,3R*,4S*)-2-(tert-butoxycarbonyl)-4-((R*)-1-(tert-butoxycarbonyl)ethyl)-3-hydroxycyclopentanecarboxylate (15): White solid; yield 85% (340 mg); mp 141–142 °C; Rf 0.46 (n-hexane/EtOAc 1:1); IR (KBr) ν/cm–1: 3426, 3378, 3333, 2979, 1688, 1718, 1703, 1522, 1176; 1H NMR (400 MHz, CDCl3) δ 1.21–1.30 (m, 6H, CH3), 1.40–1.46 (m, 18H, CH3), 1.84–1.97 (m, 2H, CH2, H-4), 2.03–2.20 (m, 2H, CH2, H-1), 2.54 (q, J = 9.10 Hz, 1H, H-2,), 3.73–3.82 (m, 1H, H-3), 3.87–4.04 (m, 2H, N-H, CH), 4.10–4.22 (m, 2H, OCH2), 4.83 (br s, 1H, N-H), OH group not observed – exchanged; 13C NMR (100 MHz, CDCl3) δ 14.1, 20.0, 27.5, 28.7, 28.8, 45.6, 46.1, 46.8, 60.9, 62.5, 78.1, 80.1, 80.3, 154.0, 156.4, 174.6; MS (ESI) m/z: 418 [M + 2H]+; Anal. calcd for C20H36N2O7: C, 57.67; H, 8.71; N, 6.73; found: C, 57.91; H, 8.46; N, 6.58.
Ethyl (1S*,2R*,3S*,4R*)-2-(tert-butoxycarbonyl)-4-((S*)-1-(tert-butoxycarbonyl)ethyl)-3-hydroxycyclopentanecarboxylate (16): White solid; yield 82% (328 mg); mp 166–167 °C; Rf 0.32 (n-hexane/EtOAc 1:1); IR (KBr) ν/cm–1: 3485, 3368, 3353, 2975, 1733, 1681, 1667, 1533, 1167; 1H NMR (400 MHz, CDCl3) δ 1.17–1.31 (m, 6H, CH3), 1.38–1.46 (m, 18H, CH3), 1.79–2.15 (m, 3H, CH2, H-1, H-4), 2.72–2.87 (m, 1H, CH2), 3.77–4.03 (m, 1H, CH), 4.06–4.23 (m, 4H, H-2, H-3, OCH2), 4.37–4.48 (m, 1H, N-H), 4.88 (br s, 1H, N-H), OH group not observed – exchanged; 13C NMR (100 MHz, CDCl3) 14.6, 21.6, 28.7, 28.8, 47.2, 49.0, 59.9, 61.2, 61.6, 69.4, 74.7, 80.0, 85.9, 117.5, 156.1, 158.8, 171.3; MS (ESI) m/z: 418 [M + 2H]+; Anal. calcd for C20H36N2O7: C, 57.67; H, 8.71; N, 6.73; found: C, 57.43; H, 8.40; N, 6.95.
X-ray crystallographic study of 12: Crystallographic data were collected at 123 K with a Nonius-Kappa CCD area detector diffractometer, using graphite-monochromatized Mo Ka radiation (λ = 0.71073 Å) as reported earlier [51]. Crystal data for 12, C20H36N2O7, Mr = 416.51, triclinic, space group P−1 (no. 2), a = 9.3765(2), b = 13.7078(4), c = 18.7792(4) Å, α = 96.609(2), β = 95.261(1), γ = 100.965(1), V = 2337.9(1) Å3, T = 123 K, Z = 4, μ(Mo Kα) = 0.089 mm–1, 9120 unique reflections (Rint = 0.034) which were used in calculations. The final R1 (for the data with F2 > 2δ(F2) was 0.042 and wR2(F2) (all data) was 0.111.
The SHELXL-97 program [52] was used to solve the structure by direct methods and to perform full-matrix, least-squares refinements on F2. The unit cell of 12 contains two molecules with slightly different conformations. The CH hydrogen atoms were included at fixed distances from their host atoms with fixed displacement parameters. The NH and OH hydrogen atoms were refined isotropically. The deposition number CCDC 845835 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge at http://www.ccdc.cam.ac.uk/conts/retrieving.html [or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: (internat.) +44-1223-336-033; Email: deposit@ccdc.cam.ac.uk].
References
-
Pinto, A.; Conti, P.; De Amici, M.; Tamborini, L.; Grazioso, G.; Colleoni, S.; Mennini, T.; Gobbi, M.; De Micheli, C. Tetrahedron: Asymmetry 2008, 19, 867–875. doi:10.1016/j.tetasy.2008.03.001
Return to citation in text: [1] -
Conti, P.; Caligiuri, A.; Pinto, A.; Roda, G.; Tamborini, L.; Nielsen, B.; Madsen, U.; Frydenvang, K.; Colombo, A.; De Micheli, C. Eur. J. Med. Chem. 2007, 42, 1059–1068. doi:10.1016/j.ejmech.2007.01.013
Return to citation in text: [1] -
Roda, G.; Conti, P.; De Amici, M.; He, J.; Polavaropu, P. L.; De Micheli, C. Tetrahedron: Asymmetry 2004, 15, 3079–3090. doi:10.1016/j.tetasy.2004.07.037
Return to citation in text: [1] -
Conti, P.; De Amici, M.; Di Ventimiglia, S. J.; Stensbøl, T. B.; Madsen, U.; Bräuner-Osborne, H.; Russo, E.; De Sarro, G.; Bruno, G.; De Micheli, C. J. Med. Chem. 2003, 46, 3102–3108. doi:10.1021/jm0308085
Return to citation in text: [1] -
Park, K.-H.; Olmstead, M. M.; Kurth, M. J. J. Org. Chem. 1998, 63, 113–117. doi:10.1021/jo9714831
Return to citation in text: [1] -
Pinto, A.; Conti, P.; Grazioso, G.; Tamborini, L.; Madsen, U.; Nielsen, B.; De Micheli, C. Eur. J. Med. Chem. 2011, 46, 787–793. doi:10.1016/j.ejmech.2010.12.020
Return to citation in text: [1] -
Bode, J. W.; Carreire, E. M. Org. Lett. 2001, 3, 1587–1590. doi:10.1021/ol015885d
Return to citation in text: [1] [2] -
Bode, J. W.; Fraefel, N.; Muri, D.; Carreira, E. M. Angew. Chem., Int. Ed. 2001, 40, 2082–2085. doi:10.1002/1521-3773(20010601)40:11<2082::AID-ANIE2082>3.0.CO;2-1
Return to citation in text: [1] [2] [3] [4] -
Jiang, D.; Chen, Y. J. Org. Chem. 2008, 73, 9181–9183. doi:10.1021/jo801831c
Return to citation in text: [1] [2] -
Tang, S.; He, J.; Sun, Y.; He, L.; She, X. J. Org. Chem. 2010, 75, 1961–1966. doi:10.1021/jo1000065
Return to citation in text: [1] [2] -
Marotta, E.; Micheloni, L. M.; Scardovi, N.; Righi, P. Org. Lett. 2001, 3, 727–729. doi:10.1021/ol0070379
Return to citation in text: [1] [2] -
Scott, J. P.; Oliver, S. F.; Brands, K. M. J.; Brewer, S. E.; Davies, A. J.; Gibb, A. D.; Hands, D.; Keen, S. P.; Sheen, F. J.; Reamer, R. A.; Wilson, R. D.; Dolling, U.-H. J. Org. Chem. 2006, 71, 3086–3092. doi:10.1021/jo060033i
Return to citation in text: [1] [2] -
Maimone, T. J.; Shi, J.; Ashida, S.; Baran, P. S. J. Am. Chem. Soc. 2009, 131, 17066–17067. doi:10.1021/ja908194b
Return to citation in text: [1] [2] -
Minter, A. R.; Fuller, A. A.; Mapp, A. K. J. Am. Chem. Soc. 2003, 125, 6846–6847. doi:10.1021/ja0298747
Return to citation in text: [1] [2] -
Fuller, A. A.; Chen, B.; Minter, A. R.; Mapp, A. K. J. Am. Chem. Soc. 2005, 127, 5376–5383. doi:10.1021/ja0431713
Return to citation in text: [1] [2] -
Sewald, N. Angew. Chem., Int. Ed. 2003, 42, 5794–5795. doi:10.1002/anie.200301692
Return to citation in text: [1] [2] -
Tokizane, M.; Sato, K.; Ohta, T.; Ito, Y. Tetrahedron: Asymmetry 2008, 19, 2519–2528. doi:10.1016/j.tetasy.2008.11.005
Return to citation in text: [1] [2] -
Palkó, M.; Kiss, L.; Fülöp, F. Curr. Med. Chem. 2005, 12, 3063–3083. doi:10.2174/092986705774933443
Return to citation in text: [1] -
Kiss, L.; Forró, E.; Fülöp, F. Synthesis of carbocyclic β-amino acids. In Amino Acids, Peptides and Proteins in Organic Chemistry; Hughes, A. B., Ed.; Wiley-VCH: Weinheim, Germany, 2009; Vol. 1, pp 367–409.
Return to citation in text: [1] -
Kiss, L.; Fülöp, F. Synlett 2010, 1302–1314. doi:10.1055/s-0029-1219821
Return to citation in text: [1] -
Fülöp, F. Chem. Rev. 2001, 101, 2181–2204. doi:10.1021/cr000456z
Return to citation in text: [1] -
Ishikawa, H.; Suzuki, T.; Orita, H.; Uchimaru, T.; Hayashi, Y. Chem.–Eur. J. 2010, 16, 12616–12626. doi:10.1002/chem.201001108
Return to citation in text: [1] -
Ko, J. S.; Keum, J. E.; Ko, S. Y. J. Org. Chem. 2010, 75, 7006–7009. doi:10.1021/jo101517g
Return to citation in text: [1] -
Karpf, M.; Trussardi, R. Angew. Chem., Int. Ed. 2009, 48, 5760–5762. doi:10.1002/anie.200901561
Return to citation in text: [1] -
Satoh, N.; Akiba, T.; Yokoshima, S.; Fukuyama, T. Tetrahedron 2009, 65, 3239–3245. doi:10.1016/j.tet.2008.09.103
Return to citation in text: [1] -
Ishikawa, H.; Suzuki, T.; Hayashi, Y. Angew. Chem., Int. Ed. 2009, 48, 1304–1307. doi:10.1002/anie.200804883
Return to citation in text: [1] -
Sullivan, B.; Carrera, I.; Drouin, M.; Hudlicky, T. Angew. Chem., Int. Ed. 2009, 48, 4229–4231. doi:10.1002/anie.200901345
Return to citation in text: [1] -
Trost, B. M.; Zhang, T. Angew. Chem., Int. Ed. 2008, 47, 3759–3761. doi:10.1002/anie.200800282
Return to citation in text: [1] -
Zhu, S.; Yu, S.; Wang, Y.; Ma, D. Angew. Chem., Int. Ed. 2010, 49, 4656–4660. doi:10.1002/anie.201001644
Return to citation in text: [1] -
Mohan, S.; McAtamney, S.; Haselhorst, T.; von Itzstein, M.; Pinto, B. M. J. Med. Chem. 2010, 53, 7377–7391. doi:10.1021/jm100822f
Return to citation in text: [1] -
Kamimura, A.; Nakano, T. J. Org. Chem. 2010, 75, 3133–3136. doi:10.1021/jo1002856
Return to citation in text: [1] -
Nie, L.-D.; Shi, X.-X.; Ko, K. H.; Lu, W.-D. J. Org. Chem. 2009, 74, 3970–3973. doi:10.1021/jo900218k
Return to citation in text: [1] -
Osato, H.; Jones, I. L.; Chen, A.; Chai, C. L. L. Org. Lett. 2010, 12, 60–63. doi:10.1021/ol9024716
Return to citation in text: [1] -
Wena, W.-H.; Wang, S.-Y.; Tsai, K.-C.; Cheng, Y.-S. E.; Yang, A.-S.; Fang, J.-M.; Wong, C.-H. Bioorg. Med. Chem. 2010, 18, 4074–4084. doi:10.1016/j.bmc.2010.04.010
Return to citation in text: [1] -
Xu, G.; Kiefel, M. J.; Wilson, J. C.; Andrew, P. W.; Oggioni, M. R.; Taylor, G. L. J. Am. Chem. Soc. 2011, 133, 1718–1721. doi:10.1021/ja110733q
Return to citation in text: [1] -
Calveras, J.; Nagai, Y.; Sultana, I.; Ueda, Y.; Higashi, T.; Shoji, M.; Sugai, T. Tetrahedron 2010, 66, 4284–4291. doi:10.1016/j.tet.2010.04.045
Return to citation in text: [1] -
Honda, T.; Kubo, S.; Masuda, T.; Arai, M.; Kobayashi, Y.; Yamashita, M. Bioorg. Med. Chem. Lett. 2009, 19, 2938–2940. doi:10.1016/j.bmcl.2009.04.067
Return to citation in text: [1] -
Soulé, J.-F.; Mathieu, A.; Norsikian, S.; Beau, J.-M. Org. Lett. 2010, 12, 5322–5325. doi:10.1021/ol102326b
Return to citation in text: [1] -
Sorbera, L. A.; Graul, A.; Castaner, J. Drugs Future 2000, 25, 249–251. doi:10.1358/dof.2000.025.03.565302
Return to citation in text: [1] [2] -
Babu, Y. S.; Chand, P.; Bantia, S.; Kotian, P.; Dehghani, A.; El-Kattan, Y.; Lin, T.-H.; Hutchison, T. L.; Elliott, A. J.; Parker, C. D.; Ananth, S. L.; Horn, L. L.; Laver, G. W.; Montgomery, J. A. J. Med. Chem. 2000, 43, 3482–3486. doi:10.1021/jm0002679
Return to citation in text: [1] [2] -
Chand, P.; Kotian, P. L.; Dehghani, A.; El-Kattan, Y.; Lin, T.-H.; Hutchison, T. L.; Babu, Y. S.; Bantia, S.; Elliott, A. J.; Montgomery, J. A. J. Med. Chem. 2001, 44, 4379–4392. doi:10.1021/jm010277p
Return to citation in text: [1] [2] -
Chand, P.; Babu, Y. S.; Bantia, S.; Rowland, S.; Dehghani, A.; Kotian, P. L.; Hutchison, T. L.; Ali, S.; Brouillette, W.; El-Kattan, Y.; Lin, T.-H. J. Med. Chem. 2004, 47, 1919–1929. doi:10.1021/jm0303406
Return to citation in text: [1] [2] -
Lü, W. J.; Chen, Y. L.; Ma, W. P.; Zhang, X. Y.; Luan, F.; Liu, M. C.; Chen, X. G.; Hu, Z. D. Eur. J. Med. Chem. 2008, 43, 569–576. doi:10.1016/j.ejmech.2007.04.011
Return to citation in text: [1] [2] -
Oakley, A. J.; Barrett, S.; Peat, T. S.; Newman, J.; Streltsov, V. A.; Waddington, L.; Saito, T.; Tashiro, M.; McKimm-Breschkin, J. L. J. Med. Chem. 2010, 53, 6421–6431. doi:10.1021/jm100621s
Return to citation in text: [1] [2] -
Chand, P.; Bantia, S.; Kotian, P. L.; El-Kattan, Y.; Lin, T.-H.; Babu, Y. S. Bioorg. Med. Chem. 2005, 13, 4071–4077. doi:10.1016/j.bmc.2005.03.048
Return to citation in text: [1] [2] -
Cui, Y.; Jiao, Z.; Gong, J.; Yu, Q.; Zheng, X.; Quan, J.; Luo, M.; Yang, Z. Org. Lett. 2010, 12, 4–7. doi:10.1021/ol902438f
Return to citation in text: [1] [2] -
Yi, X.; Guo, Z.; Chu, F. M. Bioorg. Med. Chem. 2003, 11, 1465–1474. doi:10.1016/S0968-0896(02)00602-8
Return to citation in text: [1] [2] -
Kiss, L.; Nonn, M.; Forró, E.; Sillanpää, R.; Fülöp, F. Tetrahedron Lett. 2009, 50, 2605–2608. doi:10.1016/j.tetlet.2009.03.119
Return to citation in text: [1] [2] [3] -
Nonn, M.; Kiss, L.; Forró, E.; Mucsi, Z.; Fülöp, F. Tetrahedron 2011, 67, 4079–4085. doi:10.1016/j.tet.2011.04.005
Return to citation in text: [1] [2] [3] [4] -
Jiang, H.; Elsner, P.; Jensen, K. L.; Falcicchio, A.; Marcos, V.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2009, 48, 6844–6848. doi:10.1002/anie.200901446
Return to citation in text: [1] -
Kanizsai, I.; Szakonyi, Z.; Sillanpää, R.; D'hooghe, M.; De Kimpe, N.; Fülöp, F. Tetrahedron: Asymmetry 2006, 17, 2857–2863. doi:10.1016/j.tetasy.2006.11.006
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A 2008, 64, 112–122. doi:10.1107/S0108767307043930
Return to citation in text: [1]
| 52. | Sheldrick, G. M. Acta Crystallogr., Sect. A 2008, 64, 112–122. doi:10.1107/S0108767307043930 |
| 8. | Bode, J. W.; Fraefel, N.; Muri, D.; Carreira, E. M. Angew. Chem., Int. Ed. 2001, 40, 2082–2085. doi:10.1002/1521-3773(20010601)40:11<2082::AID-ANIE2082>3.0.CO;2-1 |
| 51. | Kanizsai, I.; Szakonyi, Z.; Sillanpää, R.; D'hooghe, M.; De Kimpe, N.; Fülöp, F. Tetrahedron: Asymmetry 2006, 17, 2857–2863. doi:10.1016/j.tetasy.2006.11.006 |
| 1. | Pinto, A.; Conti, P.; De Amici, M.; Tamborini, L.; Grazioso, G.; Colleoni, S.; Mennini, T.; Gobbi, M.; De Micheli, C. Tetrahedron: Asymmetry 2008, 19, 867–875. doi:10.1016/j.tetasy.2008.03.001 |
| 2. | Conti, P.; Caligiuri, A.; Pinto, A.; Roda, G.; Tamborini, L.; Nielsen, B.; Madsen, U.; Frydenvang, K.; Colombo, A.; De Micheli, C. Eur. J. Med. Chem. 2007, 42, 1059–1068. doi:10.1016/j.ejmech.2007.01.013 |
| 3. | Roda, G.; Conti, P.; De Amici, M.; He, J.; Polavaropu, P. L.; De Micheli, C. Tetrahedron: Asymmetry 2004, 15, 3079–3090. doi:10.1016/j.tetasy.2004.07.037 |
| 4. | Conti, P.; De Amici, M.; Di Ventimiglia, S. J.; Stensbøl, T. B.; Madsen, U.; Bräuner-Osborne, H.; Russo, E.; De Sarro, G.; Bruno, G.; De Micheli, C. J. Med. Chem. 2003, 46, 3102–3108. doi:10.1021/jm0308085 |
| 5. | Park, K.-H.; Olmstead, M. M.; Kurth, M. J. J. Org. Chem. 1998, 63, 113–117. doi:10.1021/jo9714831 |
| 6. | Pinto, A.; Conti, P.; Grazioso, G.; Tamborini, L.; Madsen, U.; Nielsen, B.; De Micheli, C. Eur. J. Med. Chem. 2011, 46, 787–793. doi:10.1016/j.ejmech.2010.12.020 |
| 22. | Ishikawa, H.; Suzuki, T.; Orita, H.; Uchimaru, T.; Hayashi, Y. Chem.–Eur. J. 2010, 16, 12616–12626. doi:10.1002/chem.201001108 |
| 23. | Ko, J. S.; Keum, J. E.; Ko, S. Y. J. Org. Chem. 2010, 75, 7006–7009. doi:10.1021/jo101517g |
| 24. | Karpf, M.; Trussardi, R. Angew. Chem., Int. Ed. 2009, 48, 5760–5762. doi:10.1002/anie.200901561 |
| 25. | Satoh, N.; Akiba, T.; Yokoshima, S.; Fukuyama, T. Tetrahedron 2009, 65, 3239–3245. doi:10.1016/j.tet.2008.09.103 |
| 26. | Ishikawa, H.; Suzuki, T.; Hayashi, Y. Angew. Chem., Int. Ed. 2009, 48, 1304–1307. doi:10.1002/anie.200804883 |
| 27. | Sullivan, B.; Carrera, I.; Drouin, M.; Hudlicky, T. Angew. Chem., Int. Ed. 2009, 48, 4229–4231. doi:10.1002/anie.200901345 |
| 28. | Trost, B. M.; Zhang, T. Angew. Chem., Int. Ed. 2008, 47, 3759–3761. doi:10.1002/anie.200800282 |
| 29. | Zhu, S.; Yu, S.; Wang, Y.; Ma, D. Angew. Chem., Int. Ed. 2010, 49, 4656–4660. doi:10.1002/anie.201001644 |
| 30. | Mohan, S.; McAtamney, S.; Haselhorst, T.; von Itzstein, M.; Pinto, B. M. J. Med. Chem. 2010, 53, 7377–7391. doi:10.1021/jm100822f |
| 31. | Kamimura, A.; Nakano, T. J. Org. Chem. 2010, 75, 3133–3136. doi:10.1021/jo1002856 |
| 32. | Nie, L.-D.; Shi, X.-X.; Ko, K. H.; Lu, W.-D. J. Org. Chem. 2009, 74, 3970–3973. doi:10.1021/jo900218k |
| 33. | Osato, H.; Jones, I. L.; Chen, A.; Chai, C. L. L. Org. Lett. 2010, 12, 60–63. doi:10.1021/ol9024716 |
| 39. | Sorbera, L. A.; Graul, A.; Castaner, J. Drugs Future 2000, 25, 249–251. doi:10.1358/dof.2000.025.03.565302 |
| 40. | Babu, Y. S.; Chand, P.; Bantia, S.; Kotian, P.; Dehghani, A.; El-Kattan, Y.; Lin, T.-H.; Hutchison, T. L.; Elliott, A. J.; Parker, C. D.; Ananth, S. L.; Horn, L. L.; Laver, G. W.; Montgomery, J. A. J. Med. Chem. 2000, 43, 3482–3486. doi:10.1021/jm0002679 |
| 41. | Chand, P.; Kotian, P. L.; Dehghani, A.; El-Kattan, Y.; Lin, T.-H.; Hutchison, T. L.; Babu, Y. S.; Bantia, S.; Elliott, A. J.; Montgomery, J. A. J. Med. Chem. 2001, 44, 4379–4392. doi:10.1021/jm010277p |
| 42. | Chand, P.; Babu, Y. S.; Bantia, S.; Rowland, S.; Dehghani, A.; Kotian, P. L.; Hutchison, T. L.; Ali, S.; Brouillette, W.; El-Kattan, Y.; Lin, T.-H. J. Med. Chem. 2004, 47, 1919–1929. doi:10.1021/jm0303406 |
| 43. | Lü, W. J.; Chen, Y. L.; Ma, W. P.; Zhang, X. Y.; Luan, F.; Liu, M. C.; Chen, X. G.; Hu, Z. D. Eur. J. Med. Chem. 2008, 43, 569–576. doi:10.1016/j.ejmech.2007.04.011 |
| 44. | Oakley, A. J.; Barrett, S.; Peat, T. S.; Newman, J.; Streltsov, V. A.; Waddington, L.; Saito, T.; Tashiro, M.; McKimm-Breschkin, J. L. J. Med. Chem. 2010, 53, 6421–6431. doi:10.1021/jm100621s |
| 45. | Chand, P.; Bantia, S.; Kotian, P. L.; El-Kattan, Y.; Lin, T.-H.; Babu, Y. S. Bioorg. Med. Chem. 2005, 13, 4071–4077. doi:10.1016/j.bmc.2005.03.048 |
| 46. | Cui, Y.; Jiao, Z.; Gong, J.; Yu, Q.; Zheng, X.; Quan, J.; Luo, M.; Yang, Z. Org. Lett. 2010, 12, 4–7. doi:10.1021/ol902438f |
| 47. | Yi, X.; Guo, Z.; Chu, F. M. Bioorg. Med. Chem. 2003, 11, 1465–1474. doi:10.1016/S0968-0896(02)00602-8 |
| 18. | Palkó, M.; Kiss, L.; Fülöp, F. Curr. Med. Chem. 2005, 12, 3063–3083. doi:10.2174/092986705774933443 |
| 19. | Kiss, L.; Forró, E.; Fülöp, F. Synthesis of carbocyclic β-amino acids. In Amino Acids, Peptides and Proteins in Organic Chemistry; Hughes, A. B., Ed.; Wiley-VCH: Weinheim, Germany, 2009; Vol. 1, pp 367–409. |
| 20. | Kiss, L.; Fülöp, F. Synlett 2010, 1302–1314. doi:10.1055/s-0029-1219821 |
| 21. | Fülöp, F. Chem. Rev. 2001, 101, 2181–2204. doi:10.1021/cr000456z |
| 49. | Nonn, M.; Kiss, L.; Forró, E.; Mucsi, Z.; Fülöp, F. Tetrahedron 2011, 67, 4079–4085. doi:10.1016/j.tet.2011.04.005 |
| 11. | Marotta, E.; Micheloni, L. M.; Scardovi, N.; Righi, P. Org. Lett. 2001, 3, 727–729. doi:10.1021/ol0070379 |
| 12. | Scott, J. P.; Oliver, S. F.; Brands, K. M. J.; Brewer, S. E.; Davies, A. J.; Gibb, A. D.; Hands, D.; Keen, S. P.; Sheen, F. J.; Reamer, R. A.; Wilson, R. D.; Dolling, U.-H. J. Org. Chem. 2006, 71, 3086–3092. doi:10.1021/jo060033i |
| 13. | Maimone, T. J.; Shi, J.; Ashida, S.; Baran, P. S. J. Am. Chem. Soc. 2009, 131, 17066–17067. doi:10.1021/ja908194b |
| 14. | Minter, A. R.; Fuller, A. A.; Mapp, A. K. J. Am. Chem. Soc. 2003, 125, 6846–6847. doi:10.1021/ja0298747 |
| 15. | Fuller, A. A.; Chen, B.; Minter, A. R.; Mapp, A. K. J. Am. Chem. Soc. 2005, 127, 5376–5383. doi:10.1021/ja0431713 |
| 16. | Sewald, N. Angew. Chem., Int. Ed. 2003, 42, 5794–5795. doi:10.1002/anie.200301692 |
| 17. | Tokizane, M.; Sato, K.; Ohta, T.; Ito, Y. Tetrahedron: Asymmetry 2008, 19, 2519–2528. doi:10.1016/j.tetasy.2008.11.005 |
| 50. | Jiang, H.; Elsner, P.; Jensen, K. L.; Falcicchio, A.; Marcos, V.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2009, 48, 6844–6848. doi:10.1002/anie.200901446 |
| 7. | Bode, J. W.; Carreire, E. M. Org. Lett. 2001, 3, 1587–1590. doi:10.1021/ol015885d |
| 8. | Bode, J. W.; Fraefel, N.; Muri, D.; Carreira, E. M. Angew. Chem., Int. Ed. 2001, 40, 2082–2085. doi:10.1002/1521-3773(20010601)40:11<2082::AID-ANIE2082>3.0.CO;2-1 |
| 9. | Jiang, D.; Chen, Y. J. Org. Chem. 2008, 73, 9181–9183. doi:10.1021/jo801831c |
| 10. | Tang, S.; He, J.; Sun, Y.; He, L.; She, X. J. Org. Chem. 2010, 75, 1961–1966. doi:10.1021/jo1000065 |
| 48. | Kiss, L.; Nonn, M.; Forró, E.; Sillanpää, R.; Fülöp, F. Tetrahedron Lett. 2009, 50, 2605–2608. doi:10.1016/j.tetlet.2009.03.119 |
| 49. | Nonn, M.; Kiss, L.; Forró, E.; Mucsi, Z.; Fülöp, F. Tetrahedron 2011, 67, 4079–4085. doi:10.1016/j.tet.2011.04.005 |
| 48. | Kiss, L.; Nonn, M.; Forró, E.; Sillanpää, R.; Fülöp, F. Tetrahedron Lett. 2009, 50, 2605–2608. doi:10.1016/j.tetlet.2009.03.119 |
| 49. | Nonn, M.; Kiss, L.; Forró, E.; Mucsi, Z.; Fülöp, F. Tetrahedron 2011, 67, 4079–4085. doi:10.1016/j.tet.2011.04.005 |
| 8. | Bode, J. W.; Fraefel, N.; Muri, D.; Carreira, E. M. Angew. Chem., Int. Ed. 2001, 40, 2082–2085. doi:10.1002/1521-3773(20010601)40:11<2082::AID-ANIE2082>3.0.CO;2-1 |
| 46. | Cui, Y.; Jiao, Z.; Gong, J.; Yu, Q.; Zheng, X.; Quan, J.; Luo, M.; Yang, Z. Org. Lett. 2010, 12, 4–7. doi:10.1021/ol902438f |
| 47. | Yi, X.; Guo, Z.; Chu, F. M. Bioorg. Med. Chem. 2003, 11, 1465–1474. doi:10.1016/S0968-0896(02)00602-8 |
| 7. | Bode, J. W.; Carreire, E. M. Org. Lett. 2001, 3, 1587–1590. doi:10.1021/ol015885d |
| 8. | Bode, J. W.; Fraefel, N.; Muri, D.; Carreira, E. M. Angew. Chem., Int. Ed. 2001, 40, 2082–2085. doi:10.1002/1521-3773(20010601)40:11<2082::AID-ANIE2082>3.0.CO;2-1 |
| 9. | Jiang, D.; Chen, Y. J. Org. Chem. 2008, 73, 9181–9183. doi:10.1021/jo801831c |
| 10. | Tang, S.; He, J.; Sun, Y.; He, L.; She, X. J. Org. Chem. 2010, 75, 1961–1966. doi:10.1021/jo1000065 |
| 11. | Marotta, E.; Micheloni, L. M.; Scardovi, N.; Righi, P. Org. Lett. 2001, 3, 727–729. doi:10.1021/ol0070379 |
| 12. | Scott, J. P.; Oliver, S. F.; Brands, K. M. J.; Brewer, S. E.; Davies, A. J.; Gibb, A. D.; Hands, D.; Keen, S. P.; Sheen, F. J.; Reamer, R. A.; Wilson, R. D.; Dolling, U.-H. J. Org. Chem. 2006, 71, 3086–3092. doi:10.1021/jo060033i |
| 13. | Maimone, T. J.; Shi, J.; Ashida, S.; Baran, P. S. J. Am. Chem. Soc. 2009, 131, 17066–17067. doi:10.1021/ja908194b |
| 14. | Minter, A. R.; Fuller, A. A.; Mapp, A. K. J. Am. Chem. Soc. 2003, 125, 6846–6847. doi:10.1021/ja0298747 |
| 15. | Fuller, A. A.; Chen, B.; Minter, A. R.; Mapp, A. K. J. Am. Chem. Soc. 2005, 127, 5376–5383. doi:10.1021/ja0431713 |
| 16. | Sewald, N. Angew. Chem., Int. Ed. 2003, 42, 5794–5795. doi:10.1002/anie.200301692 |
| 17. | Tokizane, M.; Sato, K.; Ohta, T.; Ito, Y. Tetrahedron: Asymmetry 2008, 19, 2519–2528. doi:10.1016/j.tetasy.2008.11.005 |
| 39. | Sorbera, L. A.; Graul, A.; Castaner, J. Drugs Future 2000, 25, 249–251. doi:10.1358/dof.2000.025.03.565302 |
| 40. | Babu, Y. S.; Chand, P.; Bantia, S.; Kotian, P.; Dehghani, A.; El-Kattan, Y.; Lin, T.-H.; Hutchison, T. L.; Elliott, A. J.; Parker, C. D.; Ananth, S. L.; Horn, L. L.; Laver, G. W.; Montgomery, J. A. J. Med. Chem. 2000, 43, 3482–3486. doi:10.1021/jm0002679 |
| 41. | Chand, P.; Kotian, P. L.; Dehghani, A.; El-Kattan, Y.; Lin, T.-H.; Hutchison, T. L.; Babu, Y. S.; Bantia, S.; Elliott, A. J.; Montgomery, J. A. J. Med. Chem. 2001, 44, 4379–4392. doi:10.1021/jm010277p |
| 42. | Chand, P.; Babu, Y. S.; Bantia, S.; Rowland, S.; Dehghani, A.; Kotian, P. L.; Hutchison, T. L.; Ali, S.; Brouillette, W.; El-Kattan, Y.; Lin, T.-H. J. Med. Chem. 2004, 47, 1919–1929. doi:10.1021/jm0303406 |
| 43. | Lü, W. J.; Chen, Y. L.; Ma, W. P.; Zhang, X. Y.; Luan, F.; Liu, M. C.; Chen, X. G.; Hu, Z. D. Eur. J. Med. Chem. 2008, 43, 569–576. doi:10.1016/j.ejmech.2007.04.011 |
| 44. | Oakley, A. J.; Barrett, S.; Peat, T. S.; Newman, J.; Streltsov, V. A.; Waddington, L.; Saito, T.; Tashiro, M.; McKimm-Breschkin, J. L. J. Med. Chem. 2010, 53, 6421–6431. doi:10.1021/jm100621s |
| 45. | Chand, P.; Bantia, S.; Kotian, P. L.; El-Kattan, Y.; Lin, T.-H.; Babu, Y. S. Bioorg. Med. Chem. 2005, 13, 4071–4077. doi:10.1016/j.bmc.2005.03.048 |
| 34. | Wena, W.-H.; Wang, S.-Y.; Tsai, K.-C.; Cheng, Y.-S. E.; Yang, A.-S.; Fang, J.-M.; Wong, C.-H. Bioorg. Med. Chem. 2010, 18, 4074–4084. doi:10.1016/j.bmc.2010.04.010 |
| 35. | Xu, G.; Kiefel, M. J.; Wilson, J. C.; Andrew, P. W.; Oggioni, M. R.; Taylor, G. L. J. Am. Chem. Soc. 2011, 133, 1718–1721. doi:10.1021/ja110733q |
| 36. | Calveras, J.; Nagai, Y.; Sultana, I.; Ueda, Y.; Higashi, T.; Shoji, M.; Sugai, T. Tetrahedron 2010, 66, 4284–4291. doi:10.1016/j.tet.2010.04.045 |
| 37. | Honda, T.; Kubo, S.; Masuda, T.; Arai, M.; Kobayashi, Y.; Yamashita, M. Bioorg. Med. Chem. Lett. 2009, 19, 2938–2940. doi:10.1016/j.bmcl.2009.04.067 |
| 38. | Soulé, J.-F.; Mathieu, A.; Norsikian, S.; Beau, J.-M. Org. Lett. 2010, 12, 5322–5325. doi:10.1021/ol102326b |
| 48. | Kiss, L.; Nonn, M.; Forró, E.; Sillanpää, R.; Fülöp, F. Tetrahedron Lett. 2009, 50, 2605–2608. doi:10.1016/j.tetlet.2009.03.119 |
| 49. | Nonn, M.; Kiss, L.; Forró, E.; Mucsi, Z.; Fülöp, F. Tetrahedron 2011, 67, 4079–4085. doi:10.1016/j.tet.2011.04.005 |
© 2012 Nonn et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)