Abstract
Hydrozirconation of 1-hexyne, the addition to in situ prepared N-acyliminium species, and ring-closing metathesis (RCM) were key steps in the preparation of a tricyclic isoindolinone scaffold. An unusual alkene isomerization process during the RCM was identified and studied in some detail. Chemical diversification for library synthesis was achieved by a subsequent alkene epoxidation and zinc-mediated aminolysis reaction. The resulting library products provided selective hits among a large number of high-throughput screens reported in PubChem, thus illustrating the utility of the novel scaffold.

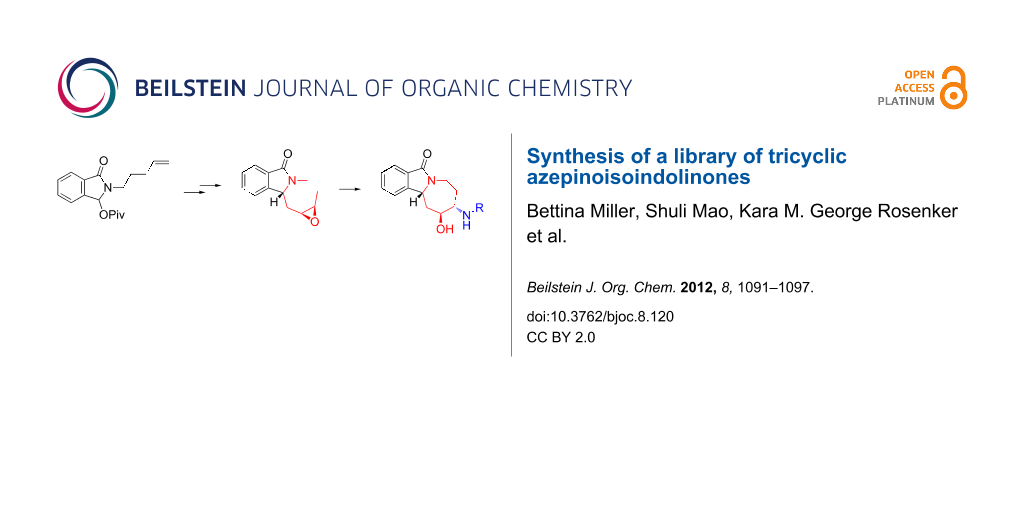
Graphical Abstract
Introduction
Isoindolinones represent a common scaffold seen in naturally occurring compounds such as magallanesine [1], lennoxamine [2] and clitocybin A [3], or drug candidates such as pagoclone [4] (Figure 1). These heterocycles have demonstrated a variety of pharmacological activities, including anti-inflammatory [5], antihypertensive [6] and vasodilatory [7], antipsychotic [8,9], and anticancer effects [10]. Due to the broad biological properties and the general utility of isoindolinones in the preparation of other synthetic building blocks, a variety of approaches for the preparation of these heterocycles have been explored [11-18]. Previously, we reported on the addition of organometallic reagents to in situ generated N-acyliminium ions [19]. This methodology applies to a variety of commercially available or easily prepared starting materials and creates many opportunities for further functionalization and chemical library synthesis. For example, a ring-closing metathesis of the alkene addition product affords structurally novel tricyclic isoindolinones with a newly formed seven-membered ring [19]. We have now developed this concept further toward a library synthesis of functionalized azepino-isoindolinone derivatives.
![[1860-5397-8-120-1]](/bjoc/content/figures/1860-5397-8-120-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Representative isoindolinone natural products and pharmaceuticals.
Figure 1: Representative isoindolinone natural products and pharmaceuticals.
Results and Discussion
N-Alkylation of phthalimide with 4-penten-1-ol under Mitsunobu conditions, followed by NaBH4 reduction and pivaloate protection of the intermediate hemiaminal, provided alkene 1 in 59% overall yield (Scheme 1). After hydrozirconation of 1-hexyne with zirconocene hydrochloride [20-23], addition of trimethylaluminium activated the in situ generated alkenylzirconocene and allowed the displacement of the pivaloate on 1 in 55% yield to afford diene 2 [19,24].
![[1860-5397-8-120-i1]](/bjoc/content/inline/1860-5397-8-120-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Formation of isomerized azepinoisoindoline 3 and oxirane 5.
Scheme 1: Formation of isomerized azepinoisoindoline 3 and oxirane 5.
Ring-closing metathesis of 2 using Grubbs 2nd generation catalyst [25] in the presence of 1 equiv of Ti(OiPr)4 [26,27] at room temperature provided, surprisingly, a modest 45% yield of the alkene-isomerized homoallylic amide 3 instead of the expected allylic amide 4 (Scheme 1). This result was reproduced with Zhan catalyst-1B [28,29], which gave 3 in 50% yield. The structure of alkene 3 was determined based on the X-ray analysis of epoxide 5 (Figure 2), obtained with NaHCO3-buffered meta-chloroperbenzoic acid (m-CPBA) in 57% yield [30,31].
![[1860-5397-8-120-2]](/bjoc/content/figures/1860-5397-8-120-2.png?scale=1.8&max-width=1024&background=FFFFFF)
The surprising formation of 3 instead of 4 under the metathesis conditions could be explained by a ruthenium-catalyzed double-bond isomerization [32]. The release of ring strain, however, can only be partially responsible for this facile isomerization. DFT calculations of the five possible alkene isomers of 4 indicated a decrease in relative energy from 4 to 3, but other isomers were even lower in energy (Figure 3). The starting geometries for the alkene isomers prior to DFT optimizations were obtained by a conformational search using the MMFF force field.
![[1860-5397-8-120-3]](/bjoc/content/figures/1860-5397-8-120-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Relative energies of alkene isomers based on RB3LYP/6-311G* calculations with MacSpartan ’06.
Figure 3: Relative energies of alkene isomers based on RB3LYP/6-311G* calculations with MacSpartan ’06.
In order to investigate the factors influencing the alkene isomerization process, we conducted a ring-closing metathesis in the absence of Ti(OiPr)4 (Scheme 2). The resulting product was different from 3, based on a TLC analysis, but proved to be quite labile during workup. Therefore, it was immediately subjected to m-CPBA epoxidation conditions to give a modest yield of the further oxidized 6, which was structurally assigned by X-ray analysis (Figure 4). The formation of 6 implies the intermediate presence of alkene 4, the product of a regular RCM of diene 2. Accordingly, the isolation of 6, and the absence of significant quantities of 5, confirmed the chelating additive Ti(OiPr)4 as the primary factor responsible for the isomerization of 4 to 3 in the previous reaction sequence. An additional contributing reason for the exclusive formation of 3 in the earlier metathesis reactions could be the decomposition of the acid-labile isomer 4 under the reaction and chromatographic-purification conditions. A possible pathway for decomposition is indicated by the benzylic/allylic methine oxidation product 6. The ability of Ti(OiPr)4 to induce alkene isomerization during the ring-closing metathesis reaction is noteworthy; while there are a number of additives known to decrease the rate of isomerization in RCM [33-35], we are unaware of any previous report on an alkene-isomerization-promoting effect of an additive in this reaction. We can speculate that the presence of Ti(OiPr)4 stabilizes the ruthenium alkylidene complex and, thus allows product isomerization to take place during and after the RCM reaction (see below).
![[1860-5397-8-120-i2]](/bjoc/content/inline/1860-5397-8-120-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Ring-closing metathesis of diene 2 in the absence of Ti(OiPr)4 and isolation of hydroxy epoxide 6 after treatment with m-CPBA.
Scheme 2: Ring-closing metathesis of diene 2 in the absence of Ti(OiPr)4 and isolation of hydroxy epoxide 6 a...
![[1860-5397-8-120-4]](/bjoc/content/figures/1860-5397-8-120-4.png?scale=1.8&max-width=1024&background=FFFFFF)
Figure 4: X-Ray structure of epoxyalcohol 6.
Figure 4: X-Ray structure of epoxyalcohol 6.
We also briefly studied the influence of the diene substitution pattern on the rate of isomerization from 4 to 3 and the corresponding product distribution (Scheme 3). Addition of in situ prepared vinyl alane to pivaloate 1 provided the diene 7 in 80% yield. RCM with Grubbs second-generation catalyst in the presence of Ti(OiPr)4 led to an exclusive conversion to alkene 4, i.e., no alkene isomerization was observed in this case, and no homoallylic amide 3 was detected in the reaction mixture. Similarly, in the absence of Ti(OiPr)4, crude 4 was obtained in 77% yield (Supporting Information File 1). The different reaction course with alkenes 2 and 7 indicates a role of the ruthenium carbene intermediate in the isomerization. Metathesis of 2 leads to an alkylidene complex, that could form a ruthenium hydride species. In contrast, metathesis of 7 provides a more reactive methylidene complex that is also likely to decompose more quickly and, thus, be unavailable for isomerization of the kinetic product 4 beyond the time span of the completion of the RCM reaction [36]. It was, however, difficult to purify product 4 due to its chemical instability. When the RCM reaction of 7 was conducted on larger scale in the absence of Ti(OiPr)4, and the crude intermediate was subjected to m-CPBA oxidation, epoxy alcohol 6 was isolated in 11% overall yield. LC–MS as well as NMR analyses suggested a 5:1 ratio of epimers at the hemiacetal carbon. Hydroxylation/oxidation at the benzylic position with m-CPBA in air in the presence of bicarbonate has been observed previously, and a radical mechanism was proposed [37].
![[1860-5397-8-120-i3]](/bjoc/content/inline/1860-5397-8-120-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation and RCM reaction of bis-terminal diene analogue 7.
Scheme 3: Preparation and RCM reaction of bis-terminal diene analogue 7.
In summary, these studies suggest that the alkene isomerization from allylic to homoallylic amides under RCM conditions is both dependent on the presence of the Lewis acidic additive Ti(OiPr)4 as well as the substitution pattern of the α,ω-diene precursor.
With alkene 3 and the corresponding epoxide 5 in hand, a ZnI2-mediated amine alkylation protocol could be employed, which introduced a variety of nitrogen nucleophiles 8{1–13} (Scheme 4) [38,39]. Co(ClO4)2 hexahydrate could also be used in place of ZnI2, but was generally less efficient (Table 1). Anilines with electron-withdrawing (CF3, CN, CO2Et), electron-donating (OCH3), and halogen substituents (F, Cl, Br) in ortho-, meta- and para-positions were used (Figure 5). Furthermore, aminopyridines 8{11} and 8{12} as well as aliphatic amine 8{13} were compatible with the reaction conditions. With the exception of 8{13}, two regioisomeric products were formed: the major isomer 9{1–12} was obtained by an attack on the distal carbon atom of the epoxide, while the minor isomer 10{1–13} was obtained by proximal ring opening. These isomers were separated by chromatography on SiO2, and an X-ray analysis confirmed the structural assignment for 10{7} (Figure 6). The remainder of the library products were assigned based on the characteristic chemical-shift data for 10{7} and its congener 9{7} [40]. Yields and purities of amino alcohols 9{1–12} and 10{1–13} are summarized in Table 1.
![[1860-5397-8-120-i4]](/bjoc/content/inline/1860-5397-8-120-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Conversion of epoxide 5 to 1,2-amino alcohols.
Scheme 4: Conversion of epoxide 5 to 1,2-amino alcohols.
Table 1: Library matrix of products 9{1–13} and 10{1–13} [isolated yield (%) and purity by ELSD (%)].
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-120-i5.svg?max-width=637&scale=1.0)
|
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-120-i6.svg?max-width=637&scale=1.0)
|
|
| Amine segment R | 9{1–13} | 10{1–13} |
|---|---|---|
| 8{1} | 56 (>99) | 19 (94)a |
| 8{2} | 81 (95) | 15 (>99) |
| 8{3} | 24 (>99) | 10 (99) |
| 8{4} | 72 (99) | 17 (99) |
| 8{5} | 44 (99) | 33 (95) |
| 8{6} | 69 (>99) | 17 (95) |
| 8{7} | 41 (>99) | 12 (>99) |
| 8{8} | 58 (99) | 26 (98) |
| 8{9} | 85 (99) | 15 (99) |
| 8{10} | 59 (>99) | 15 (72) |
| 8{11} | 59 (>99) | 32 (>99) |
| 8{12} | 20 (99) | –b |
| 8{13} | –b | 46 (>99) |
aCo(ClO4)2·6H2O was used in place of ZnI2; bProduct was not isolated.
![[1860-5397-8-120-5]](/bjoc/content/figures/1860-5397-8-120-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Amine building blocks for library synthesis.
Figure 5: Amine building blocks for library synthesis.
![[1860-5397-8-120-6]](/bjoc/content/figures/1860-5397-8-120-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: X-ray structure of amino alcohol 10{7}.
Figure 6: X-ray structure of amino alcohol 10{7}.
The 9{1–11}/10{1–11} isolated product ratios varied between 5:1 and 1.5:1, with no obvious trends discernable. Interestingly, for adamantyl amine 8{13}, only the corresponding 10{13} was isolated, most likely due to the steric bulk of the adamantyl group: molecular mechanics calculations indicate that the aminolysis of 5 to regioisomer 10 proceeds with minimal isomerization of the seven-membered ring geometry in the lowest-energy product conformer, whereas the formation of 9 requires a substantial ring flip [41].
Conclusion
A library of novel tricyclic isoindolinone amino alcohols was prepared in seven steps from commercially available starting materials. Key transformations include the addition of in situ generated alkenylalanes to an N-acyliminium ion derived from pivaloate 1, a tandem ring-closing metathesis–isomerization sequence and a ZnI2-mediated epoxide aminolysis. We investigated the factors influencing the alkene isomerization during the RCM process, and identified the presence of the additive Ti(OiPr)4, the substitution pattern on the alkene, and the chemical reactivity of the benzylic allylic methine carbon to be significant contributors. Regioisomeric library products 9 and 10 were submitted to the NIH Small Molecule Repository (SMR) [42], screened in the Molecular Libraries Probe Center Network (MLPCN) [43], and biological results were deposited in PubChem [44]. For example, 9{2} was tested in 188 assays and identified as an active hit (based on the hit criteria in the individual test systems) in 4 assays, including a cell-based assay to identify antagonists of the orexin 1 receptor; an assay to identify inhibitors of Apaf-1 (apoptotic peptidase activating factor 1); a cell-based assay to identify antagonists of the human M1 muscarinic receptor; and a cellular assay to identify human immunodeficiency virus 1 inhibitors. Amino alcohol 9{7} was tested in 185 bioassays reported in PubChem, and found to serve as an inhibitor of human platelet activating factor acetylhydrolase 2 (PAFAH2). It is clear from these and other screening data disclosed for this series in the PubChem database that the tricyclic isoindolinone scaffolds hold strong potential for the development of selective and potent lead structures.
Supporting Information
Supporting information contains experimental procedures for newly synthesized compounds and NMR spectra.
| Supporting Information File 1: Experimental procedures and characterization details of synthesized compounds. | ||
| Format: PDF | Size: 3.0 MB | Download |
References
-
Valencia, E.; Fajardo, V.; Freyer, A. J.; Shamma, M. Tetrahedron Lett. 1985, 26, 993–996. doi:10.1016/S0040-4039(00)98494-6
Return to citation in text: [1] -
Valencia, E.; Freyer, A. J.; Shamma, M.; Fajardo, V. Tetrahedron Lett. 1984, 25, 599–602. doi:10.1016/S0040-4039(00)99948-9
Return to citation in text: [1] -
Yoo, K.-D.; Park, E.-S.; Lim, Y.; Kang, S.-I.; Yoo, S.-H.; Won, H.-H.; Kim, Y.-H.; Yoo, I.-D.; Yoo, H.-S.; Hong, J. T.; Yun, Y.-P. J. Pharmacol. Sci. 2012, 118, 171–177. doi:10.1254/jphs.11159FP
Return to citation in text: [1] -
Sorbera, L. A.; Leeson, P. A.; Silvestre, J.; Castaner, J. Drugs Future 2001, 26, 651–657. doi:10.1358/dof.2001.026.07.630003
Return to citation in text: [1] -
Ajitha, M.; Rajnarayana, K. Int. J. Pharma Bio Sci. 2011, 2 (4), 81–90.
Return to citation in text: [1] -
Shirasaka, T.; Kunitake, T.; Tsuneyoshi, I. Brain Res. 2009, 1300, 105–113. doi:10.1016/j.brainres.2009.08.092
Return to citation in text: [1] -
Kato, Y.; Ebiike, H.; Achiwa, K.; Ashizawa, N.; Kurihara, T.; Kobayashi, F. Chem. Pharm. Bull. 1990, 38, 2060–2062. doi:10.1248/cpb.38.2060
Return to citation in text: [1] -
Ito, S.; Hirata, Y.; Nagatomi, Y.; Satoh, A.; Suzuki, G.; Kimura, T.; Satow, A.; Maehara, S.; Hikichi, H.; Hata, M.; Ohta, H.; Kawamoto, H. Bioorg. Med. Chem. Lett. 2009, 19, 5310–5313. doi:10.1016/j.bmcl.2009.07.145
Return to citation in text: [1] -
Favor, D. A.; Powers, J. J.; White, A. D.; Fitzgerald, L. W.; Groppi, V.; Serpa, K. A. Bioorg. Med. Chem. Lett. 2010, 20, 5666–5669. doi:10.1016/j.bmcl.2010.08.023
Return to citation in text: [1] -
Hardcastle, I. R.; Liu, J.; Valeur, E.; Watson, A.; Ahmed, S. U.; Blackburn, T. J.; Bennaceur, K.; Clegg, W.; Drummond, C.; Endicott, J. A.; Golding, B. T.; Griffin, R. J.; Gruber, J.; Haggerty, K.; Harrington, R. W.; Hutton, C.; Kemp, S.; Lu, X.; McDonnell, J. M.; Newell, D. R.; Noble, M. E. M.; Payne, S. L.; Revill, C. H.; Riedinger, C.; Xu, Q.; Lunec, J. J. Med. Chem. 2011, 54, 1233–1243. doi:10.1021/jm1011929
Return to citation in text: [1] -
Yang, G.; Zhang, W. Org. Lett. 2012, 14, 268–271. doi:10.1021/ol203043h
Return to citation in text: [1] -
Eastwood, P.; González, J.; Gómez, E.; Caturla, F.; Aguilar, N.; Mir, M.; Aiguadé, J.; Matassa, V.; Balagué, C.; Orellana, A.; Domínguez, M. Bioorg. Med. Chem. Lett. 2011, 21, 6253–6257. doi:10.1016/j.bmcl.2011.09.006
Return to citation in text: [1] -
Luo, Y.; Xiao, F.; Qian, S.; He, Q.; Lu, W.; Yang, B. Med. Chem. Commun. 2011, 2, 1054–1057. doi:10.1039/c1md00105a
Return to citation in text: [1] -
Zou, H.; Zhang, L.; Ouyang, J.; Giulianotti, M. A.; Yu, Y. Eur. J. Med. Chem. 2011, 46, 5970–5977. doi:10.1016/j.ejmech.2011.10.009
Return to citation in text: [1] -
Kilikli, A. A.; Dengiz, C.; Özcan, S.; Balci, M. Synthesis 2011, 3697–3705. doi:10.1055/s-0030-1260235
Return to citation in text: [1] -
Augner, D.; Gerbino, D. C.; Slavov, N.; Neudörfl, J.-M.; Schmalz, H.-G. Org. Lett. 2011, 13, 5374–5377. doi:10.1021/ol202271k
Return to citation in text: [1] -
Zhu, C.; Falck, J. R. Org. Lett. 2011, 13, 1214–1217. doi:10.1021/ol200093f
Return to citation in text: [1] -
Angelin, M.; Rahm, M.; Fischer, A.; Brinck, T.; Ramström, O. J. Org. Chem. 2010, 75, 5882–5887. doi:10.1021/jo100868z
Return to citation in text: [1] -
Pierce, J. G.; Waller, D. L.; Wipf, P. J. Organomet. Chem. 2007, 692, 4618–4629. doi:10.1016/j.jorganchem.2007.05.035
Return to citation in text: [1] [2] [3] -
Wailes, P. C.; Weigold, H.; Bell, A. P. J. Organomet. Chem. 1971, 27, 373–378. doi:10.1016/S0022-328X(00)82168-3
Return to citation in text: [1] -
Schwartz, J.; Labinger, J. A. Angew. Chem., Int. Ed. Engl. 1976, 15, 333–340. doi:10.1002/anie.197603331
Return to citation in text: [1] -
Wipf, P.; Kendall, C. Chem.–Eur. J. 2002, 8, 1778–1784. doi:10.1002/1521-3765(20020415)8:8<1778::AID-CHEM1778>3.0.CO;2-H
Return to citation in text: [1] -
Wipf, P.; Jahn, H. Tetrahedron 1996, 52, 12853–12910. doi:10.1016/0040-4020(96)00754-5
Return to citation in text: [1] -
Wipf, P.; Nunes, R. L. Tetrahedron 2004, 60, 1269–1279. doi:10.1016/j.tet.2003.12.018
Return to citation in text: [1] -
Trnka, T. M.; Grubbs, R. H. Acc. Chem. Res. 2001, 34, 18–29. doi:10.1021/ar000114f
Return to citation in text: [1] -
Fürstner, A.; Langemann, K. J. Am. Chem. Soc. 1997, 119, 9130–9136. doi:10.1021/ja9719945
Return to citation in text: [1] -
Ghosh, A. K.; Cappiello, J.; Shin, D. Tetrahedron Lett. 1998, 39, 4651–4654. doi:10.1016/S0040-4039(98)00887-9
Return to citation in text: [1] -
Zhan, Z.-Y. Ruthenium complex ligand, ruthenium complex, carried ruthenium complex catalyst and the preparing methods and the use thereof. WO Patent 2007003135, Jan 11, 2007.
Return to citation in text: [1] -
Rix, D.; Caijo, F.; Laurent, I.; Boeda, F.; Clavier, H.; Nolan, S. P.; Mauduit, M. J. Org. Chem. 2008, 73, 4225–4228. doi:10.1021/jo800203d
Return to citation in text: [1] -
Ahn, J.-B.; Yun, C.-S.; Kim, K. H.; Ha, D.-C. J. Org. Chem. 2000, 65, 9249–9251. doi:10.1021/jo0012187
Return to citation in text: [1] -
The structure of 3 was erroneously assigned as 4 in Ref. [19].
Return to citation in text: [1] -
Joe, D.; Overman, L. E. Tetrahedron Lett. 1997, 38, 8635–8638. doi:10.1016/S0040-4039(97)10373-2
Return to citation in text: [1] -
Gimeno, N.; Formentín, P.; Steinke, J. H. G.; Vilar, R. Eur. J. Org. Chem. 2007, 918–924. doi:10.1002/ejoc.200600908
Return to citation in text: [1] -
Hong, S. H.; Sanders, D. P.; Lee, C. W.; Grubbs, R. H. J. Am. Chem. Soc. 2005, 127, 17160–17161. doi:10.1021/ja052939w
Return to citation in text: [1] -
Schmidt, B. J. Mol. Catal. A: Chem. 2006, 254, 53–57. doi:10.1016/j.molcata.2006.03.026
Return to citation in text: [1] -
Sanford, M. S.; Love, J. A.; Grubbs, R. H. J. Am. Chem. Soc. 2001, 123, 6543–6554. doi:10.1021/ja010624k
Return to citation in text: [1] -
Ma, D.; Xia, C.; Tian, H. Tetrahedron Lett. 1999, 40, 8915–8917. doi:10.1016/S0040-4039(99)01886-9
Return to citation in text: [1] -
Pachón, L. D.; Gamez, P.; van Brussel, J. J. M.; Reedijk, J. Tetrahedron Lett. 2003, 44, 6025–6027. doi:10.1016/S0040-4039(03)01480-1
Return to citation in text: [1] -
Shivani; Pujala, B.; Chakraborti, A. K. J. Org. Chem. 2007, 72, 3713–3722. doi:10.1021/jo062674j
Return to citation in text: [1] -
The characteristic proton used for assignment of all library products is the benzylic methine proton, which appears between 4.5 and 5.0 ppm as an apparent triplet with J ~ 5 Hz in compounds 9{1–12} and as a doublet with J ~ 10 Hz in compounds 10{1–13}. These splitting patterns and coupling constants (as a function of the dihedral angles) are confirmed by examination of the X-ray structure of 10{7} (showing dihedral angles of 75.9° and −168.3° for the benzylic methine hydrogen at 4.69 ppm (d, 1H, J = 9.9 Hz) and its two vicinal methylene hydrogens). The lowest energy conformer of 9{7} calculated with MMFF in Spartan 10 shows dihedral angles of −36.6° and 77.2° for the benzylic methine hydrogen at 4.85 ppm (t, 1H, J = 5.0 Hz) and its two vicinal methylene hydrogens.
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-120-i7.png?max-width=637&scale=1.18182)
Return to citation in text: [1] -
Lowest energy conformers calculated with MMFF in Spartan 10:
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-120-i8.png?max-width=637&scale=1.18182)
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-120-i9.png?max-width=637&scale=1.18182)
Return to citation in text: [1] -
SMR. http://mli.nih.gov/mli/compound-repository.
Return to citation in text: [1] -
MLPCN. http://mli.nih.gov/mli/.
Return to citation in text: [1] -
PubChem. http://pubchem.ncbi.nlm.nih.gov/.
Return to citation in text: [1]
| 19. | Pierce, J. G.; Waller, D. L.; Wipf, P. J. Organomet. Chem. 2007, 692, 4618–4629. doi:10.1016/j.jorganchem.2007.05.035 |
| 1. | Valencia, E.; Fajardo, V.; Freyer, A. J.; Shamma, M. Tetrahedron Lett. 1985, 26, 993–996. doi:10.1016/S0040-4039(00)98494-6 |
| 25. | Trnka, T. M.; Grubbs, R. H. Acc. Chem. Res. 2001, 34, 18–29. doi:10.1021/ar000114f |
| 4. | Sorbera, L. A.; Leeson, P. A.; Silvestre, J.; Castaner, J. Drugs Future 2001, 26, 651–657. doi:10.1358/dof.2001.026.07.630003 |
| 26. | Fürstner, A.; Langemann, K. J. Am. Chem. Soc. 1997, 119, 9130–9136. doi:10.1021/ja9719945 |
| 27. | Ghosh, A. K.; Cappiello, J.; Shin, D. Tetrahedron Lett. 1998, 39, 4651–4654. doi:10.1016/S0040-4039(98)00887-9 |
| 3. | Yoo, K.-D.; Park, E.-S.; Lim, Y.; Kang, S.-I.; Yoo, S.-H.; Won, H.-H.; Kim, Y.-H.; Yoo, I.-D.; Yoo, H.-S.; Hong, J. T.; Yun, Y.-P. J. Pharmacol. Sci. 2012, 118, 171–177. doi:10.1254/jphs.11159FP |
| 20. | Wailes, P. C.; Weigold, H.; Bell, A. P. J. Organomet. Chem. 1971, 27, 373–378. doi:10.1016/S0022-328X(00)82168-3 |
| 21. | Schwartz, J.; Labinger, J. A. Angew. Chem., Int. Ed. Engl. 1976, 15, 333–340. doi:10.1002/anie.197603331 |
| 22. | Wipf, P.; Kendall, C. Chem.–Eur. J. 2002, 8, 1778–1784. doi:10.1002/1521-3765(20020415)8:8<1778::AID-CHEM1778>3.0.CO;2-H |
| 23. | Wipf, P.; Jahn, H. Tetrahedron 1996, 52, 12853–12910. doi:10.1016/0040-4020(96)00754-5 |
| 2. | Valencia, E.; Freyer, A. J.; Shamma, M.; Fajardo, V. Tetrahedron Lett. 1984, 25, 599–602. doi:10.1016/S0040-4039(00)99948-9 |
| 19. | Pierce, J. G.; Waller, D. L.; Wipf, P. J. Organomet. Chem. 2007, 692, 4618–4629. doi:10.1016/j.jorganchem.2007.05.035 |
| 24. | Wipf, P.; Nunes, R. L. Tetrahedron 2004, 60, 1269–1279. doi:10.1016/j.tet.2003.12.018 |
| 10. | Hardcastle, I. R.; Liu, J.; Valeur, E.; Watson, A.; Ahmed, S. U.; Blackburn, T. J.; Bennaceur, K.; Clegg, W.; Drummond, C.; Endicott, J. A.; Golding, B. T.; Griffin, R. J.; Gruber, J.; Haggerty, K.; Harrington, R. W.; Hutton, C.; Kemp, S.; Lu, X.; McDonnell, J. M.; Newell, D. R.; Noble, M. E. M.; Payne, S. L.; Revill, C. H.; Riedinger, C.; Xu, Q.; Lunec, J. J. Med. Chem. 2011, 54, 1233–1243. doi:10.1021/jm1011929 |
| 19. | Pierce, J. G.; Waller, D. L.; Wipf, P. J. Organomet. Chem. 2007, 692, 4618–4629. doi:10.1016/j.jorganchem.2007.05.035 |
| 8. | Ito, S.; Hirata, Y.; Nagatomi, Y.; Satoh, A.; Suzuki, G.; Kimura, T.; Satow, A.; Maehara, S.; Hikichi, H.; Hata, M.; Ohta, H.; Kawamoto, H. Bioorg. Med. Chem. Lett. 2009, 19, 5310–5313. doi:10.1016/j.bmcl.2009.07.145 |
| 9. | Favor, D. A.; Powers, J. J.; White, A. D.; Fitzgerald, L. W.; Groppi, V.; Serpa, K. A. Bioorg. Med. Chem. Lett. 2010, 20, 5666–5669. doi:10.1016/j.bmcl.2010.08.023 |
| 19. | Pierce, J. G.; Waller, D. L.; Wipf, P. J. Organomet. Chem. 2007, 692, 4618–4629. doi:10.1016/j.jorganchem.2007.05.035 |
| 7. | Kato, Y.; Ebiike, H.; Achiwa, K.; Ashizawa, N.; Kurihara, T.; Kobayashi, F. Chem. Pharm. Bull. 1990, 38, 2060–2062. doi:10.1248/cpb.38.2060 |
| 6. | Shirasaka, T.; Kunitake, T.; Tsuneyoshi, I. Brain Res. 2009, 1300, 105–113. doi:10.1016/j.brainres.2009.08.092 |
| 11. | Yang, G.; Zhang, W. Org. Lett. 2012, 14, 268–271. doi:10.1021/ol203043h |
| 12. | Eastwood, P.; González, J.; Gómez, E.; Caturla, F.; Aguilar, N.; Mir, M.; Aiguadé, J.; Matassa, V.; Balagué, C.; Orellana, A.; Domínguez, M. Bioorg. Med. Chem. Lett. 2011, 21, 6253–6257. doi:10.1016/j.bmcl.2011.09.006 |
| 13. | Luo, Y.; Xiao, F.; Qian, S.; He, Q.; Lu, W.; Yang, B. Med. Chem. Commun. 2011, 2, 1054–1057. doi:10.1039/c1md00105a |
| 14. | Zou, H.; Zhang, L.; Ouyang, J.; Giulianotti, M. A.; Yu, Y. Eur. J. Med. Chem. 2011, 46, 5970–5977. doi:10.1016/j.ejmech.2011.10.009 |
| 15. | Kilikli, A. A.; Dengiz, C.; Özcan, S.; Balci, M. Synthesis 2011, 3697–3705. doi:10.1055/s-0030-1260235 |
| 16. | Augner, D.; Gerbino, D. C.; Slavov, N.; Neudörfl, J.-M.; Schmalz, H.-G. Org. Lett. 2011, 13, 5374–5377. doi:10.1021/ol202271k |
| 17. | Zhu, C.; Falck, J. R. Org. Lett. 2011, 13, 1214–1217. doi:10.1021/ol200093f |
| 18. | Angelin, M.; Rahm, M.; Fischer, A.; Brinck, T.; Ramström, O. J. Org. Chem. 2010, 75, 5882–5887. doi:10.1021/jo100868z |
| 32. | Joe, D.; Overman, L. E. Tetrahedron Lett. 1997, 38, 8635–8638. doi:10.1016/S0040-4039(97)10373-2 |
| 28. | Zhan, Z.-Y. Ruthenium complex ligand, ruthenium complex, carried ruthenium complex catalyst and the preparing methods and the use thereof. WO Patent 2007003135, Jan 11, 2007. |
| 29. | Rix, D.; Caijo, F.; Laurent, I.; Boeda, F.; Clavier, H.; Nolan, S. P.; Mauduit, M. J. Org. Chem. 2008, 73, 4225–4228. doi:10.1021/jo800203d |
| 30. | Ahn, J.-B.; Yun, C.-S.; Kim, K. H.; Ha, D.-C. J. Org. Chem. 2000, 65, 9249–9251. doi:10.1021/jo0012187 |
| 31. | The structure of 3 was erroneously assigned as 4 in Ref. [19]. |
| 40. |
The characteristic proton used for assignment of all library products is the benzylic methine proton, which appears between 4.5 and 5.0 ppm as an apparent triplet with J ~ 5 Hz in compounds 9{1–12} and as a doublet with J ~ 10 Hz in compounds 10{1–13}. These splitting patterns and coupling constants (as a function of the dihedral angles) are confirmed by examination of the X-ray structure of 10{7} (showing dihedral angles of 75.9° and −168.3° for the benzylic methine hydrogen at 4.69 ppm (d, 1H, J = 9.9 Hz) and its two vicinal methylene hydrogens). The lowest energy conformer of 9{7} calculated with MMFF in Spartan 10 shows dihedral angles of −36.6° and 77.2° for the benzylic methine hydrogen at 4.85 ppm (t, 1H, J = 5.0 Hz) and its two vicinal methylene hydrogens.
|
| 37. | Ma, D.; Xia, C.; Tian, H. Tetrahedron Lett. 1999, 40, 8915–8917. doi:10.1016/S0040-4039(99)01886-9 |
| 38. | Pachón, L. D.; Gamez, P.; van Brussel, J. J. M.; Reedijk, J. Tetrahedron Lett. 2003, 44, 6025–6027. doi:10.1016/S0040-4039(03)01480-1 |
| 39. | Shivani; Pujala, B.; Chakraborti, A. K. J. Org. Chem. 2007, 72, 3713–3722. doi:10.1021/jo062674j |
| 33. | Gimeno, N.; Formentín, P.; Steinke, J. H. G.; Vilar, R. Eur. J. Org. Chem. 2007, 918–924. doi:10.1002/ejoc.200600908 |
| 34. | Hong, S. H.; Sanders, D. P.; Lee, C. W.; Grubbs, R. H. J. Am. Chem. Soc. 2005, 127, 17160–17161. doi:10.1021/ja052939w |
| 35. | Schmidt, B. J. Mol. Catal. A: Chem. 2006, 254, 53–57. doi:10.1016/j.molcata.2006.03.026 |
| 36. | Sanford, M. S.; Love, J. A.; Grubbs, R. H. J. Am. Chem. Soc. 2001, 123, 6543–6554. doi:10.1021/ja010624k |
© 2012 Miller et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)