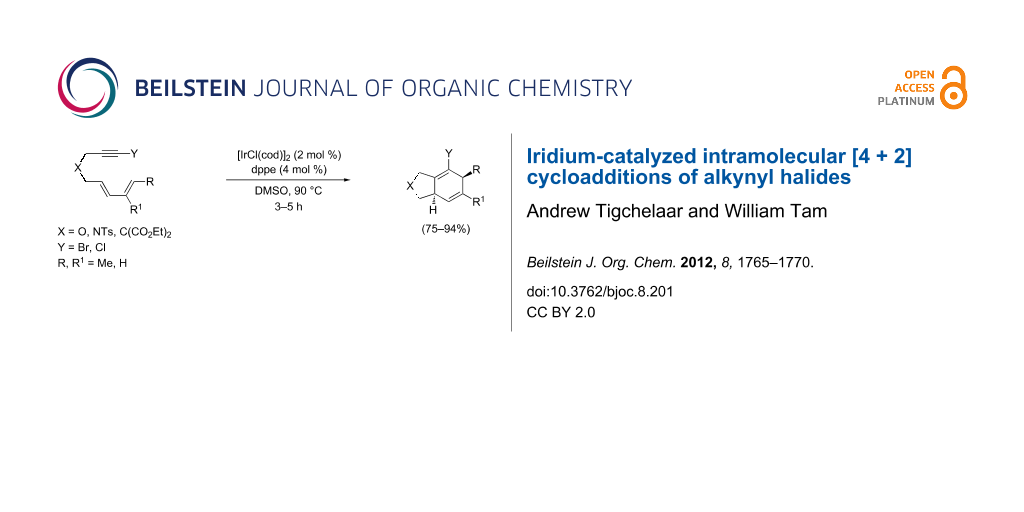
Abstract
Iridium-catalyzed intramolecular [4 + 2] cycloadditions of diene-tethered alkynyl halides were investigated by using [IrCl(cod)]2 as catalyst, and dppe was found to be the most suitable phosphine ligand for the reaction. No oxidative insertion of the iridium into the carbon–halide bond was observed, and the reactions proceeded to provide the halogenated cycloadducts in good yield (75–94%). These results are the first examples of cycloadditions of alkynyl halides using an iridium catalyst.

Graphical Abstract
Introduction
Iridium complexes have been used as catalysts for a wide variety of reactions, including homogeneous hydrogenation [1,2], C–H activation [3-5], asymmetric ring-opening reactions [6], and a variety of cycloisomerizations [7-10], and cycloadditions [11-20]. Traditionally these types of cycloisomerization and cycloaddition reactions are possible by making use of other metal complexes such as Pd [21,22] and Co [23-25], but recent advances in iridium chemistry have expanded the scope of the metal complexes that can be used. Common Ir-catalyzed cycloadditions include [2 + 2 + 2] cycloadditions of diynes [11-13] or enynes [16] with alkynes, [2 + 2 + 2] cyclotrimerizations of alkynes [14,15], and Pauson–Khand-type [2 + 2 + 1] cycloadditions of enynes [17-19] and allenynes [20]; however, Ir-catalyzed [4 + 2] cycloadditions are rare in the literature [26,27].
Transition-metal-catalyzed (TMC) [4 + 2] cycloadditions are an efficient way of making 6-membered rings, particularly in the case of electronically similar dienes and dienophiles, which can require high temperatures and long reaction times for the thermal cycloaddition to occur [28]. Many different transition-metal catalysts have been described for these types of reactions, employing a variety of different metals including Ni [29], Co [30], Rh [31-34], Pd [35,36] and Ir [26,27]. Much research has been done on intramolecular TMC [4 + 2] cycloadditions of a variety of diene-tethered alkynes to form substituted bicyclic products 2 (Scheme 1); however, the majority of these examples demonstrate Y = alkyl or aryl, and R = methyl. An interesting variation on this reaction is to utilize diene-tethered alkynyl halides (Y = halogen) in order to form a halogenated bicyclic product.
![[1860-5397-8-201-i1]](/bjoc/content/inline/1860-5397-8-201-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Literature examples of intramolecular TMC [4 + 2] cycloadditions of diene-tethered alkynes.
Scheme 1: Literature examples of intramolecular TMC [4 + 2] cycloadditions of diene-tethered alkynes.
Alkynyl halides are a versatile moiety in organic synthesis, and can be prepared under mild conditions [37]. They can be seen as a dual functionalized molecule in TMC reactions, with the two main modes of reactivity proceeding by insertion of a metal into the carbon–halide bond (Scheme 2, type 1), or coordination of the π-system of the acetylene to the metal in a η2 fashion (Scheme 2, type 2). The most extensive studies done on alkynyl halides in TMC reactions are on cross-coupling reactions that proceed by oxidative insertion of the metal into the carbon–halide bond (type 1), and these types of reactions have been used to synthesize building blocks, such as enynes [38,39], diynes [40,41], and triynes [42-44]. Conversely, formation of intermediate 5 is rare, likely due to competition with oxidative insertion of the metal into the carbon–halide bond; however, there are reported cases of Co-catalyzed [2 + 2 + 1] [45], Ru-catalyzed [2 + 2] [46], and Rh-catalyzed [4 + 2] [33] cycloadditions that proceed via intermediate 5.
![[1860-5397-8-201-i2]](/bjoc/content/inline/1860-5397-8-201-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction pathways of alkynyl halides in transition-metal-catalyzed reactions.
Scheme 2: Reaction pathways of alkynyl halides in transition-metal-catalyzed reactions.
Results and Discussion
To the best of our knowledge, there are no known Ir-catalyzed [4 + 2] cycloadditions involving alkynyl halides. In this article, we report our studies on [IrCl(cod)]2/dppe-catalyzed intramolecular [4 + 2] cycloadditions of diene-tethered alkynyl halides. In order to begin the study, alkynyl bromide 1a was synthesized as previously described [33], and several different catalytic iridium systems were screened (Table 1).
Table 1: Optimization of iridium-catalyst for [4 + 2] cycloaddition of alkynyl bromide 1a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-201-i6.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Catalyst | Additive/liganda (4 mol %)b | Solvent | Temp. (°C) | Time (h) | Yield (%)c | |
|---|---|---|---|---|---|---|---|
| 2a | 6 | ||||||
| 1 | A | none | toluene | 90 | 18 | 35 | 12 |
| 2 | A | none | toluene | 90 | 3 | 60 | 26 |
| 3 | B | AgSbF6 | acetone | 25 | 1 | 0d | 0 |
| 4 | B | AgSbF6 | toluene | 90 | 3 | 0d | 0 |
| 5 | B | dppm | toluene | 90 | 3 | 25 | 0 |
| 6 | B | dppe | toluene | 90 | 3 | 69 | 0 |
| 7 | B | dppp | toluene | 90 | 3 | 50 | 3 |
| 8 | B | dppe | toluene | 25 | 3 | 0e | 0 |
| 9 | B | PPh3 | toluene | 90 | 3 | 23 | 17 |
| 10 | B | BINAP | toluene | 90 | 3 | 28 | 18 |
aAbbreviations: dppm = 1,1-bis(diphenylphosphino)methane, dppe = 1,2-bis(diphenylphosphino)ethane, dppp = 1,3-bis(diphenylphosphino)propane, BINAP = 2,2'-bis(diphenylphosphino)-1,1'-binaphthyl. bExcept for entry 9, where 8 mol % was used for the mono-phosphine ligand. cIsolated yields after column chromatography. dDecomposition of starting material was observed. eOnly starting material was recovered.
Initial attempts were undertaken by using the catalyst system described by Shibata [26] employing Vaska’s complex in toluene (Table 1, entries 1 and 2) and cycloadduct 2a was formed in good yield; however, the formation of the aromatized product 6 was also observed. Addition of the silver(I) salt AgSbF6 to the commercially available [IrCl(cod)]2 was attempted in order to generate the cationic Ir(I) species (Table 1, entries 3 and 4), but this resulted in decomposition of the starting material, both at room temperature and at 90 °C. A variety of phosphine ligands were screened in conjunction with [IrCl(cod)]2 [16,26], and these provided low to moderate yields of 2a, with little to no formation of the aromatized cycloadduct 6 (Table 1, entries 5–8). Using racemic BINAP and PPh3 ligands also provided 2 in low yield, but a significant amount of 6 was also formed. From these observations, [IrCl(cod)]2 (2 mol %) and dppe (4 mol %) were carried forward, and a variety of solvents were screened at different temperatures in an attempt to further optimize the cycloaddition for cycloadduct 2a (Table 2).
Table 2: Optimization of solvent and temperature for the [IrCl(cod)]2/dppe-catalyzed [4 + 2] cycloaddition of 1a.
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-201-i7.svg?max-width=637&scale=1.0)
|
|||
| Entry | Solvent | Temp. (°C) | Yield (%)a |
|---|---|---|---|
| 1 | toluene | 90 | 69 |
| 2 | 1,4-dioxane | 90 | 61b |
| 3 | DCE | 75 | 70 |
| 4 | acetone | 45 | 0c |
| 5 | NMP | 90 | 74 |
| 6 | NMP | 75 | 46c |
| 7 | DMSO | 90 | 94 |
| 8 | DMSO | 65 | 25c |
| 9 | DMF | 90 | 87 |
aIsolated yields after column chromatography. b9% of aromatized cycloadduct 6 also recovered. cStarting material recovered.
Employing nonpolar solvents 1,4-dioxane and 1,2-dichloroethane (Table 2, entries 2 and 3) provided very similar results to toluene, and the lower-boiling-point solvent acetone (Table 2, entry 4) was ineffective when heated to 45 °C. High-boiling-point, polar aprotic solvents NMP, DMSO, and DMF (Table 2, entries 5, 7, 9) all improved the yield of 2a when the reaction was run at 90 °C, with DMSO being the most effective. Attempts to lower the reaction temperature by using NMP and DMSO (Table 2, entries 6 and 8) as the solvent resulted in incomplete reaction after 3 h, so the most efficient reaction conditions for the intramolecular [4 + 2] cycloaddition of diene-tethered alkynyl bromide 1a were found to be [IrCl(COD)]2 (2 mol %), dppe (4 mol %) in DMSO at 90 ºC.
In order to illustrate the general applicability of this catalytic system, a variety of diene-tethered alkynyl halides were synthesized, including three previously unreported substrates 1c, 1e, and 1g (Table 3). Alcohol 8 was prepared in two steps from tiglic aldehyde (7) as described by Gilbertson [47], and dienynes 9 and 11 could be synthesized from 8 (Scheme 3). Deprotonation of 8 with sodium hydride, followed by trapping with propargyl bromide provided 9 in 60% yield, and a Mitsunobu reaction between 8 and sulfonamide 10 (prepared as per reference [47]) provided 11 in 74% yield. Bromination of 9 and 11 provided diene-tethered alkynyl halides 1c (57%) and 1e (83%), respectively.
![[1860-5397-8-201-i3]](/bjoc/content/inline/1860-5397-8-201-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of diene-tethered alkynyl halides 1c and 1e.
Scheme 3: Synthesis of diene-tethered alkynyl halides 1c and 1e.
Synthesis of substrate 1g commenced with deprotonation of diethyl malonate, followed by addition of mesylate 12, and subsequent deprotonation and addition of propargyl bromide as described by Gilbertson [31] to provide dieneyne 13 (Scheme 4). Diene 13 was then treated under the standard bromination conditions to provide 1g in 77% yield.
![[1860-5397-8-201-i4]](/bjoc/content/inline/1860-5397-8-201-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of diene-tethered alkynyl halide 1g.
Scheme 4: Synthesis of diene-tethered alkynyl halide 1g.
Compounds 1a–1g were then subjected to the optimized cycloaddition conditions (Table 3). In general, the Ir-catalyzed [4 + 2] cycloadditions occurred smoothly for all substrates, forming bicyclic products 2 in good yield (75–94%) (Table 3), and in each case a single stereoisomer was formed, with the H at the ring junction and the R group anti to each other. The stereochemistry of the cycloadducts was assigned by comparison with spectral data from previous work. This stereochemistry is consistent with that of previously reported intramolecular [4 + 2] cycloadditions in which the relative stereochemistry was assigned by X-ray diffraction analysis [32] or GOESY NMR [33]. For both alkynyl bromide 1a and alkynyl chloride 1b (Table 3, entries 1 and 2) the reaction went to completion quite quickly; however, a decreased yield was seen in the cycloaddition of 1b. Attempts at the cycloaddition with the analogous alkynyl iodide provided a complex mixture of products. Changing R1 = H to R1 = Me (compare entries 1 and 3, Table 3) increased the reaction time slightly to 4 h, and a lower yield was seen for the cycloaddition of 1c. A similar trend was seen for the nitrogen-tethered substrates (Table 3, entries 4 and 5) when changing R1 = H to R1 = Me, as the reaction time increased from 3 to 4 h and the yield decreased slightly. The all-carbon tethered substrates (Table 3, entries 6 and 7) required longer reaction times, but the cycloadditions occurred in good yields. Changing R = H to R = Me had little effect on the yield (Table 3, entries 6 and 7).
Table 3: [IrCl(cod)]2/dppe-catalyzed [4 + 2] cycloadditions of various alkynyl halides.
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-201-i8.svg?max-width=637&scale=1.0)
|
||||||||
| Entry | Alkynyl halide | X | Y | R | R1 | Time (h) | Cycloadduct | Yielda |
|---|---|---|---|---|---|---|---|---|
| 1 | 1a | O | Br | Me | H | 3 | 2a | 94 |
| 2 | 1b | O | Cl | Me | H | 3 | 2b | 77 |
| 3 | 1c | O | Br | Me | Me | 4 | 2c | 75b |
| 4 | 1d | NTs | Br | Me | H | 4 | 2d | 89 |
| 5 | 1e | NTs | Br | Me | Me | 4 | 2e | 77c |
| 6 | 1f | C(CO2Et)2 | Br | H | H | 5 | 2f | 80d |
| 7 | 1g | C(CO2Et)2 | Br | Me | H | 5 | 2g | 85 |
aIsolated yields after column chromatography. bThe aromatized product was also formed in trace amounts. cIsolated yield after recrystallization from hexanes. dThe aromatized product was also formed in 6% yield.
Attempts at the cycloaddition reaction with substrates bearing a 4-atom tether were unsuccessful (Scheme 5). Two different substrates were synthesized and submitted to the optimized reaction conditions, but only the starting material was recovered. Changing the reaction conditions to the original system of Vaska’s complex in toluene (conditions B, Scheme 5) did not change this result. The previous Ir-catalyzed [4 + 2] cycloaddition work by Shibata and co-workers did not include any examples of similar substrates [26]; however, previous work in our group has shown that Rh-catalyzed [4 + 2] cycloadditions of this type of substrate occur in good yield [33].
![[1860-5397-8-201-i5]](/bjoc/content/inline/1860-5397-8-201-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Unsuccessful cycloaddition attempts with substrates with a 4-atom tether.
Scheme 5: Unsuccessful cycloaddition attempts with substrates with a 4-atom tether.
These cycloadducts 2 are interesting due to the retention of the halogen in the cycloaddition reaction. This opens up the possibility of further coupling reactions to gain access to complex bicyclic molecules that would not be accessible directly by cycloaddition. Previous work has demonstrated this synthetic utility by the further functionalization of 2a by Pd-catalyzed Suzuki and Heck coupling reactions [33]. In theory any of the cycloadducts 2 could undergo similar transformations, making them valuable intermediates that can be easily extended by metal-catalyzed coupling reactions.
Conclusion
In conclusion, we have shown the first successful examples of Ir-catalyzed intramolecular [4 + 2] cycloadditions of alkynyl halides. The cycloadditions proceeded smoothly at 90 °C to afford the bicyclic products in good yields, and oxidative insertion of the iridium into the carbon–halide bond did not appear to be a problem. In addition to the known compounds synthesized, two previously unreported alkynyl halide substrates were synthesized, and were also found to undergo [4 + 2] cycloaddition under the reported conditions. The Ir-catalyzed reactions provided the cycloadducts in similar yields to the previous Rh-catalyzed reactions of alkynyl halides; however, the Ir-catalyzed reactions required higher reaction temperatures (90 versus 25 °C).
Supporting Information
| Supporting Information File 1: Experimental procedures for new compounds 1c, 1e, 1g, and cycloadducts 2a–g. | ||
| Format: PDF | Size: 285.4 KB | Download |
| Supporting Information File 2: Copies of 1H and 13C NMR spectra for new compounds 1c, 1e, 1g, and cycloadducts 2a–g. | ||
| Format: PDF | Size: 780.8 KB | Download |
References
-
Crabtree, R. Acc. Chem. Res. 1979, 12, 331–337. doi:10.1021/ar50141a005
Return to citation in text: [1] -
Brown, J. M. Angew. Chem., Int. Ed. Engl. 1987, 26, 190–203. doi:10.1002/anie.198701901
Return to citation in text: [1] -
Burford, R. J.; Piers, W. E.; Parvez, M. Organometallics 2012, 31, 2949–2952. doi:10.1021/om300127g
Return to citation in text: [1] -
Hashiguchi, B. G.; Bischof, S. M.; Konnick, M. M.; Periana, R. A. Acc. Chem. Res. 2012, 45, 885–898. doi:10.1021/ar200250r
Return to citation in text: [1] -
Werner, H. Angew. Chem., Int. Ed. 2010, 49, 4714–4728. doi:10.1002/anie.201000306
Return to citation in text: [1] -
Fang, S.; Liang, X.; Long, Y.-H.; Li, X.; Yang, D.; Wang, S.; Li, C. Organometallics 2012, 31, 3113–3118. doi:10.1021/om3000295
Return to citation in text: [1] -
Chatani, N.; Inoue, H.; Morimoto, T.; Muto, T.; Murai, S. J. Org. Chem. 2001, 66, 4433–4436. doi:10.1021/jo010091y
Return to citation in text: [1] -
Yamamoto, Y.; Hayashi, H.; Saigoku, T.; Nishiyama, H. J. Am. Chem. Soc. 2005, 127, 10804–10805. doi:10.1021/ja053408a
Return to citation in text: [1] -
Shibata, T.; Yamasaki, M.; Kadowaki, S.; Takagi, L. Synlett 2004, 2812–2814. doi:10.1055/s-2004-835631
Return to citation in text: [1] -
Shibata, T.; Kobayashi, Y.; Maekawa, S.; Toshida, N.; Takagi, K. Tetrahedron 2005, 61, 9018–9024. doi:10.1016/j.tet.2005.07.039
Return to citation in text: [1] -
Takeuchi, R.; Tanaka, S.; Nakaya, Y. Tetrahedron Lett. 2001, 42, 2991–2994. doi:10.1016/S0040-4039(01)00338-0
Return to citation in text: [1] [2] -
Kezuka, S.; Tanaka, S.; Ohe, T.; Nakaya, Y.; Takeuchi, R. J. Org. Chem. 2006, 71, 543–552. doi:10.1021/jo051874c
Return to citation in text: [1] [2] -
Matsuda, T.; Kadowaki, S.; Goya, T.; Murakami, M. Org. Lett. 2007, 9, 133–136. doi:10.1021/ol062732n
Return to citation in text: [1] [2] -
Takeuchi, R.; Nakaya, Y. Org. Lett. 2003, 5, 3659–3662. doi:10.1021/ol035330d
Return to citation in text: [1] [2] -
Farnetti, E.; Marsich, N. J. Organomet. Chem. 2004, 689, 14–17. doi:10.1016/j.jorganchem.2003.10.004
Return to citation in text: [1] [2] -
Kezuka, S.; Okado, T.; Niou, E.; Takeuchi, R. Org. Lett. 2005, 7, 1711–1714. doi:10.1021/ol050085e
Return to citation in text: [1] [2] [3] -
Shibata, T.; Takagi, K. J. Am. Chem. Soc. 2000, 122, 9852–9853. doi:10.1021/ja000899k
Return to citation in text: [1] [2] -
Shibata, T.; Toshida, N.; Yamazaki, M.; Maekawa, S.; Takagi, K. Tetrahedron 2005, 61, 9974–9979. doi:10.1016/j.tet.2005.08.016
Return to citation in text: [1] [2] -
Jeong, N.; Kim, D. H.; Choi, J. H. Chem. Commun. 2004, 1134–1135. doi:10.1039/B401288G
Return to citation in text: [1] [2] -
Shibata, T.; Kadowaki, S.; Hirase, M.; Takagi, K. Synlett 2003, 573–575. doi:10.1055/s-2003-37532
Return to citation in text: [1] [2] -
Trost, B. M. Angew. Chem., Int. Ed. Engl. 1995, 34, 259–281. doi:10.1002/anie.199502591
Return to citation in text: [1] -
Ojima, I.; Tzamarioudaki, M.; Li, Z.; Donovan, R. J. Chem. Rev. 1996, 96, 635–662. doi:10.1021/cr950065y
Return to citation in text: [1] -
Vollhardt, K. P. C. Angew. Chem., Int. Ed. Engl. 1984, 23, 539–556. doi:10.1002/anie.198405393
Return to citation in text: [1] -
Khand, I. U.; Knox, G. R.; Pauson, P. L.; Watts, W. E.; Foreman, M. I. J. Chem. Soc., Perkin Trans. 1 1973, 977–981. doi:10.1039/P19730000977
Return to citation in text: [1] -
Kotha, S.; Brahmachary, E.; Lahiri, K. Eur. J. Org. Chem. 2005, 4741–4767. doi:10.1002/ejoc.200500411
Return to citation in text: [1] -
Shibata, T.; Takasaku, K.; Takesue, Y.; Hirata, N.; Takagi, K. Synlett 2002, 1681–1682. doi:10.1055/s-2002-34217
Return to citation in text: [1] [2] [3] [4] [5] -
Shibata, T.; Nishizawa, G.; Endo, K. Synlett 2008, 765–768. doi:10.1055/s-2008-1032112
Return to citation in text: [1] [2] -
Lautens, M.; Klute, W.; Tam, W. Chem. Rev. 1996, 96, 49–92. doi:10.1021/cr950016l
Return to citation in text: [1] -
Wender, P. A.; Smith, T. E. Tetrahedron 1998, 54, 1255–1275. doi:10.1016/S0040-4020(97)10223-X
Return to citation in text: [1] -
Hilt, G.; Lüers, S.; Smolko, K. I. Org. Lett. 2005, 7, 251–253. doi:10.1021/ol0477879
Return to citation in text: [1] -
Gilbertson, S. R.; Hoge, G. S.; Genov, D. G. J. Org. Chem. 1998, 63, 10077–10080. doi:10.1021/jo981870q
Return to citation in text: [1] [2] -
Jolly, R. S.; Luedtke, G.; Sheehan, D.; Livinghouse, T. J. Am. Chem. Soc. 1990, 112, 4965–4966. doi:10.1021/ja00168a055
Return to citation in text: [1] [2] -
Yoo, W.-J.; Allen, A.; Villeneuve, K.; Tam, W. Org. Lett. 2005, 7, 5853–5856. doi:10.1021/ol052412o
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Shibata, T.; Chiba, T.; Hirashima, H.; Ueno, Y.; Endo, K. Angew. Chem., Int. Ed. 2009, 48, 8066–8069. doi:10.1002/anie.200903715
Return to citation in text: [1] -
Murakami, M.; Itami, K.; Ito, Y. J. Am. Chem. Soc. 1997, 119, 7163–7164. doi:10.1021/ja970626y
Return to citation in text: [1] -
Murakami, M.; Minamida, R.; Itami, K.; Sawamura, M.; Ito, Y. Chem. Commun. 2000, 2293–2294. doi:10.1039/B007781J
Return to citation in text: [1] -
Hofmeister, H.; Annen, K.; Laurent, H.; Wiechert, R. Angew. Chem., Int. Ed. Engl. 1984, 23, 727–729. doi:10.1002/anie.198407271
Return to citation in text: [1] -
Yoshida, H.; Shirakawa, E.; Kurahashi, T.; Nakao, Y.; Hiyama, T. Organometallics 2000, 19, 5671–5678. doi:10.1021/om000828u
Return to citation in text: [1] -
Liu, Y.; Zhong, Z.; Nakajima, K.; Takahashi, T. J. Org. Chem. 2002, 67, 7451–7456. doi:10.1021/jo026037e
Return to citation in text: [1] -
Alami, M.; Ferri, F. Tetrahedron Lett. 1996, 37, 2763–2766. doi:10.1016/0040-4039(96)00414-5
Return to citation in text: [1] -
Nishihara, Y.; Ikegashira, K.; Hirabayashi, K.; Ando, J.; Mori, A.; Hiyama, T. J. Org. Chem. 2000, 65, 1780–1787. doi:10.1021/jo991686k
Return to citation in text: [1] -
Gung, B. W.; Kumi, G. J. Org. Chem. 2003, 68, 5956–5960. doi:10.1021/jo0344900
Return to citation in text: [1] -
Kim, S.; Kim, S.; Lee, T.; Ko, H.; Kim, D. Org. Lett. 2004, 6, 3601–3604. doi:10.1021/ol0484963
Return to citation in text: [1] -
Jiang, M. X.-W.; Rawat, M.; Wulff, W. D. J. Am. Chem. Soc. 2004, 126, 5970–5971. doi:10.1021/ja049836i
Return to citation in text: [1] -
Balsells, J.; Moyano, A.; Riera, A.; Pericàs, M. A. Org. Lett. 1999, 1, 1981–1984. doi:10.1021/ol991145h
Return to citation in text: [1] -
Villeneuve, K.; Riddell, N.; Jordan, R. W.; Tsui, G. C.; Tam, W. Org. Lett. 2004, 6, 4543–4546. doi:10.1021/ol048111g
Return to citation in text: [1] -
DeBoef, B.; Counts, W. R.; Gilbertson, S. R. J. Org. Chem. 2007, 72, 799–804. doi:10.1021/jo0620462
Return to citation in text: [1] [2]
| 26. | Shibata, T.; Takasaku, K.; Takesue, Y.; Hirata, N.; Takagi, K. Synlett 2002, 1681–1682. doi:10.1055/s-2002-34217 |
| 16. | Kezuka, S.; Okado, T.; Niou, E.; Takeuchi, R. Org. Lett. 2005, 7, 1711–1714. doi:10.1021/ol050085e |
| 26. | Shibata, T.; Takasaku, K.; Takesue, Y.; Hirata, N.; Takagi, K. Synlett 2002, 1681–1682. doi:10.1055/s-2002-34217 |
| 47. | DeBoef, B.; Counts, W. R.; Gilbertson, S. R. J. Org. Chem. 2007, 72, 799–804. doi:10.1021/jo0620462 |
| 1. | Crabtree, R. Acc. Chem. Res. 1979, 12, 331–337. doi:10.1021/ar50141a005 |
| 2. | Brown, J. M. Angew. Chem., Int. Ed. Engl. 1987, 26, 190–203. doi:10.1002/anie.198701901 |
| 11. | Takeuchi, R.; Tanaka, S.; Nakaya, Y. Tetrahedron Lett. 2001, 42, 2991–2994. doi:10.1016/S0040-4039(01)00338-0 |
| 12. | Kezuka, S.; Tanaka, S.; Ohe, T.; Nakaya, Y.; Takeuchi, R. J. Org. Chem. 2006, 71, 543–552. doi:10.1021/jo051874c |
| 13. | Matsuda, T.; Kadowaki, S.; Goya, T.; Murakami, M. Org. Lett. 2007, 9, 133–136. doi:10.1021/ol062732n |
| 14. | Takeuchi, R.; Nakaya, Y. Org. Lett. 2003, 5, 3659–3662. doi:10.1021/ol035330d |
| 15. | Farnetti, E.; Marsich, N. J. Organomet. Chem. 2004, 689, 14–17. doi:10.1016/j.jorganchem.2003.10.004 |
| 16. | Kezuka, S.; Okado, T.; Niou, E.; Takeuchi, R. Org. Lett. 2005, 7, 1711–1714. doi:10.1021/ol050085e |
| 17. | Shibata, T.; Takagi, K. J. Am. Chem. Soc. 2000, 122, 9852–9853. doi:10.1021/ja000899k |
| 18. | Shibata, T.; Toshida, N.; Yamazaki, M.; Maekawa, S.; Takagi, K. Tetrahedron 2005, 61, 9974–9979. doi:10.1016/j.tet.2005.08.016 |
| 19. | Jeong, N.; Kim, D. H.; Choi, J. H. Chem. Commun. 2004, 1134–1135. doi:10.1039/B401288G |
| 20. | Shibata, T.; Kadowaki, S.; Hirase, M.; Takagi, K. Synlett 2003, 573–575. doi:10.1055/s-2003-37532 |
| 29. | Wender, P. A.; Smith, T. E. Tetrahedron 1998, 54, 1255–1275. doi:10.1016/S0040-4020(97)10223-X |
| 33. | Yoo, W.-J.; Allen, A.; Villeneuve, K.; Tam, W. Org. Lett. 2005, 7, 5853–5856. doi:10.1021/ol052412o |
| 7. | Chatani, N.; Inoue, H.; Morimoto, T.; Muto, T.; Murai, S. J. Org. Chem. 2001, 66, 4433–4436. doi:10.1021/jo010091y |
| 8. | Yamamoto, Y.; Hayashi, H.; Saigoku, T.; Nishiyama, H. J. Am. Chem. Soc. 2005, 127, 10804–10805. doi:10.1021/ja053408a |
| 9. | Shibata, T.; Yamasaki, M.; Kadowaki, S.; Takagi, L. Synlett 2004, 2812–2814. doi:10.1055/s-2004-835631 |
| 10. | Shibata, T.; Kobayashi, Y.; Maekawa, S.; Toshida, N.; Takagi, K. Tetrahedron 2005, 61, 9018–9024. doi:10.1016/j.tet.2005.07.039 |
| 30. | Hilt, G.; Lüers, S.; Smolko, K. I. Org. Lett. 2005, 7, 251–253. doi:10.1021/ol0477879 |
| 6. | Fang, S.; Liang, X.; Long, Y.-H.; Li, X.; Yang, D.; Wang, S.; Li, C. Organometallics 2012, 31, 3113–3118. doi:10.1021/om3000295 |
| 26. | Shibata, T.; Takasaku, K.; Takesue, Y.; Hirata, N.; Takagi, K. Synlett 2002, 1681–1682. doi:10.1055/s-2002-34217 |
| 27. | Shibata, T.; Nishizawa, G.; Endo, K. Synlett 2008, 765–768. doi:10.1055/s-2008-1032112 |
| 26. | Shibata, T.; Takasaku, K.; Takesue, Y.; Hirata, N.; Takagi, K. Synlett 2002, 1681–1682. doi:10.1055/s-2002-34217 |
| 3. | Burford, R. J.; Piers, W. E.; Parvez, M. Organometallics 2012, 31, 2949–2952. doi:10.1021/om300127g |
| 4. | Hashiguchi, B. G.; Bischof, S. M.; Konnick, M. M.; Periana, R. A. Acc. Chem. Res. 2012, 45, 885–898. doi:10.1021/ar200250r |
| 5. | Werner, H. Angew. Chem., Int. Ed. 2010, 49, 4714–4728. doi:10.1002/anie.201000306 |
| 28. | Lautens, M.; Klute, W.; Tam, W. Chem. Rev. 1996, 96, 49–92. doi:10.1021/cr950016l |
| 33. | Yoo, W.-J.; Allen, A.; Villeneuve, K.; Tam, W. Org. Lett. 2005, 7, 5853–5856. doi:10.1021/ol052412o |
| 16. | Kezuka, S.; Okado, T.; Niou, E.; Takeuchi, R. Org. Lett. 2005, 7, 1711–1714. doi:10.1021/ol050085e |
| 17. | Shibata, T.; Takagi, K. J. Am. Chem. Soc. 2000, 122, 9852–9853. doi:10.1021/ja000899k |
| 18. | Shibata, T.; Toshida, N.; Yamazaki, M.; Maekawa, S.; Takagi, K. Tetrahedron 2005, 61, 9974–9979. doi:10.1016/j.tet.2005.08.016 |
| 19. | Jeong, N.; Kim, D. H.; Choi, J. H. Chem. Commun. 2004, 1134–1135. doi:10.1039/B401288G |
| 32. | Jolly, R. S.; Luedtke, G.; Sheehan, D.; Livinghouse, T. J. Am. Chem. Soc. 1990, 112, 4965–4966. doi:10.1021/ja00168a055 |
| 11. | Takeuchi, R.; Tanaka, S.; Nakaya, Y. Tetrahedron Lett. 2001, 42, 2991–2994. doi:10.1016/S0040-4039(01)00338-0 |
| 12. | Kezuka, S.; Tanaka, S.; Ohe, T.; Nakaya, Y.; Takeuchi, R. J. Org. Chem. 2006, 71, 543–552. doi:10.1021/jo051874c |
| 13. | Matsuda, T.; Kadowaki, S.; Goya, T.; Murakami, M. Org. Lett. 2007, 9, 133–136. doi:10.1021/ol062732n |
| 20. | Shibata, T.; Kadowaki, S.; Hirase, M.; Takagi, K. Synlett 2003, 573–575. doi:10.1055/s-2003-37532 |
| 33. | Yoo, W.-J.; Allen, A.; Villeneuve, K.; Tam, W. Org. Lett. 2005, 7, 5853–5856. doi:10.1021/ol052412o |
| 23. | Vollhardt, K. P. C. Angew. Chem., Int. Ed. Engl. 1984, 23, 539–556. doi:10.1002/anie.198405393 |
| 24. | Khand, I. U.; Knox, G. R.; Pauson, P. L.; Watts, W. E.; Foreman, M. I. J. Chem. Soc., Perkin Trans. 1 1973, 977–981. doi:10.1039/P19730000977 |
| 25. | Kotha, S.; Brahmachary, E.; Lahiri, K. Eur. J. Org. Chem. 2005, 4741–4767. doi:10.1002/ejoc.200500411 |
| 47. | DeBoef, B.; Counts, W. R.; Gilbertson, S. R. J. Org. Chem. 2007, 72, 799–804. doi:10.1021/jo0620462 |
| 21. | Trost, B. M. Angew. Chem., Int. Ed. Engl. 1995, 34, 259–281. doi:10.1002/anie.199502591 |
| 22. | Ojima, I.; Tzamarioudaki, M.; Li, Z.; Donovan, R. J. Chem. Rev. 1996, 96, 635–662. doi:10.1021/cr950065y |
| 14. | Takeuchi, R.; Nakaya, Y. Org. Lett. 2003, 5, 3659–3662. doi:10.1021/ol035330d |
| 15. | Farnetti, E.; Marsich, N. J. Organomet. Chem. 2004, 689, 14–17. doi:10.1016/j.jorganchem.2003.10.004 |
| 31. | Gilbertson, S. R.; Hoge, G. S.; Genov, D. G. J. Org. Chem. 1998, 63, 10077–10080. doi:10.1021/jo981870q |
| 26. | Shibata, T.; Takasaku, K.; Takesue, Y.; Hirata, N.; Takagi, K. Synlett 2002, 1681–1682. doi:10.1055/s-2002-34217 |
| 27. | Shibata, T.; Nishizawa, G.; Endo, K. Synlett 2008, 765–768. doi:10.1055/s-2008-1032112 |
| 31. | Gilbertson, S. R.; Hoge, G. S.; Genov, D. G. J. Org. Chem. 1998, 63, 10077–10080. doi:10.1021/jo981870q |
| 32. | Jolly, R. S.; Luedtke, G.; Sheehan, D.; Livinghouse, T. J. Am. Chem. Soc. 1990, 112, 4965–4966. doi:10.1021/ja00168a055 |
| 33. | Yoo, W.-J.; Allen, A.; Villeneuve, K.; Tam, W. Org. Lett. 2005, 7, 5853–5856. doi:10.1021/ol052412o |
| 34. | Shibata, T.; Chiba, T.; Hirashima, H.; Ueno, Y.; Endo, K. Angew. Chem., Int. Ed. 2009, 48, 8066–8069. doi:10.1002/anie.200903715 |
| 35. | Murakami, M.; Itami, K.; Ito, Y. J. Am. Chem. Soc. 1997, 119, 7163–7164. doi:10.1021/ja970626y |
| 36. | Murakami, M.; Minamida, R.; Itami, K.; Sawamura, M.; Ito, Y. Chem. Commun. 2000, 2293–2294. doi:10.1039/B007781J |
| 33. | Yoo, W.-J.; Allen, A.; Villeneuve, K.; Tam, W. Org. Lett. 2005, 7, 5853–5856. doi:10.1021/ol052412o |
| 33. | Yoo, W.-J.; Allen, A.; Villeneuve, K.; Tam, W. Org. Lett. 2005, 7, 5853–5856. doi:10.1021/ol052412o |
| 45. | Balsells, J.; Moyano, A.; Riera, A.; Pericàs, M. A. Org. Lett. 1999, 1, 1981–1984. doi:10.1021/ol991145h |
| 46. | Villeneuve, K.; Riddell, N.; Jordan, R. W.; Tsui, G. C.; Tam, W. Org. Lett. 2004, 6, 4543–4546. doi:10.1021/ol048111g |
| 40. | Alami, M.; Ferri, F. Tetrahedron Lett. 1996, 37, 2763–2766. doi:10.1016/0040-4039(96)00414-5 |
| 41. | Nishihara, Y.; Ikegashira, K.; Hirabayashi, K.; Ando, J.; Mori, A.; Hiyama, T. J. Org. Chem. 2000, 65, 1780–1787. doi:10.1021/jo991686k |
| 42. | Gung, B. W.; Kumi, G. J. Org. Chem. 2003, 68, 5956–5960. doi:10.1021/jo0344900 |
| 43. | Kim, S.; Kim, S.; Lee, T.; Ko, H.; Kim, D. Org. Lett. 2004, 6, 3601–3604. doi:10.1021/ol0484963 |
| 44. | Jiang, M. X.-W.; Rawat, M.; Wulff, W. D. J. Am. Chem. Soc. 2004, 126, 5970–5971. doi:10.1021/ja049836i |
| 37. | Hofmeister, H.; Annen, K.; Laurent, H.; Wiechert, R. Angew. Chem., Int. Ed. Engl. 1984, 23, 727–729. doi:10.1002/anie.198407271 |
| 38. | Yoshida, H.; Shirakawa, E.; Kurahashi, T.; Nakao, Y.; Hiyama, T. Organometallics 2000, 19, 5671–5678. doi:10.1021/om000828u |
| 39. | Liu, Y.; Zhong, Z.; Nakajima, K.; Takahashi, T. J. Org. Chem. 2002, 67, 7451–7456. doi:10.1021/jo026037e |
© 2012 Tigchelaar and Tam; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)