Abstract
A mild, efficient and versatile method has been developed for the construction of a functionalized natural product, meridianin, and its post conversion to pyrimido-β-carboline by cationic π- cyclization. The strategy involves the introduction of an amino group at the C-5 of the pyrimidine ring and utilizing the nucleophilictiy of the C-2 in the indole ring to facilitate cationic π-cyclization.

Graphical Abstract
Introduction
Indole is an important pharmacophore present in many natural and designed polyheterocyclic synthetic products of therapeutic importance [1-3]. The range of applications for these therapeutically relevant indole-based polyheterocycles includes protein kinase C inhibitors, 5-HT agonists, melatonin agonists, and glucocorticoid receptor modulators, displaying cytotoxic, antiviral, antimicrobial, antiparasitic, anti-inflammatory, antiserotonin, Ca2+/calmodulin antagonistic [4] and antitopoisomerase-I activities [5-16].
The presence of a heterocyclic ring at the position 3 of the indole represents an important class of marine alkaloids, such as oxazole (martefragin [17], amazol [18]), imidazole (topsentins [19,20] and nortopsentins [21-23]), dihydroimidazole (discodermindole [24]), oxadiazine (alboinon [25]), piperazine (dragmacidon [26]), maleimide (didemidines [25]), and pyrimidine (meridianins) [27-29]. Among these, meridianins (Figure 1), the tethered biheterocycles isolated from the south Atlantic tunicate Aplidium meridianum, represent an interesting synthetic target owing to their biological implication involving cytotoxicity towards murine-tumor cell lines [30,31] and as potent protein-kinase inhibitor [32].
![[1860-5397-8-220-1]](/bjoc/content/figures/1860-5397-8-220-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
In continuation of our ongoing studies pertaining to the synthesis of the indole-based natural products, i.e., isocryptolepine, δ-carbolines, and α-carbolines; and the indole-based polyheterocycles, i.e., indolo-quinolines, pericyclic indolo-benzazepines, iodo-indoloazepinones, indoloindazole, and azepino-indole [33-43], we were prompted to transform the indole-based alkaloid meridianins into annulated indole-based polyheterocycles as novel chemprobes.
For the synthesis of meridianin-inspired indole-based annulated polyheterocycles, we proposed to transform tethered biheterocycles into β-carboline-based polyheterocycles, a new prototype hitherto not reported in the literature. β-Carbolines are some of the most widely distributed alkaloids, associated with activities ranging from antineoplastic (tubulin binding) [44-46], anticonvulsive, hypnotic and anxiolytic (benzodiazepine receptor ligands) [47], antimicrobial as well as topoisomerase-II inhibition [48] to inhibition of cGMP-dependent processes [49,50].
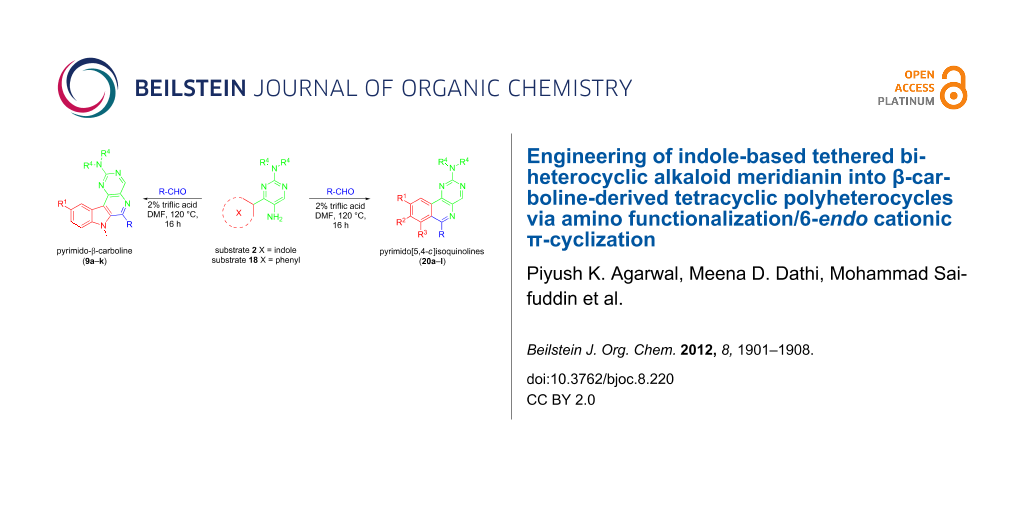
In this communication, we report engineering of naturally occurring tethered indole-based biheterocyclic alkaloid meridianins into β-carboline-derived tetracyclic polyheterocycles by amino functionalization of the pyrimidine ring followed by 6-endo cationic π-cyclization.
Results and Discussion
Our studies commenced with the functionalization of the pyrimidine ring in the meridianin followed by the application of cationic π-cyclization [51-60] to generate an additional pyridine ring as part of β-carboline-derived tetracyclic polyheterocycles. Initial attempts to synthesize amino-functionalized substrate 2a by introducing a nitro or nitroso group at the para position (position 5) of the 2-aminopyrimidine linked to the indole at C-3 position, using various reported protocols, either failed to produce the desired nitro compound or resulted in an inseparable mixture of compounds (Scheme 1).
![[1860-5397-8-220-i1]](/bjoc/content/inline/1860-5397-8-220-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of functionalized meridianin with an amino group at position 5.
Scheme 1: Synthesis of functionalized meridianin with an amino group at position 5.
This led us to attempt the synthesis of substrate 2a using an alternate strategy in a manner that may lead to the generation of a nitro pyrimidine ring tethered to the indole at position 3 (Scheme 2). For the generation of pyrimidine rings we used the modified procedure described previously by us [56]. Initial attempts to synthesize α-nitroketone 5a by using different protocols failed. To achieve this, the carboxylic group of 1-methyl-1H-indole-3-carboxylic acid (3a) was activated by preparing N-acylbenzotriazole [61]. Next, 1H-benzo[d][1,2,3]triazol-1-yl-(1-methyl-1H-indol-3-yl)methanone (4a) was converted into its α-nitroketone 5a by treating with nitromethane in the presence of potassium tert-butoxide in DMSO [62]. 1-(1-Methyl-1H-indol-3-yl)-2-nitroethanone (5a) was reacted with N,N-dimethylformamide dimethyl acetal to form (E)-3-(dimethylamino)-1-(1-methyl-1H-indol-3-yl)-2-nitroprop-2-en-1-one (6a) [63-66], which was then converted to 4-(1-methyl-1H-indol-3-yl)-5-nitropyrimidin-2-amine (7a) in the presence of guanidine hydrochloride. Finally, the desired substrate 4-(1-methyl-1H-indol-3-yl)pyrimidin-2,5-diamine (2a) was obtained by reducing 7a with H2/Pd in methanol. Synthesis of substrate 2b with diversity in the phenyl ring of the indole was carried out in a similar manner as described for 2a. In order to introduce diversity in the pyrimidine ring of the designed substrate 2, compound 6a was treated with 1,1-dimethylguanidine sulfate salt to get N,N-dimethyl-4-(1-methyl-1H-indol-3-yl)-5-nitropyrimidin-2-amine (8). The nitro group in 8 was finally reduced with H2/Pd in methanol to give N2,N2-dimethyl-4-(1-methyl-1H-indol-3-yl)pyrimidine-2,5-diamine (2c) as a new substrate (Scheme 3).
![[1860-5397-8-220-i2]](/bjoc/content/inline/1860-5397-8-220-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of a functionalized meridianin with an amino group at position 5.
Scheme 2: Synthesis of a functionalized meridianin with an amino group at position 5.
![[1860-5397-8-220-i3]](/bjoc/content/inline/1860-5397-8-220-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of substrate for the modified Pictet–Spengler reaction.
Scheme 3: Synthesis of substrate for the modified Pictet–Spengler reaction.
After successfully accomplishing the synthesis of amino-functionalized meridianins 2, we next examined their abilities to undergo 6-endo cyclization in the presence of aldehydes (Scheme 4). Initially we treated the substrate 2a with 4-chlorobenzaldehyde using a variety of traditional Pictet–Spengler protocols involving 1% TFA in DCM at rt, with p-TsOH in toluene under reflux, and AcOH in ethanol under reflux. The conditions failed to favor cationic π-cyclization even after prolonged stirring and resulted in imines as the only isolated product (Table 1, entries 1–6). Since the role of Brønsted acids are considered to be an important factor [67] in promoting cationic π-cyclization by enhancing the electrophilicity of the imines, we envisaged that employing stronger acids may facilitate 6-endo cyclization. Accordingly, the amine 2a was treated with 4-chlorobenzaldehyde by using strong Brønsted acids, such as triflic acid and methanesulfonic acid (MSA), to facilitate π-cyclization, and progress of the reaction was monitored by TLC.
![[1860-5397-8-220-i4]](/bjoc/content/inline/1860-5397-8-220-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: The Pictet–Spengler reaction involving substrate 2a. Reagents and conditions: (i) RCHO, 2% triflic acid in DMF, 120 °C, 16 h.
Scheme 4: The Pictet–Spengler reaction involving substrate 2a. Reagents and conditions: (i) RCHO, 2% triflic ...
Table 1: Optimization of the reaction conditions for conversion of substrate 2a to 9a.
| Entry | Brønsted acid | Solvent | Temp (oC) | Time (h) | Producta |
|---|---|---|---|---|---|
| 1 | 1% TFA | DCM | rt | 24 | 0 |
| 2 | 1% triflic acid | DMF | rt | 24 | 0 |
| 3 | p-TsOH | Toluene | 120 | 24 | 0 |
| 4 | AcOH | Ethanol | 100 | 24 | 0 |
| 5 | 2% MSA | ACN | 80 | 24 | 0 |
| 6 | 2% MSA | DMF | 120 | 16 | 0 |
| 7 | 2% triflic acid | ACN | 80 | 24 | 27 |
| 8 | 2% triflic acid | DMF | 120 | 16 | 87 |
| 9 | 5% triflic acid | DMF | 120 | 16 | 80 |
| 10 | 1% triflic acid | DMF | 120 | 16 | 75 |
| 11 | 0.5% triflic acid | DMF | 120 | 16 | 35 |
abased on HPLC.
Although no conversion was observed for 2a in the presence of MSA (Table 1, entry 5 and entry 6) in CH3CN and DMF at 80 °C and 120 °C, respectively, the presence of 2% triflic acid in DMF (Table 1, entry 8) favored complete conversion of 2a into 9a in >87% purity based on HPLC. The crude product obtained after workup was purified by silica gel column chromatography with EtOAc/hexane as an eluent in 84% isolated yield. An increase or decrease in the concentration of triflic acid was found to be detrimental (Table 1, entries 9–11). Similarly, switching solvents from DMF to ACN in the presence of 2% triflic acid reduced the yield to 27% (Table 1, entry 7). The scope and limitation of the strategy with substrate 2a and 2c was established by synthesizing 11 compounds based on pyrimido-β-carbolines 9a–k (Table 2), using eight aromatic aldehydes.
Table 2: Pyrimido-β-carbolines based on 9.
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-220-i8.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Substrate | R-CHO | Pictet–Spengler products 9, R | Yield (%)a | tR (min)b | |
|---|---|---|---|---|---|---|
| 1 | 2a | 4-Cl-C6H4-CHO | 9a | 4-Cl-C6H4 | 84 | 14.92 |
| 2 | 2a | 4-OEt-C6H4-CHO | 9b | 4-OEt-C6H4 | 82 | 14.14 |
| 3 | 2a | 4-OMe-C6H4-CHO | 9c | 4-OMe-C6H4 | 78 | 13.25 |
| 4 | 2a | 4-Br-C6H4-CHO | 9d | 4-Br-C6H4 | 85 | 13.90 |
| 5 | 2a | 3,4-diCl-C6H-CHO | 9e | 3,4-diCl-C6H3 | 80 | 14.50 |
| 6 | 2c | 4-Br-C6H4-CHO | 9f | 4-Br-C6H4 | 72 | 15.89 |
| 7 | 2c | 4-NO2-C6H4-CHO | 9g | 4-NO2-C6H4-CHO | 76 | 15.82 |
| 8 | 2c | 4-OMe-C6H4-CHO | 9h | 4-OMe-C6H4-CHO | 79 | 13.45 |
| 9 | 2c | 3,4-diOMe-C6H3-CHO | 9i | 3,4-diOMe-C6H3 | 81 | 12.95 |
| 10 | 2c | 4-Cl-C6H4-CHO | 9j | 4-Cl-C6H4 | 86 | 14.82 |
| 11 | 2c | 2-Cl-C6H4-CHO | 9k | 2-Cl-C6H4 | 78 | 14.37 |
aIsolated yield. bRetention time on HPLC (C18 reversed-phase column; 150 mm × 4.8 mm; 5 µm) with a linear gradient of 0–100% CH3CN in water over 30 min. Flow rate of 1.0 mL/min and UV detection at 220/254 nm.
In general for all reactions, the protocol involving 2% triflic acid in DMF at 120 °C for 16 h was employed, furnishing products 9 in good isolated yields (72–86%). Pleasingly, aldehydes with either an electron-donating or -withdrawing group had no adverse effect and afforded 9 with minimal variation in yields. However, the substrate 2b on application of the protocol involving 2% triflic acid in DMF at 120 °C produced a dihydroproduct instead of the oxidized compound. Continuing the reaction for a further 24 h failed to produce the oxidized product (Scheme 5).
![[1860-5397-8-220-i5]](/bjoc/content/inline/1860-5397-8-220-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of dihydropyrimido-β-carbolines: (i) R-CHO, 2% triflic acid in DMF, 120 °C, 16 h.
Scheme 5: Synthesis of dihydropyrimido-β-carbolines: (i) R-CHO, 2% triflic acid in DMF, 120 °C, 16 h.
After successfully establishing the strategy on 2a-c, we then decided to replace the indole nucleus by another activated nucleus such as trimethoxy- and dimethoxybenzene and designed a substrate 18 (Scheme 6) for the Pictet–Spengler reaction using a similar approach as was used for the synthesis of substrates 2.
![[1860-5397-8-220-i6]](/bjoc/content/inline/1860-5397-8-220-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of substrates 18a–c for the modified Pictet–Spengler reaction.
Scheme 6: Synthesis of substrates 18a–c for the modified Pictet–Spengler reaction.
The synthesis of substrates 18 is depicted in Scheme 6. Initially, the carboxylic group of benzoic acids 13a,b was activated by preparing N-acylbenzotriazoles 14, which were then converted into their α-nitroketones 15 by treatment with nitromethane in the presence of potassium tert-butoxide in DMSO. Reaction of 15 with N,N-dimethylformamide dimethyl acetal provided 16, which was cyclized in the presence of guanidine hydrochloride and N,N-dimethylguanidine sulfate to give the nitro products 17a,b and 17c. Finally, the desired substrates 18 were obtained by reducing 17 with H2/Pd in methanol. Next, substrates 18 were exposed to the cationic π-cyclization with a variety of aldehydes using the protocol involving 2% triflic acid in DMF at 120 °C (Scheme 7). The crude endo-cyclized product obtained after workup was purified by silica-gel column chromatography furnishing pyrimido[5,4-c]isoquinolines 20a–l (Table 3) in 79–92% isolated yields.
![[1860-5397-8-220-i7]](/bjoc/content/inline/1860-5397-8-220-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: General strategy for the Pictet–Spengler reaction involving substrates 18. Reagents and conditions: (i) R-CHO, 2% triflic acid in DMF, 120 °C, 16 h.
Scheme 7: General strategy for the Pictet–Spengler reaction involving substrates 18. Reagents and conditions:...
Table 3: Pyrimido[5,4-c]isoquinolines based on 20.
| Entry | Substrate | R3-CHO | Pictet–Spengler products 20, R3 | Yield (%)a | tR (min)b | |
|---|---|---|---|---|---|---|
| 1 | 18a | 4-F-C6H4-CHO | 20a | 4-F-C6H4-CHO | 85 | 20.12 |
| 2 | 18a | 4-Br-C6H4-CHO | 20b | 4-Br-C6H4-CHO | 87 | 21.95 |
| 3 | 18a | 4-Cl-C6H4-CHO | 20c | 4-Cl-C6H4-CHO | 83 | 25.25 |
| 4 | 18b | 4-OMe-C6H4-CHO | 20d | 4-OMe-C6H4-CHO | 89 | 21.42 |
| 5 | 18b | 4-Cl-C6H4-CHO | 20e | 4-Cl-C6H4-CHO | 92 | 20.65 |
| 6 | 18b | 4-F-C6H4-CHO | 20f | 4-F-C6H4-CHO | 91 | 21.98 |
| 7 | 18b | 4-Br-C6H4-CHO | 20g | 4-Br-C6H4-CHO | 87 | 22.85 |
| 8 | 18c | 4-NO2-C6H4-CHO | 20h | 4-NO2-C6H4-CHO | 81 | 20.46 |
| 9 | 18c | 3,4-diOMe-C6H3-CHO | 20i | 3,4-diOMe-C6H3-CHO | 84 | 21.56 |
| 10 | 18c | 3,4-diCl-C6H3-CHO | 20j | 3,4-diCl-C6H3-CHO | 83 | 21.45 |
| 11 | 18c | 4-Br-C6H4-CHO | 20k | 4-Br-C6H4-CHO | 79 | 20.12 |
| 12 | 18c | 4-OMe-C6H4-CHO | 20l | 4-OMe-C6H4-CHO | 87 | 19.89 |
aIsolated yield.
bRetention time on HPLC (C18 reversed-phase column; 150 mm × 4.8 mm; 5 µm) with a linear gradient of 0–100% CH3CN in water over 30 min. Flow rate of 1.0 mL/min and UV detection at 220/254 nm.
Conclusion
In conclusion, we have developed a mild and efficient protocol for the synthesis of pyrimido[5,4-c]isoquinolones and pyrimido-β-carbolines using a modified Pictet–Spengler strategy. Our method offers a unique opportunity to introduce rigidity in these flexible molecules, as well as diversity, which enables the design of a library based on the natural product. Our methodology further demonstrated a broad substrate scope and reactivity, and thus it can be applied for the synthesis of a variety of novel polycyclic skeletons based on natural products.
Supporting Information
| Supporting Information File 1: Experimental section, 1H and 13C NMR spectra of the compounds 2a–c, 4a–7a, 4b–7b, 8, 9a–k, 12a–d, 14a–18a, 14b–16b, 17–18c, 17b–18b, 20a–l. | ||
| Format: PDF | Size: 7.2 MB | Download |
References
-
Faulkner, D. J. Nat. Prod. Rep. 2001, 18, R1–R49. doi:10.1039/b006897g
Return to citation in text: [1] -
Humphrey, G. R.; Kuethe, J. T. Chem. Rev. 2006, 106, 2875–2911. doi:10.1021/cr0505270
Return to citation in text: [1] -
Takayama, H.; Tsutsumi, S.-i.; Kitajima, M.; Santiarworn, D.; Liawruangrath, B.; Aimi, N. Chem. Pharm. Bull. 2003, 51, 232–233. doi:10.1248/cpb.51.232
Return to citation in text: [1] -
Lanteri, M. L.; Pagnussat, G. C.; Lamattina, L. J. Exp. Bot. 2006, 57, 1341–1351. doi:10.1093/jxb/erj109
Return to citation in text: [1] -
Xie, G.; Zimmermann, J.; Meyer, T.; Lown, J. W. Bioorg. Med. Chem. Lett. 1995, 5, 497–500. doi:10.1016/0960-894X(95)00060-7
Return to citation in text: [1] -
Chambers, M. S.; Street, L. J.; Goodacre, S.; Hobbs, S. C.; Hunt, P. H.; Jelley, R. A.; Matassa, V. G.; Reeve, A. J.; Sternfeld, F.; Beer, M. S.; Stanton, J. A.; Rathbone, D.; Watt, A. P.; MacLeod, A. M. J. Med. Chem. 1999, 42, 691–705. doi:10.1021/jm980569z
Return to citation in text: [1] -
Faust, R.; Garratt, P. J.; Jones, R.; Yeh, L.-K. J. Med. Chem. 2000, 43, 1050–1061. doi:10.1021/jm980684+
Return to citation in text: [1] -
Roach, S. L.; Higuchi, R. I.; Adams, M. E.; Liu, Y.; Karanewsky, D. S.; Marschke, K. B.; Mais, D. E.; Miner, J. N.; Zhi, L. Bioorg. Med. Chem. Lett. 2008, 18, 3504–3508. doi:10.1016/j.bmcl.2008.05.029
Return to citation in text: [1] -
Chaturvedula, V. S. P.; Sprague, S.; Schilling, J. K.; Kingston, D. G. I. J. Nat. Prod. 2003, 66, 528–531. doi:10.1021/np020548e
Return to citation in text: [1] -
Zhao, Z.; Wolkenberg, S. E.; Lu, M.; Munshi, V.; Moyer, G.; Feng, M.; Carella, A. V.; Ecto, L. T.; Gabryelski, L. J.; Lai, M.-T.; Prasad, S. G.; Yan, Y.; McGaughey, G. B.; Miller, M. D.; Lindsley, C. W.; Hartman, G. D.; Vacca, J. P.; Williams, T. M. Bioorg. Med. Chem. Lett. 2008, 18, 554–559. doi:10.1016/j.bmcl.2007.11.085
Return to citation in text: [1] -
Scribner, A.; Moore, J. A., III; Ouvry, G.; Fisher, M.; Wyvratt, M.; Leavitt, P.; Liberator, P.; Gurnett, A.; Brown, C.; Mathew, J.; Thompson, D.; Schmatz, D.; Biftu, T. Bioorg. Med. Chem. Lett. 2009, 19, 1517–1521. doi:10.1016/j.bmcl.2009.01.001
Return to citation in text: [1] -
Doebel, K. J.; Wasley, J. W. F. J. Med. Chem. 1972, 15, 1081–1082. doi:10.1021/jm00280a027
Return to citation in text: [1] -
Remy, D. C.; Rittle, K. E.; Hunt, C. A.; Anderson, P. S.; Engelhardt, E. L.; Clineschmidt, B. V.; Scriabine, A. J. Med. Chem. 1977, 20, 1681–1684. doi:10.1021/jm00222a031
Return to citation in text: [1] -
Saifuddin, M.; Agarwal, P. K.; Sharma, S. K.; Madadapu, A. K.; Gupta, S.; Harit, V. K.; Kundu, B. Eur. J. Org. Chem. 2010, 5108–5117. doi:10.1002/ejoc.201000596
Return to citation in text: [1] -
Quintanar-Audelo, M.; Fernández-Carvajal, A.; Van Den Nest, W.; Carreño, C.; Ferrer-Montiel, A.; Albericio, F. J. Med. Chem. 2007, 50, 6133–6143. doi:10.1021/jm070612v
Return to citation in text: [1] -
Ferlin, M. G.; Chiarelotto, G.; Gasparotto, V.; Via, L. D.; Pezzi, V.; Barzon, L.; Palù, G.; Castagliuolo, I. J. Med. Chem. 2005, 48, 3417–3427. doi:10.1021/jm049387x
Return to citation in text: [1] -
Vervoort, H. C.; Richards-Gross, S. E.; Fenical, W.; Lee, A. Y.; Clardy, J. J. Org. Chem. 1997, 62, 1486–1490. doi:10.1021/jo961789s
Return to citation in text: [1] -
Takahashi, S.; Matsunaga, T.; Hasegawa, C.; Saito, H.; Fujita, D.; Kiuchi, F.; Tsuda, Y. Chem. Pharm. Bull. 1998, 46, 1527–1529. doi:10.1248/cpb.46.1527
Return to citation in text: [1] -
Kawasaki, Y.; Yamashita, M.; Ohta, S. J. Chem. Soc., Chem. Commun. 1994, 2085–2086. doi:10.1039/C39940002085
Return to citation in text: [1] -
Kawasaki, Y.; Yamashita, M.; Ohta, S. Chem. Pharm. Bull. 1996, 44, 1831–1839. doi:10.1248/cpb.44.1831
Return to citation in text: [1] -
Bartik, K.; Braekman, J.-C.; Daloze, D.; Stoller, C.; Huysecom, J.; Vandevyver, G.; Ottinger, R. Can. J. Chem. 1987, 65, 2118–2121. doi:10.1139/v87-352
Return to citation in text: [1] -
Tsuji, S.; Rinehart, K. L.; Gunasekera, S. P.; Kashman, Y.; Cross, S. S.; Lui, M. S.; Pomponi, S. A.; Diaz, M. C. J. Org. Chem. 1988, 53, 5446–5453. doi:10.1021/jo00258a009
Return to citation in text: [1] -
Kawasaki, I.; Katsuma, H.; Nakayama, Y.; Yamashita, M.; Otha, S. Heterocycles 1998, 48, 1887–1901. doi:10.3987/COM-98-8225
Return to citation in text: [1] -
Sun, H. H.; Sakemi, S. J. Org. Chem. 1991, 56, 4307–4308. doi:10.1021/jo00013a045
Return to citation in text: [1] -
Bergmann, T.; Schories, D.; Steffan, B. Tetrahedron 1997, 53, 2055–2060. doi:10.1016/S0040-4020(96)01168-4
Return to citation in text: [1] [2] -
Kohmoto, S.; Kashman, Y.; McConnell, O. J.; Rinehart, K. L., Jr.; Wright, A.; Koehn, F. J. Org. Chem. 1988, 53, 3116–3118. doi:10.1021/jo00248a040
Return to citation in text: [1] -
Franco, L. H.; Joffe, E. B. K.; Puricelly, L.; Tatian, M.; Seldes, A. M.; Palermo, J. A. J. Nat. Prod. 1998, 61, 1130–1132. doi:10.1021/np970493u
Return to citation in text: [1] -
Seldes, A. M.; Brasco, M. F. R.; Hernández Franco, L.; Palermo, J. A. Nat. Prod. Res. 2007, 21, 555–563. doi:10.1080/14786410601133517
Return to citation in text: [1] -
Sperry, J. Tetrahedron Lett. 2011, 52, 4537–4538. doi:10.1016/j.tetlet.2011.06.073
Return to citation in text: [1] -
Jiang, B.; Yang, C.-G.; Xiong, W.-N.; Wang, J. Bioorg. Med. Chem. 2001, 9, 1149–1154. doi:10.1016/S0968-0896(00)00337-0
Return to citation in text: [1] -
Rossignol, E.; Debiton, E.; Fabbro, D.; Moreau, P.; Prudhomme, M.; Anizon, F. Anti-Cancer Drugs 2008, 19, 789–792. doi:10.1097/CAD.0b013e32830ce4d8
Return to citation in text: [1] -
Gompel, M.; Leost, M.; Joffe, E. B. K.; Puricelli, L.; Franco, L. H.; Palermo, J.; Meijer, L. Bioorg. Med. Chem. Lett. 2004, 14, 1703–1707. doi:10.1016/j.bmcl.2004.01.050
Return to citation in text: [1] -
Saifuddin, M.; Agarwal, P. K.; Sharma, S. K.; Mandadapu, A. K.; Gupta, S.; Harit, V. K.; Kundu, B. Eur. J. Org. Chem. 2010, 5108–5117. doi:10.1002/ejoc.201000596
Return to citation in text: [1] -
Sharma, S. K.; Gupta, S.; Saifuddin, M.; Mandadapu, A. K.; Agarwal, P. K.; Gauniyal, H. M.; Kundu, B. Tetrahedron Lett. 2011, 52, 65–68. doi:10.1016/j.tetlet.2010.10.147
Return to citation in text: [1] -
Sharma, S. K.; Mandadapu, A. K.; Saifuddin, M.; Gupta, S.; Agarwal, P. K.; Mandwal, A. K.; Gauniyal, H. M.; Kundu, B. Tetrahedron Lett. 2010, 51, 6022–6024. doi:10.1016/j.tetlet.2010.09.054
Return to citation in text: [1] -
Agarwal, P. K.; Sawant, D.; Sharma, S.; Kundu, B. Eur. J. Org. Chem. 2009, 292–303. doi:10.1002/ejoc.200800929
Return to citation in text: [1] -
Sharma, S. K.; Sharma, S.; Agarwal, P. K.; Kundu, B. Eur. J. Org. Chem. 2009, 1309–1312. doi:10.1002/ejoc.200801201
Return to citation in text: [1] -
Saha, B.; Sharma, S.; Sawant, D.; Kundu, B. Tetrahedron Lett. 2007, 48, 1379–1383. doi:10.1016/j.tetlet.2006.12.112
Return to citation in text: [1] -
Sharma, S.; Kundu, B. Tetrahedron Lett. 2008, 49, 7062–7065. doi:10.1016/j.tetlet.2008.09.154
Return to citation in text: [1] -
Gupta, S.; Kumar, B.; Kundu, B. J. Org. Chem. 2011, 76, 10154–10162. doi:10.1021/jo201994v
Return to citation in text: [1] -
Gupta, S.; Sharma, S. K.; Mandadapu, A. K.; Gauniyal, H. M.; Kundu, B. Tetrahedron Lett. 2011, 52, 4288–4291. doi:10.1016/j.tetlet.2011.06.021
Return to citation in text: [1] -
Sharma, S. K.; Mandadapu, A. K.; Kumar, B.; Kundu, B. J. Org. Chem. 2011, 76, 6798–6805. doi:10.1021/jo201228t
Return to citation in text: [1] -
Saha, B.; Kumar, R.; Maulik, P. R.; Kundu, B. Tetrahedron Lett. 2006, 47, 2765–2769. doi:10.1016/j.tetlet.2006.02.072
Return to citation in text: [1] -
Takasu, K.; Shimogama, T.; Saiin, C.; Kim, H.-S.; Wataya, Y.; Ihara, M. Bioorg. Med. Chem. Lett. 2004, 14, 1689–1692. doi:10.1016/j.bmcl.2004.01.055
Return to citation in text: [1] -
Boursereau, Y.; Coldham, I. Bioorg. Med. Chem. Lett. 2004, 14, 5841–5844. doi:10.1016/j.bmcl.2004.09.036
Return to citation in text: [1] -
Wu, Y.; Zhao, M.; Wang, C.; Peng, S. Bioorg. Med. Chem. Lett. 2002, 12, 2331–2333. doi:10.1016/S0960-894X(02)00403-1
Return to citation in text: [1] -
Behforouz, M.; Merriman, R. L. Methods of using lavendamycin analogs. U.S. Patent 5,646,150, July 8, 1997.
Return to citation in text: [1] -
Batch, A.; Dodd, R. H. J. Org. Chem. 1998, 63, 872–877. doi:10.1021/jo971485l
Return to citation in text: [1] -
Pommier, Y.; MacDonald, T. L.; Madalengoitia, J. S. U.S. Patent 5,622,960, April 22, 1997.
Return to citation in text: [1] -
Daugan, C. M.; Labaudiniere, R. F. Tetrahydroimidazopyridoindolediones and their use as medicaments. WO/1996/032003, Oct 17, 1996.
Return to citation in text: [1] -
Kundu, B.; Sawant, D.; Chhabra, R. A. J. Comb. Chem. 2005, 7, 317–321. doi:10.1021/cc049851j
Return to citation in text: [1] -
Kundu, B.; Sawant, D.; Partani, P.; Kesarwani, A. P. J. Org. Chem. 2005, 70, 4889–4892. doi:10.1021/jo050384h
Return to citation in text: [1] -
Duggineni, S.; Sawant, D.; Saha, B.; Kundu, B. Tetrahedron 2006, 62, 3228–3241. doi:10.1016/j.tet.2006.01.063
Return to citation in text: [1] -
Sharma, S.; Saha, B.; Sawant, D.; Kundu, B. J. Comb. Chem. 2007, 9, 783–792. doi:10.1021/cc0700445
Return to citation in text: [1] -
Saha, B.; Sharma, S.; Sawant, D.; Kundu, B. Tetrahedron 2008, 64, 8676–8684. doi:10.1016/j.tet.2008.07.003
Return to citation in text: [1] -
Agarwal, P. K.; Sharma, S. K.; Sawant, D.; Kundu, B. Tetrahedron 2009, 65, 1153–1161. doi:10.1016/j.tet.2008.11.067
Return to citation in text: [1] [2] -
Agarwal, P. K.; Saifuddin, M.; Kundu, B. Tetrahedron 2010, 66, 862–870. doi:10.1016/j.tet.2009.11.115
Return to citation in text: [1] -
Mandadapu, A. K.; Saifuddin, M.; Agarwal, P. K.; Kundu, B. Org. Biomol. Chem. 2009, 7, 2796–2803. doi:10.1039/b905696c
Return to citation in text: [1] -
Kundu, B.; Partani, P.; Duggineni, S.; Sawant, D. J. Comb. Chem. 2005, 7, 909–915. doi:10.1021/cc0500699
Return to citation in text: [1] -
Sharma, S.; Kundu, B. J. Comb. Chem. 2009, 11, 720–731. doi:10.1021/cc9000345
Return to citation in text: [1] -
Katritzky, A. R.; Zhang, Y.; Singh, S. K. Synthesis 2003, 2795–2798. doi:10.1055/s-2003-42462
Return to citation in text: [1] -
Katritzky, A. R.; Abdel-Fattah, A. A. A.; Gromova, A. V.; Witek, R.; Steel, P. J. J. Org. Chem. 2005, 70, 9211–9214. doi:10.1021/jo051231x
Return to citation in text: [1] -
Bredereck, H.; Effenberger, F.; Botsch, H.; Rehn, H. Chem. Ber. 1965, 98, 1081–1806. doi:10.1002/cber.19650980412
Return to citation in text: [1] -
Fresneda, P. M.; Molina, P.; Delgado, S.; Bleda, J. A. Tetrahedron Lett. 2000, 41, 4777–4780. doi:10.1016/S0040-4039(00)00728-0
Return to citation in text: [1] -
Karpov, A. S.; Merkul, E.; Rominger, F.; Müller, T. J. J. Angew. Chem., Int. Ed. 2005, 44, 6951–6956. doi:10.1002/anie.200501703
Return to citation in text: [1] -
Rossignol, E.; Youssef, A.; Moreau, P.; Prudhomme, M.; Anizon, F. Tetrahedron 2007, 63, 10169–10176. doi:10.1016/j.tet.2007.07.095
Return to citation in text: [1] -
Cox, E. D.; Cook, J. Chem. Rev. 1995, 95, 1797–1842. doi:10.1021/cr00038a004
Return to citation in text: [1]
| 1. | Faulkner, D. J. Nat. Prod. Rep. 2001, 18, R1–R49. doi:10.1039/b006897g |
| 2. | Humphrey, G. R.; Kuethe, J. T. Chem. Rev. 2006, 106, 2875–2911. doi:10.1021/cr0505270 |
| 3. | Takayama, H.; Tsutsumi, S.-i.; Kitajima, M.; Santiarworn, D.; Liawruangrath, B.; Aimi, N. Chem. Pharm. Bull. 2003, 51, 232–233. doi:10.1248/cpb.51.232 |
| 18. | Takahashi, S.; Matsunaga, T.; Hasegawa, C.; Saito, H.; Fujita, D.; Kiuchi, F.; Tsuda, Y. Chem. Pharm. Bull. 1998, 46, 1527–1529. doi:10.1248/cpb.46.1527 |
| 33. | Saifuddin, M.; Agarwal, P. K.; Sharma, S. K.; Mandadapu, A. K.; Gupta, S.; Harit, V. K.; Kundu, B. Eur. J. Org. Chem. 2010, 5108–5117. doi:10.1002/ejoc.201000596 |
| 34. | Sharma, S. K.; Gupta, S.; Saifuddin, M.; Mandadapu, A. K.; Agarwal, P. K.; Gauniyal, H. M.; Kundu, B. Tetrahedron Lett. 2011, 52, 65–68. doi:10.1016/j.tetlet.2010.10.147 |
| 35. | Sharma, S. K.; Mandadapu, A. K.; Saifuddin, M.; Gupta, S.; Agarwal, P. K.; Mandwal, A. K.; Gauniyal, H. M.; Kundu, B. Tetrahedron Lett. 2010, 51, 6022–6024. doi:10.1016/j.tetlet.2010.09.054 |
| 36. | Agarwal, P. K.; Sawant, D.; Sharma, S.; Kundu, B. Eur. J. Org. Chem. 2009, 292–303. doi:10.1002/ejoc.200800929 |
| 37. | Sharma, S. K.; Sharma, S.; Agarwal, P. K.; Kundu, B. Eur. J. Org. Chem. 2009, 1309–1312. doi:10.1002/ejoc.200801201 |
| 38. | Saha, B.; Sharma, S.; Sawant, D.; Kundu, B. Tetrahedron Lett. 2007, 48, 1379–1383. doi:10.1016/j.tetlet.2006.12.112 |
| 39. | Sharma, S.; Kundu, B. Tetrahedron Lett. 2008, 49, 7062–7065. doi:10.1016/j.tetlet.2008.09.154 |
| 40. | Gupta, S.; Kumar, B.; Kundu, B. J. Org. Chem. 2011, 76, 10154–10162. doi:10.1021/jo201994v |
| 41. | Gupta, S.; Sharma, S. K.; Mandadapu, A. K.; Gauniyal, H. M.; Kundu, B. Tetrahedron Lett. 2011, 52, 4288–4291. doi:10.1016/j.tetlet.2011.06.021 |
| 42. | Sharma, S. K.; Mandadapu, A. K.; Kumar, B.; Kundu, B. J. Org. Chem. 2011, 76, 6798–6805. doi:10.1021/jo201228t |
| 43. | Saha, B.; Kumar, R.; Maulik, P. R.; Kundu, B. Tetrahedron Lett. 2006, 47, 2765–2769. doi:10.1016/j.tetlet.2006.02.072 |
| 17. | Vervoort, H. C.; Richards-Gross, S. E.; Fenical, W.; Lee, A. Y.; Clardy, J. J. Org. Chem. 1997, 62, 1486–1490. doi:10.1021/jo961789s |
| 44. | Takasu, K.; Shimogama, T.; Saiin, C.; Kim, H.-S.; Wataya, Y.; Ihara, M. Bioorg. Med. Chem. Lett. 2004, 14, 1689–1692. doi:10.1016/j.bmcl.2004.01.055 |
| 45. | Boursereau, Y.; Coldham, I. Bioorg. Med. Chem. Lett. 2004, 14, 5841–5844. doi:10.1016/j.bmcl.2004.09.036 |
| 46. | Wu, Y.; Zhao, M.; Wang, C.; Peng, S. Bioorg. Med. Chem. Lett. 2002, 12, 2331–2333. doi:10.1016/S0960-894X(02)00403-1 |
| 5. | Xie, G.; Zimmermann, J.; Meyer, T.; Lown, J. W. Bioorg. Med. Chem. Lett. 1995, 5, 497–500. doi:10.1016/0960-894X(95)00060-7 |
| 6. | Chambers, M. S.; Street, L. J.; Goodacre, S.; Hobbs, S. C.; Hunt, P. H.; Jelley, R. A.; Matassa, V. G.; Reeve, A. J.; Sternfeld, F.; Beer, M. S.; Stanton, J. A.; Rathbone, D.; Watt, A. P.; MacLeod, A. M. J. Med. Chem. 1999, 42, 691–705. doi:10.1021/jm980569z |
| 7. | Faust, R.; Garratt, P. J.; Jones, R.; Yeh, L.-K. J. Med. Chem. 2000, 43, 1050–1061. doi:10.1021/jm980684+ |
| 8. | Roach, S. L.; Higuchi, R. I.; Adams, M. E.; Liu, Y.; Karanewsky, D. S.; Marschke, K. B.; Mais, D. E.; Miner, J. N.; Zhi, L. Bioorg. Med. Chem. Lett. 2008, 18, 3504–3508. doi:10.1016/j.bmcl.2008.05.029 |
| 9. | Chaturvedula, V. S. P.; Sprague, S.; Schilling, J. K.; Kingston, D. G. I. J. Nat. Prod. 2003, 66, 528–531. doi:10.1021/np020548e |
| 10. | Zhao, Z.; Wolkenberg, S. E.; Lu, M.; Munshi, V.; Moyer, G.; Feng, M.; Carella, A. V.; Ecto, L. T.; Gabryelski, L. J.; Lai, M.-T.; Prasad, S. G.; Yan, Y.; McGaughey, G. B.; Miller, M. D.; Lindsley, C. W.; Hartman, G. D.; Vacca, J. P.; Williams, T. M. Bioorg. Med. Chem. Lett. 2008, 18, 554–559. doi:10.1016/j.bmcl.2007.11.085 |
| 11. | Scribner, A.; Moore, J. A., III; Ouvry, G.; Fisher, M.; Wyvratt, M.; Leavitt, P.; Liberator, P.; Gurnett, A.; Brown, C.; Mathew, J.; Thompson, D.; Schmatz, D.; Biftu, T. Bioorg. Med. Chem. Lett. 2009, 19, 1517–1521. doi:10.1016/j.bmcl.2009.01.001 |
| 12. | Doebel, K. J.; Wasley, J. W. F. J. Med. Chem. 1972, 15, 1081–1082. doi:10.1021/jm00280a027 |
| 13. | Remy, D. C.; Rittle, K. E.; Hunt, C. A.; Anderson, P. S.; Engelhardt, E. L.; Clineschmidt, B. V.; Scriabine, A. J. Med. Chem. 1977, 20, 1681–1684. doi:10.1021/jm00222a031 |
| 14. | Saifuddin, M.; Agarwal, P. K.; Sharma, S. K.; Madadapu, A. K.; Gupta, S.; Harit, V. K.; Kundu, B. Eur. J. Org. Chem. 2010, 5108–5117. doi:10.1002/ejoc.201000596 |
| 15. | Quintanar-Audelo, M.; Fernández-Carvajal, A.; Van Den Nest, W.; Carreño, C.; Ferrer-Montiel, A.; Albericio, F. J. Med. Chem. 2007, 50, 6133–6143. doi:10.1021/jm070612v |
| 16. | Ferlin, M. G.; Chiarelotto, G.; Gasparotto, V.; Via, L. D.; Pezzi, V.; Barzon, L.; Palù, G.; Castagliuolo, I. J. Med. Chem. 2005, 48, 3417–3427. doi:10.1021/jm049387x |
| 30. | Jiang, B.; Yang, C.-G.; Xiong, W.-N.; Wang, J. Bioorg. Med. Chem. 2001, 9, 1149–1154. doi:10.1016/S0968-0896(00)00337-0 |
| 31. | Rossignol, E.; Debiton, E.; Fabbro, D.; Moreau, P.; Prudhomme, M.; Anizon, F. Anti-Cancer Drugs 2008, 19, 789–792. doi:10.1097/CAD.0b013e32830ce4d8 |
| 4. | Lanteri, M. L.; Pagnussat, G. C.; Lamattina, L. J. Exp. Bot. 2006, 57, 1341–1351. doi:10.1093/jxb/erj109 |
| 32. | Gompel, M.; Leost, M.; Joffe, E. B. K.; Puricelli, L.; Franco, L. H.; Palermo, J.; Meijer, L. Bioorg. Med. Chem. Lett. 2004, 14, 1703–1707. doi:10.1016/j.bmcl.2004.01.050 |
| 25. | Bergmann, T.; Schories, D.; Steffan, B. Tetrahedron 1997, 53, 2055–2060. doi:10.1016/S0040-4020(96)01168-4 |
| 25. | Bergmann, T.; Schories, D.; Steffan, B. Tetrahedron 1997, 53, 2055–2060. doi:10.1016/S0040-4020(96)01168-4 |
| 24. | Sun, H. H.; Sakemi, S. J. Org. Chem. 1991, 56, 4307–4308. doi:10.1021/jo00013a045 |
| 27. | Franco, L. H.; Joffe, E. B. K.; Puricelly, L.; Tatian, M.; Seldes, A. M.; Palermo, J. A. J. Nat. Prod. 1998, 61, 1130–1132. doi:10.1021/np970493u |
| 28. | Seldes, A. M.; Brasco, M. F. R.; Hernández Franco, L.; Palermo, J. A. Nat. Prod. Res. 2007, 21, 555–563. doi:10.1080/14786410601133517 |
| 29. | Sperry, J. Tetrahedron Lett. 2011, 52, 4537–4538. doi:10.1016/j.tetlet.2011.06.073 |
| 21. | Bartik, K.; Braekman, J.-C.; Daloze, D.; Stoller, C.; Huysecom, J.; Vandevyver, G.; Ottinger, R. Can. J. Chem. 1987, 65, 2118–2121. doi:10.1139/v87-352 |
| 22. | Tsuji, S.; Rinehart, K. L.; Gunasekera, S. P.; Kashman, Y.; Cross, S. S.; Lui, M. S.; Pomponi, S. A.; Diaz, M. C. J. Org. Chem. 1988, 53, 5446–5453. doi:10.1021/jo00258a009 |
| 23. | Kawasaki, I.; Katsuma, H.; Nakayama, Y.; Yamashita, M.; Otha, S. Heterocycles 1998, 48, 1887–1901. doi:10.3987/COM-98-8225 |
| 19. | Kawasaki, Y.; Yamashita, M.; Ohta, S. J. Chem. Soc., Chem. Commun. 1994, 2085–2086. doi:10.1039/C39940002085 |
| 20. | Kawasaki, Y.; Yamashita, M.; Ohta, S. Chem. Pharm. Bull. 1996, 44, 1831–1839. doi:10.1248/cpb.44.1831 |
| 26. | Kohmoto, S.; Kashman, Y.; McConnell, O. J.; Rinehart, K. L., Jr.; Wright, A.; Koehn, F. J. Org. Chem. 1988, 53, 3116–3118. doi:10.1021/jo00248a040 |
| 49. | Pommier, Y.; MacDonald, T. L.; Madalengoitia, J. S. U.S. Patent 5,622,960, April 22, 1997. |
| 50. | Daugan, C. M.; Labaudiniere, R. F. Tetrahydroimidazopyridoindolediones and their use as medicaments. WO/1996/032003, Oct 17, 1996. |
| 47. | Behforouz, M.; Merriman, R. L. Methods of using lavendamycin analogs. U.S. Patent 5,646,150, July 8, 1997. |
| 48. | Batch, A.; Dodd, R. H. J. Org. Chem. 1998, 63, 872–877. doi:10.1021/jo971485l |
| 63. | Bredereck, H.; Effenberger, F.; Botsch, H.; Rehn, H. Chem. Ber. 1965, 98, 1081–1806. doi:10.1002/cber.19650980412 |
| 64. | Fresneda, P. M.; Molina, P.; Delgado, S.; Bleda, J. A. Tetrahedron Lett. 2000, 41, 4777–4780. doi:10.1016/S0040-4039(00)00728-0 |
| 65. | Karpov, A. S.; Merkul, E.; Rominger, F.; Müller, T. J. J. Angew. Chem., Int. Ed. 2005, 44, 6951–6956. doi:10.1002/anie.200501703 |
| 66. | Rossignol, E.; Youssef, A.; Moreau, P.; Prudhomme, M.; Anizon, F. Tetrahedron 2007, 63, 10169–10176. doi:10.1016/j.tet.2007.07.095 |
| 61. | Katritzky, A. R.; Zhang, Y.; Singh, S. K. Synthesis 2003, 2795–2798. doi:10.1055/s-2003-42462 |
| 62. | Katritzky, A. R.; Abdel-Fattah, A. A. A.; Gromova, A. V.; Witek, R.; Steel, P. J. J. Org. Chem. 2005, 70, 9211–9214. doi:10.1021/jo051231x |
| 51. | Kundu, B.; Sawant, D.; Chhabra, R. A. J. Comb. Chem. 2005, 7, 317–321. doi:10.1021/cc049851j |
| 52. | Kundu, B.; Sawant, D.; Partani, P.; Kesarwani, A. P. J. Org. Chem. 2005, 70, 4889–4892. doi:10.1021/jo050384h |
| 53. | Duggineni, S.; Sawant, D.; Saha, B.; Kundu, B. Tetrahedron 2006, 62, 3228–3241. doi:10.1016/j.tet.2006.01.063 |
| 54. | Sharma, S.; Saha, B.; Sawant, D.; Kundu, B. J. Comb. Chem. 2007, 9, 783–792. doi:10.1021/cc0700445 |
| 55. | Saha, B.; Sharma, S.; Sawant, D.; Kundu, B. Tetrahedron 2008, 64, 8676–8684. doi:10.1016/j.tet.2008.07.003 |
| 56. | Agarwal, P. K.; Sharma, S. K.; Sawant, D.; Kundu, B. Tetrahedron 2009, 65, 1153–1161. doi:10.1016/j.tet.2008.11.067 |
| 57. | Agarwal, P. K.; Saifuddin, M.; Kundu, B. Tetrahedron 2010, 66, 862–870. doi:10.1016/j.tet.2009.11.115 |
| 58. | Mandadapu, A. K.; Saifuddin, M.; Agarwal, P. K.; Kundu, B. Org. Biomol. Chem. 2009, 7, 2796–2803. doi:10.1039/b905696c |
| 59. | Kundu, B.; Partani, P.; Duggineni, S.; Sawant, D. J. Comb. Chem. 2005, 7, 909–915. doi:10.1021/cc0500699 |
| 60. | Sharma, S.; Kundu, B. J. Comb. Chem. 2009, 11, 720–731. doi:10.1021/cc9000345 |
| 56. | Agarwal, P. K.; Sharma, S. K.; Sawant, D.; Kundu, B. Tetrahedron 2009, 65, 1153–1161. doi:10.1016/j.tet.2008.11.067 |
© 2012 Agarwal et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)