Abstract
We have designed a new sequential carbocupration and sulfur–lithium exchange that leads stereo- and regioselectively to trisubstituted alkenyllithiums. Subsequent trapping with various electrophiles yields tetrasubstituted olefins with good control of the double-bond geometry (E/Z ratio up to 99:1). The novel sulfur–lithium exchange could be extended to the stereoselective preparation of Z-styryl lithium derivatives with almost complete retention of the double-bond geometry.

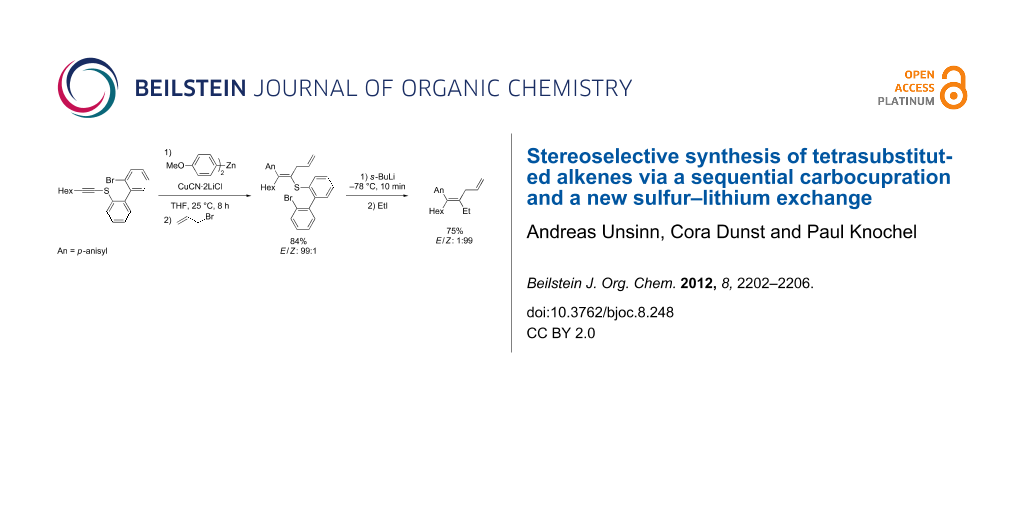
Graphical Abstract
Introduction
The stereoselective synthesis of tetrasubstituted alkenes is an important synthetic goal, which may be achieved by carbometalation methods [1-9]. The Normant carbocupration of terminal acetylenes allows the stereoselective preparation of trisubstituted alkenes with excellent E/Z ratio [10-12]. However, in order to obtain tetrasubstituted alkenes, a carbometalation of an internal alkene is required. This reaction is usually difficult due to steric hindrance and proceeds only if electron-withdrawing groups are attached to the alkyne unit to facilitate the carbometalation step. Recently, we studied the chemistry of alkenyl sulfides and their use for carbometalation extensively [13].
Therefore, we envisioned using an alkynyl thioether such as 1 as an activated alkyne. After a carbocupration of the alkynyl thioether 1 with the organozinc reagent 2 in the presence of CuCN·2LiCl [14], the alkenylcopper species 3 should be obtained. Stereoselective quenching with an electrophile (E1) should afford the tetrasubstituted alkenyl thioether 4. Extensive experimentation showed that thioethers 4 do not undergo Ni- or Pd-catalyzed cross couplings leading to products of type 5 (R = Me, Ph) [15,16]. Thus, we designed a new sulfur–lithium exchange (Scheme 1).
![[1860-5397-8-248-i1]](/bjoc/content/inline/1860-5397-8-248-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of tetrasubstituted olefins by a successive carbocupration and S–Li exchange.
Scheme 1: Synthesis of tetrasubstituted olefins by a successive carbocupration and S–Li exchange.
Sulfur–lithium exchanges proceed only readily with sulfoxides [17-19] and these reactions are often complicated by radical side reactions [20,21]. This new, direct sulfur–lithium exchange on an alkenyl thioether of type 4 involves the use of a bromobiphenyl R-group, which by treatment with BuLi at low temperatures, undergoes first a fast bromine–lithium exchange leading to an intermediate biphenyllithium derivative of type 6, followed by an intramolecular ring-closing sulfur–lithium exchange [22] leading to the desired alkenyllithium 7 (Scheme 2).
![[1860-5397-8-248-i2]](/bjoc/content/inline/1860-5397-8-248-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed mechanism of the sulfur–lithium exchange starting with the alkenyl thioether 4.
Scheme 2: Proposed mechanism of the sulfur–lithium exchange starting with the alkenyl thioether 4.
Subsequent quenching with a different electrophile E2 should afford the tetrasubstituted alkene of type 5; (Scheme 1). Herein, we demonstrate the feasibility of this methodology and thus prepare tetrasubstituted alkenes with E/Z stereoselectivities up to 99:1. Furthermore, we show that this sulfur–lithium exchange can be extended to the stereoselective preparation of Z-styryl derivatives.
Results and Discussion
First, we wish to report the synthesis of the alkynyl biphenyl thioether 1a required for the carbometalation step. Thus, octyne was deprotonated with butyllithium (1.1 equiv, THF, −78 °C, 2 h) followed by the addition of the diaryl disulfide [23] (8: 1.1 equiv, −78 °C to 25 °C, 3 h) providing the bromothioether 9 in 77% yield. Direct Pd-catalyzed Negishi cross-coupling [24-28] of 9 with an arylzinc derivative failed. However, the bromide 9 could be readily converted to the corresponding iodide 10 by a bromine–magnesium exchange using iPrMgCl·LiCl [29-35] followed by iodolysis leading to the iodide 10 in 93% yield. Treatment of 1,2-dibromobenzene with iPrMgCl·LiCl at −15 °C for 2 h followed by a transmetalation with ZnCl2 gives the required zinc reagent 11, which undergoes a Negishi cross-coupling with the iodide 10 at 50 °C (5 h) leading to the alkynyl thioether 1a in 80% yield (Scheme 3).
![[1860-5397-8-248-i3]](/bjoc/content/inline/1860-5397-8-248-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
The harsh cross-coupling conditions may be due booth to the presence of the ortho-bromo substitution in the zinc reagent 11, which considerably reduces the nucleophilicity of this arylzinc reagent by inductive effects, and also to the sulfur atom of the electrophile, which poisons the Pd catalyst. With the thioether 1a in hand, we have performed the Normant carbocupration with di-para-anisylzinc (An2Zn: 2a) according to a procedure previously developed by us [36]. Thus, the reaction of 1a (1.0 equiv) with An2Zn (1.5 equiv, THF) in the presence of CuCN·2LiCl (1.5 equiv) at 25 °C for 8 h produces the intermediate copper reagent 3a, which, after allylation with allyl bromide, provides the thioether 4a in 84% yield and an E/Z ratio of 99:1 (Scheme 4). The reaction of 3a with other typical electrophiles is possible, but proceeds in moderate yields due to the low reactivity of copper reagent 3a.
![[1860-5397-8-248-i4]](/bjoc/content/inline/1860-5397-8-248-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Carbocupration of the thioether 1a leading to the tetrasubstituted alkene 4a.
Scheme 4: Carbocupration of the thioether 1a leading to the tetrasubstituted alkene 4a.
The bromothioether 4a was then treated with s-BuLi (1.3 equiv, −78 °C, 10 min), leading to the formation of the intermediate aryllithium 6a, which undergoes the desired intramolecular sulfur–lithium exchange affording the alkenyllithium reagent 7a (Scheme 5).
![[1860-5397-8-248-i5]](/bjoc/content/inline/1860-5397-8-248-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of the alkenyllithium reagent 7a by an S–Li exchange.
Scheme 5: Synthesis of the alkenyllithium reagent 7a by an S–Li exchange.
This alkenyllithium was quenched with typical electrophiles with a high retention of the double-bond geometry. Thus, the treatment of 7a with EtI (2 equiv, −78 °C, 15 min) provides the tetrasubstituted alkene 5a in 75% yield and an E/Z ratio of 1:99. Direct carboxylation by the reaction with ethyl chloroformate (1.1 equiv, −78 °C, 15 min) furnishes the corresponding unsaturated ethylester 5b in 55% isolated yield and an E/Z ratio of 95:5. Finally, a copper-catalyzed allylation with ethyl 2-(bromomethyl)acrylate [37] (1.5 equiv, −78 to 0 °C, 2 h) affords the triene 5c in 55% yield and an E/Z ratio of 99:1 (Scheme 6).
![[1860-5397-8-248-i6]](/bjoc/content/inline/1860-5397-8-248-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Quenching of the alkynyllithium 7a. (Product ratios and diastereoselectivities were determined by 1H- and 2D-NMR.)
Scheme 6: Quenching of the alkynyllithium 7a. (Product ratios and diastereoselectivities were determined by 1...
These quenching experiments demonstrate that this new method based on a successive carbocupration and sulfur–lithium exchange allows the stereoselective preparation of various tetrasubstituted alkenes. Since Normant has shown that various alkylcopper species add to alkynyl thioethers [38-40], the use of a bromobiphenyl substituent (R2) on the sulfur may allow a general stereoselective synthesis of tetra-substituted alkenes.
In order to prove that this new sulfur–lithium exchange has further applications in the stereoselective synthesis of alkenes, we prepared the Z-alkenyl thioether 12 starting from 2,2’-dibromobiphenyl. Thus, the performance of a double bromine–lithium exchange with BuLi (1.1 equiv, −78 °C, 0.25 h) followed by a quenching with tetramethylthiuram disulfide (1.1 equiv, −78 to 25 °C, 12 h) furnishes the dithiocarbamate 13 in 82% yield. Since the reduction to the free thiol is hard to achieve due to dibenzothiophene formation [41], we performed an in situ deprotection and stereoselective addition to phenylacetylene [42] (1.5 equiv, 1.25 equiv NaOEt, EtOH, reflux, 15 h) yielding the Z-alkenyl thioether 12 in 74% yield (Scheme 7).
![[1860-5397-8-248-i7]](/bjoc/content/inline/1860-5397-8-248-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis and quenching of Z-styryllithium.
Scheme 7: Synthesis and quenching of Z-styryllithium.
Treatment of 12 with t-BuLi (1.6 equiv, −78 °C, 10 min) provides directly the Z-styryllithium 14, which stereoselectively adds to α,α,α-trifluoroacetophenone (0.8 equiv, −78 °C, 0.5 h) and cyclopentanone (0.8 equiv, −78 °C, 0.5 h) to afford the expected tertiary allylic alcohols 15a–b in 71–82% yield and E/Z ratios of >1:99.
Conclusion
In summary, we have reported tetrasubstituted olefins with excellent E/Z ratios using a sequential carbocupration and a new sulfur–lithium exchange involving an alkenyl thioether bearing a 2’-bromobiphenyl substituent, which triggers efficiently the sulfur–lithium exchange. Extension to the stereoselective preparation of Z-styryllithium was shown.
Supporting Information
| Supporting Information File 1: Experimental details and characterization data of new compounds. | ||
| Format: PDF | Size: 286.3 KB | Download |
References
-
Itami, K.; Kamei, T.; Yoshida, J. J. Am. Chem. Soc. 2003, 125, 14670. doi:10.1021/ja037566i
Return to citation in text: [1] -
Das, J. P.; Chechik, H.; Marek, I. Nat. Chem. 2009, 1, 128. doi:10.1038/nchem.131
Return to citation in text: [1] -
Zhou, C.; Larock, R. C. Org. Lett. 2005, 7, 259. doi:10.1021/ol047759q
Return to citation in text: [1] -
Alonso, F.; Beletskaya, I. P.; Yus, M. Chem. Rev. 2004, 104, 3079. doi:10.1021/cr0201068
Return to citation in text: [1] -
Shirakawa, E.; Ikeda, D.; Masui, S.; Yoshida, M.; Hayashi, T. J. Am. Chem. Soc. 2012, 134, 272. doi:10.1021/ja206745w
Return to citation in text: [1] -
Dutta, B.; Gilboa, N.; Marek, I. J. Am. Chem. Soc. 2010, 132, 5588. doi:10.1021/ja101371x
Return to citation in text: [1] -
Flynn, A. B.; Ogilvie, W. W. Chem. Rev. 2007, 107, 4698. doi:10.1021/cr050051k
Return to citation in text: [1] -
Basheer, A.; Marek, I. Beilstein J. Org. Chem. 2010, 6, No. 77. doi:10.3762/bjoc.6.77
Return to citation in text: [1] -
Achyutha Rao, S.; Knochel, P. J. Am. Chem. Soc. 1991, 113, 5735. doi:10.1021/ja00015a030
Return to citation in text: [1] -
Normant, J. F.; Bourgain, M. Tetrahedron Lett. 1971, 12, 2583. doi:10.1016/S0040-4039(01)96925-4
Return to citation in text: [1] -
Normant, J. F.; Alexakis, A. Synthesis 1981, 841. doi:10.1055/s-1981-29622
Return to citation in text: [1] -
Knochel, P. Carbometallation of Alkenes and Alkynes. In Comprehensive Organic Syntheses: Selectivity, Strategy and Efficiency. In Modern Organic Chemistry; Trost, B. M.; Fleming, I.; Semmelhack, M. F., Eds.; Pergamon Press: Oxford, U.K., 1992; Vol. 4, pp 865–911.
Return to citation in text: [1] -
Dunst, C. Functionalization of Arenes and Heteroarenes by Metalation with TMP-Bases or Metal Insertion and Synthesis of Tetrasubstituted Alkenyl Sulfides via a Cu(I)-Mediated Carbometalation. Ph.D. Thesis, Ludwig-Maximilians-Universität München, Munich, Germany, 2011.
Return to citation in text: [1] -
Knochel, P.; Yeh, M. C. P.; Berk, S. C.; Talbert, J. J. Org. Chem. 1988, 53, 2390. doi:10.1021/jo00245a057
Return to citation in text: [1] -
Metzger, A.; Melzig, L.; Despotopoulou, C.; Knochel, P. Org. Lett. 2009, 11, 4228. doi:10.1021/ol9017003
Return to citation in text: [1] -
Melzig, L.; Metzger, A.; Knochel, P. J. Org. Chem. 2010, 75, 2131. doi:10.1021/jo1001615
Return to citation in text: [1] -
Satoh, T.; Takano, K.; Someya, H.; Matsuda, K. Tetrahedron Lett. 1995, 36, 7097. doi:10.1016/0040-4039(95)01435-K
Return to citation in text: [1] -
Satoh, T.; Takano, K.; Ota, H.; Someya, H.; Matsuda, K.; Koyama, M. Tetrahedron 1998, 54, 5557. doi:10.1016/S0040-4020(98)00243-9
Return to citation in text: [1] -
Satoh, T. Chem. Soc. Rev. 2007, 36, 1561. doi:10.1039/b615630b
Return to citation in text: [1] -
Rauhut, C. B.; Melzig, L.; Knochel, P. Org. Lett. 2008, 10, 3891. doi:10.1021/ol801431z
Return to citation in text: [1] -
Melzig, L.; Rauhut, C. B.; Knochel, P. Chem. Commun. 2009, 3536. doi:10.1039/b907330b
Return to citation in text: [1] -
Stoll, A. H.; Krasovskiy, A.; Knochel, P. Angew. Chem. 2006, 118, 621. doi:10.1002/ange.200501882
Angew. Chem. Int. Ed. 2006, 45, 606. doi:10.1002/anie.200501882
Return to citation in text: [1] -
Korn, T. J.; Knochel, P. Synlett 2005, 1185. doi:10.1055/s-2005-865230
Return to citation in text: [1] -
Negishi, E.; King, A. O.; Okukado, N. J. Org. Chem. 1977, 42, 1821. doi:10.1021/jo00430a041
Return to citation in text: [1] -
Negishi, E.; Okukado, N.; King, A. O.; Van Horn, D. E.; Spiegel, B. I. J. Am. Chem. Soc. 1978, 100, 2254. doi:10.1021/ja00475a059
Return to citation in text: [1] -
Negishi, E.; Takahashi, T.; Baba, S.; Van Horn, D. E.; Okukado, N. J. Am. Chem. Soc. 1987, 109, 2393. doi:10.1021/ja00242a024
Return to citation in text: [1] -
Negishi, E. Acc. Chem. Res. 1982, 15, 340. doi:10.1021/ar00083a001
Return to citation in text: [1] -
Negishi, E. Angew. Chem. 2011, 123, 6870. doi:10.1002/ange.201101380
Angew. Chem., Int. Ed. 2011, 50, 6738. doi:10.1002/anie.201101380
Return to citation in text: [1] -
Krasovskiy, A.; Knochel, P. Angew. Chem. 2004, 116, 3396. doi:10.1002/ange.200454084
Angew. Chem., Int. Ed. 2004, 43, 3333. doi:10.1002/anie.200454084
Return to citation in text: [1] -
Sämann, C.; Haag, B.; Knochel, P. Chem.–Eur. J. 2012, 18, 16145. doi:10.1002/chem.201202230
Return to citation in text: [1] -
Rauhut, C. B.; Knochel, P.; Vu, V. A.; Fleming, F. F. Org. Lett. 2008, 10, 1187. doi:10.1021/ol8000987
Return to citation in text: [1] -
Shi, L.; Chu, Y.; Knochel, P.; Mayr, H. Angew. Chem. 2008, 120, 208. doi:10.1002/ange.200704100
Angew. Chem. Int. Ed. 2008, 47, 202; doi:10.1002/anie.200704100
Return to citation in text: [1] -
Kopp, F.; Knochel, P. Org. Lett. 2007, 9, 1639. doi:10.1021/ol063136w
Return to citation in text: [1] -
Kopp, F.; Wunderlich, S.; Knochel, P. Chem. Commun. 2007, 2075. doi:10.1039/b618923g
Return to citation in text: [1] -
Ila, H.; Baron, O.; Wagner, A. J.; Knochel, P. Chem. Lett. 2006, 35, 2. doi:10.1246/cl.2006.2
Return to citation in text: [1] -
Dunst, C.; Metzger, A.; Zaburdaeva, E. A.; Knochel, P. Synthesis 2011, 3453. doi:10.1055/s-0030-1260210
Return to citation in text: [1] -
Villieras, J.; Rambaud, M. Synthesis 1982, 924. doi:10.1055/s-1982-29998
Return to citation in text: [1] -
Masure, D.; Coutrot, P.; Normant, J. F. J. Organomet. Chem. 1982, 226, C55. doi:10.1016/S0022-328X(00)83415-4
Return to citation in text: [1] -
Alexakis, A.; Cahiez, G.; Normant, J. F. Tetrahedron 1980, 36, 1961. doi:10.1016/0040-4020(80)80209-2
Return to citation in text: [1] -
Normant, J. F.; Quirion, J. C.; Alexakis, A.; Masuda, Y. Tetrahedron Lett. 1989, 30, 3955. doi:10.1016/S0040-4039(00)99293-1
Return to citation in text: [1] -
Kienle, M.; Unsinn, A.; Knochel, P. Angew. Chem. 2010, 122, 4860. doi:10.1002/ange.201001025
Angew. Chem. Int. Ed. 2010, 49, 4751. doi:10.1002/anie.201001025
Return to citation in text: [1] -
Truce, W. E.; Simms, J. A. J. Am. Chem. Soc. 1956, 78, 2756. doi:10.1021/ja01593a029
Return to citation in text: [1]
| 1. | Itami, K.; Kamei, T.; Yoshida, J. J. Am. Chem. Soc. 2003, 125, 14670. doi:10.1021/ja037566i |
| 2. | Das, J. P.; Chechik, H.; Marek, I. Nat. Chem. 2009, 1, 128. doi:10.1038/nchem.131 |
| 3. | Zhou, C.; Larock, R. C. Org. Lett. 2005, 7, 259. doi:10.1021/ol047759q |
| 4. | Alonso, F.; Beletskaya, I. P.; Yus, M. Chem. Rev. 2004, 104, 3079. doi:10.1021/cr0201068 |
| 5. | Shirakawa, E.; Ikeda, D.; Masui, S.; Yoshida, M.; Hayashi, T. J. Am. Chem. Soc. 2012, 134, 272. doi:10.1021/ja206745w |
| 6. | Dutta, B.; Gilboa, N.; Marek, I. J. Am. Chem. Soc. 2010, 132, 5588. doi:10.1021/ja101371x |
| 7. | Flynn, A. B.; Ogilvie, W. W. Chem. Rev. 2007, 107, 4698. doi:10.1021/cr050051k |
| 8. | Basheer, A.; Marek, I. Beilstein J. Org. Chem. 2010, 6, No. 77. doi:10.3762/bjoc.6.77 |
| 9. | Achyutha Rao, S.; Knochel, P. J. Am. Chem. Soc. 1991, 113, 5735. doi:10.1021/ja00015a030 |
| 15. | Metzger, A.; Melzig, L.; Despotopoulou, C.; Knochel, P. Org. Lett. 2009, 11, 4228. doi:10.1021/ol9017003 |
| 16. | Melzig, L.; Metzger, A.; Knochel, P. J. Org. Chem. 2010, 75, 2131. doi:10.1021/jo1001615 |
| 41. |
Kienle, M.; Unsinn, A.; Knochel, P. Angew. Chem. 2010, 122, 4860. doi:10.1002/ange.201001025
Angew. Chem. Int. Ed. 2010, 49, 4751. doi:10.1002/anie.201001025 |
| 14. | Knochel, P.; Yeh, M. C. P.; Berk, S. C.; Talbert, J. J. Org. Chem. 1988, 53, 2390. doi:10.1021/jo00245a057 |
| 42. | Truce, W. E.; Simms, J. A. J. Am. Chem. Soc. 1956, 78, 2756. doi:10.1021/ja01593a029 |
| 13. | Dunst, C. Functionalization of Arenes and Heteroarenes by Metalation with TMP-Bases or Metal Insertion and Synthesis of Tetrasubstituted Alkenyl Sulfides via a Cu(I)-Mediated Carbometalation. Ph.D. Thesis, Ludwig-Maximilians-Universität München, Munich, Germany, 2011. |
| 10. | Normant, J. F.; Bourgain, M. Tetrahedron Lett. 1971, 12, 2583. doi:10.1016/S0040-4039(01)96925-4 |
| 11. | Normant, J. F.; Alexakis, A. Synthesis 1981, 841. doi:10.1055/s-1981-29622 |
| 12. | Knochel, P. Carbometallation of Alkenes and Alkynes. In Comprehensive Organic Syntheses: Selectivity, Strategy and Efficiency. In Modern Organic Chemistry; Trost, B. M.; Fleming, I.; Semmelhack, M. F., Eds.; Pergamon Press: Oxford, U.K., 1992; Vol. 4, pp 865–911. |
| 38. | Masure, D.; Coutrot, P.; Normant, J. F. J. Organomet. Chem. 1982, 226, C55. doi:10.1016/S0022-328X(00)83415-4 |
| 39. | Alexakis, A.; Cahiez, G.; Normant, J. F. Tetrahedron 1980, 36, 1961. doi:10.1016/0040-4020(80)80209-2 |
| 40. | Normant, J. F.; Quirion, J. C.; Alexakis, A.; Masuda, Y. Tetrahedron Lett. 1989, 30, 3955. doi:10.1016/S0040-4039(00)99293-1 |
| 29. |
Krasovskiy, A.; Knochel, P. Angew. Chem. 2004, 116, 3396. doi:10.1002/ange.200454084
Angew. Chem., Int. Ed. 2004, 43, 3333. doi:10.1002/anie.200454084 |
| 30. | Sämann, C.; Haag, B.; Knochel, P. Chem.–Eur. J. 2012, 18, 16145. doi:10.1002/chem.201202230 |
| 31. | Rauhut, C. B.; Knochel, P.; Vu, V. A.; Fleming, F. F. Org. Lett. 2008, 10, 1187. doi:10.1021/ol8000987 |
| 32. |
Shi, L.; Chu, Y.; Knochel, P.; Mayr, H. Angew. Chem. 2008, 120, 208. doi:10.1002/ange.200704100
Angew. Chem. Int. Ed. 2008, 47, 202; doi:10.1002/anie.200704100 |
| 33. | Kopp, F.; Knochel, P. Org. Lett. 2007, 9, 1639. doi:10.1021/ol063136w |
| 34. | Kopp, F.; Wunderlich, S.; Knochel, P. Chem. Commun. 2007, 2075. doi:10.1039/b618923g |
| 35. | Ila, H.; Baron, O.; Wagner, A. J.; Knochel, P. Chem. Lett. 2006, 35, 2. doi:10.1246/cl.2006.2 |
| 22. |
Stoll, A. H.; Krasovskiy, A.; Knochel, P. Angew. Chem. 2006, 118, 621. doi:10.1002/ange.200501882
Angew. Chem. Int. Ed. 2006, 45, 606. doi:10.1002/anie.200501882 |
| 36. | Dunst, C.; Metzger, A.; Zaburdaeva, E. A.; Knochel, P. Synthesis 2011, 3453. doi:10.1055/s-0030-1260210 |
| 20. | Rauhut, C. B.; Melzig, L.; Knochel, P. Org. Lett. 2008, 10, 3891. doi:10.1021/ol801431z |
| 21. | Melzig, L.; Rauhut, C. B.; Knochel, P. Chem. Commun. 2009, 3536. doi:10.1039/b907330b |
| 17. | Satoh, T.; Takano, K.; Someya, H.; Matsuda, K. Tetrahedron Lett. 1995, 36, 7097. doi:10.1016/0040-4039(95)01435-K |
| 18. | Satoh, T.; Takano, K.; Ota, H.; Someya, H.; Matsuda, K.; Koyama, M. Tetrahedron 1998, 54, 5557. doi:10.1016/S0040-4020(98)00243-9 |
| 19. | Satoh, T. Chem. Soc. Rev. 2007, 36, 1561. doi:10.1039/b615630b |
| 24. | Negishi, E.; King, A. O.; Okukado, N. J. Org. Chem. 1977, 42, 1821. doi:10.1021/jo00430a041 |
| 25. | Negishi, E.; Okukado, N.; King, A. O.; Van Horn, D. E.; Spiegel, B. I. J. Am. Chem. Soc. 1978, 100, 2254. doi:10.1021/ja00475a059 |
| 26. | Negishi, E.; Takahashi, T.; Baba, S.; Van Horn, D. E.; Okukado, N. J. Am. Chem. Soc. 1987, 109, 2393. doi:10.1021/ja00242a024 |
| 27. | Negishi, E. Acc. Chem. Res. 1982, 15, 340. doi:10.1021/ar00083a001 |
| 28. |
Negishi, E. Angew. Chem. 2011, 123, 6870. doi:10.1002/ange.201101380
Angew. Chem., Int. Ed. 2011, 50, 6738. doi:10.1002/anie.201101380 |
© 2012 Unsinn et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)