Abstract
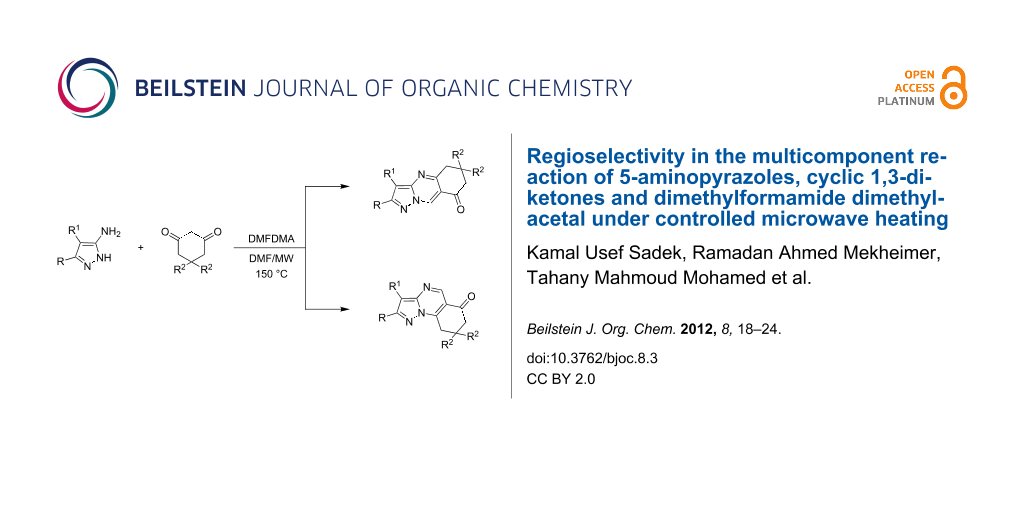
The multicomponent reaction of 5-aminopyrazole derivatives with cyclic 1,3-dicarbonyl compounds and dimethylformamide dimethylacetal (DMFDMA) in DMF at 150 °C under controlled microwave heating afforded regioselectively 8,9-dihydropyrazolo[1,5-a]quinazolin-6(7H)-ones 6 rather than the corresponding dihydropyrazolo[5,1-b]quinazolin-8(5H)-ones 4.

Graphical Abstract
Introduction
Several naturally occurring and synthetic compounds containing quinazoline derivatives are of considerable interest in fields related to the organic and medicinal chemistry of natural products [1,2]. The quinazoline ring system represents the core skeleton of an important class of heterocyclic compounds possessing a wide range of biological activities [3,4]. Multicomponent reactions (MCR) occupy an interesting position in organic synthesis because of their atom economy, simple procedures and convergent character [5-7]. An unresolved issue in multicomponent reactions is whether their selectivity is chemo- or regioselectivity, or both, due to the several possible parallel reaction pathways, which result in the formation of different products [8-10]. Many factors modulate the selectivity of synthetic transformations, such as temperature, pressure, solvent, catalyst and type of reaction control, i.e., either kinetic or thermodynamic [11-13]. It has been reported that the use of microwave or ultrasound irradiation provides an additional parameter for synthetic selectivity [14-17].
Results and Discussion
The multicomponent reaction of 5-aminopyrazoles, dimedone and aromatic aldehydes was reported to afford several different tricyclic products. Thus, in an early report [18], the reaction of the three components in ethanol under conventional heating afforded mainly the corresponding pyrazolo[3,4-b]quinolin-5-ones. This finding was later supported by other authors [19]. Recently, the results of an interesting study dealing with such reactions were described by Chebanov et al. [20] Specifically, these researchers performed the reaction at 150 °C in the presence of triethylamine by employing a sealed vessel under microwave or conventional heating, and which thus afforded pyrazoloquinolinones (Hantzsch-type dihydropyridines). On the other hand, the use of sonication at room temperature under neutral conditions favours the formation of isomeric pyrazolo[5,1-b]quinazolin-8(4H)-ones (Biginelli-type dihydro-pyrimidines) [9]. Employing more nucleophilic bases to catalyse the reaction afforded the corresponding pyrazolo[4,3-c]quinazolin-9-ones [20]. It was concluded that, under ambient and neutral conditions, the reaction proceeds under kinetic control, and the Biginelli-type dihydropyrimidines are the predominant isomers. Increasing the reaction temperature in the presence of triethylamine as base produces the more thermodynamically stable dihydropyridine (Hantzsch-type product). In addition, the nature of the catalyst plays an important role [20]. A one-pot three component reaction of 5-amino-1H-pyrazole-4-carbonitrile, dimedone and triethylorthoesters in toluene under reflux was recently reported to afford the corresponding pyrazolo[1,5-a]-quinazolin-6-one derivatives [21]. Although it is well established that 5-amino-pyrazoles have nonequivalent nucleophilic reaction centres in the aminopyrazole scaffold (N1, C4, NH2), which can lead to the formation of several different tricyclic reaction products, no general basis on which to determine the preferred tautomeric form of the final product has been established.
In continuation of our studies in which we performed multicomponent reactions using controlled microwave heating [22-24], we report herein the results of our investigation concerning the regioselectivity in multicomponent reactions of 5-aminopyrazoles, cyclic 1,3-diketones and dimethylformamide dimethylacetal (DMFDMA) under controlled microwave heating.
We began this study by treating 5-amino-3-methylpyrazole (1a) and dimedone (2a) with DMFDMA (3) in DMF under microwave heating at 150 °C for 15 min. After being cooled to room temperature, the precipitated solid product was isolated in 88% yield (Table 1). The mass spectrum of the reaction product showed a molecular ion peak m/z = 229.12 (100%). The 1H NMR revealed a singlet signal at δ = 6.70 ppm integrated for one proton, which was assigned to the pyrazoloquinazolone C3 proton, and which indicates the lack of involvement of such a proton in the condensation leading to the tricyclic system. Although, it was previously reported [20] that, due to reduced steric hindrance, the multicomponent reaction of 5-amino-3-methyl-pyrazole, aromatic aldehydes and dimedone under controlled microwave irradiation at 150 °C involves the participation of C3-H of the pyrazole ring in such a cyclocondensation reaction, this is not favoured in our case. In addition two signals were assigned to two CH2 groups and three methyl functions, and a singlet at δ = 8.75 ppm corresponding to one proton at C5. The pyrazolo[1,5-a]-quinazolin-8(5H)-one 6a was established as the reaction product, and 13C NMR was in agreement with the proposed structure, rather than with isomeric 4a, which was prepared by first reacting 1a with dimedone (2a) in DMF under microwave heating at 150 °C for 10 min to afford 5. Subsequently, treating compound 5 with DMFDMA (3), under the same experimental conditions, gave compound 6a in excellent yield (Scheme 1 and Table 1). Furthermore, the structures of compounds 5 and 6a were unambiguously confirmed by single-crystal X-ray diffraction [25,26] (Figure 1, Figure 2 and Table 1, Table 2, Table 3).
![[1860-5397-8-3-i1]](/bjoc/content/inline/1860-5397-8-3-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Microwave-assisted synthesis of 4 and 6.
Scheme 1: Microwave-assisted synthesis of 4 and 6.
Table 1: Microwave-assisted synthesis of 4 and 6.
| entry | compound | 5-aminopyrazole, 1; | cyclic 1,3-diketone, 2; | product | yield (%) |
|---|---|---|---|---|---|
| 1 | 1a |
R = CH3,
R1 = H |
2a; R2 = CH3 | 6a | 88 |
| 2 | 1a |
R = CH3,
R1 = H |
2b; R2 = H | 6b | 85 |
| 3 | 1b |
R = NH2,
R1 = CO2Et |
2b; R2 = H | 6c | 89 |
| 4 | 1c |
R = CH3,
R1 = C6H5 |
2a; R2 = CH3 | 6d | 83 |
| 5 | 1d |
R = C6H5,
R1 = H |
2b; R2 = H | 6e | 82 |
| 6 | 1e |
R = C6H5,
R1 = H |
2a; R2 = CH3 | 6f | 83 |
| 7 | 1f |
R = OH,
R1 = C6H5N=N– |
2a; R2 = CH3 | 6g | 84 |
![[1860-5397-8-3-1]](/bjoc/content/figures/1860-5397-8-3-1.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-8-3-2]](/bjoc/content/figures/1860-5397-8-3-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Table 2: Selected bond lengths and bond angles for compound 6a.
| bond lengths | bond angles | ||
|---|---|---|---|
| atom numbers | geometric parameter (Å) | atom numbers | geometric parameter (°) |
|
N1–C8
N1–C7 N2–C10 N3–C8 N3–C1 N1–C6 N6–C7 |
1.372 (3)
1.309(3) 1.344 (3) 1.397 (3) 1.490 (3) 1.377 (3) 1.421 (3) |
C7–N1–C8
N2–N3–C1 C1–N3–C8 N3–C1–C6 C8–C9–C10 C1–C6–C5 N1–C7–C6 N1–C8–C9 N3–N2–C10 N2–N3–C8 N1–C8–N3 |
116.15 (19)
125.03 (16) 122.51 (18) 116.10 (17) 106.29 (17) 119.42 (19) 124.5 (3) 133.29 (19) 103.65 (17) 112.41(16) 121.56 (18) |
Table 3: Selected bond lengths and bond angles for compound 6e.
| bond lengths | bond angles | ||
|---|---|---|---|
| atoms numbers | geometric parameter (Å) | atom numbers | geometric parameter (°) |
|
N3–C9
N3–C8 N1–C1 N2–C9 N2–C2 C2–C7 C7–C8 |
1.360 (3)
1.3147(3) 1.346 (3) 1.396 (3) 1.364 (3) 1.363 (3) 1.428 (3) |
C8–N3–C9
N1–N2–C2 N2–C1–C3 N2–C2–C7 C1–C10–C9 C2–C7–C8 N3–C8–C7 N3–C9–C10 N2–N1–C1 N1–N2–C9 N2–C9–N3 |
116.10 (19)
124.94 (19) 124.71 (18) 116.23 (18) 120.9 (17) 124.7 (2) 105.78 (17) 133.37 (19) 103.94 (14) 112.01(15) 120.99 (18) |
With this result in hand, we went on to study the scope of such multicomponent reactions with several substituted 5-aminopyrazoles and cyclic 1,3-diketones. Thus, the reaction of 1b–f with 2a,b and 3, under the same experimental conditions, afforded the corresponding pyrazolo[5,1-b]quinazolin-8(5H)-ones 6b–g, respectively. The structures of 6b–g were deduced from their 1H NMR, 13C NMR, mass spectra and elemental analyses.
Compound 6g was also obtained by an alternative route: Compound 8 was prepared by reacting enaminone 7 with 5-aminopyrazole derivative 1f in DMF under microwave heating at 150 °C for 2 min (Table 1). When this compound was refluxed in DMF under microwave heating for 13 min it underwent cyclization to give 6g (Scheme 1). Moreover, the structure of compounds 6b–g was unequivocally established by single-crystal X-ray diffraction of compounds 6e,g (Figure 3, Figure 4 and Table 3, Table 4) [27,28].
![[1860-5397-8-3-3]](/bjoc/content/figures/1860-5397-8-3-3.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-8-3-4]](/bjoc/content/figures/1860-5397-8-3-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Table 4: Selected bond lengths and bond angles for compound 6g.
| bond lengths | bond angles | ||
|---|---|---|---|
| atoms numbers | geometric parameter (Å) | atom numbers | geometric parameter (°) |
|
N3–C4
N3–C3 N1–C6 N2–C4 N2–C1 C1–C7 C1–C2 |
1.330 (2)
1.321(19) 1.343 (17) 1.393 (18) 1.343 (19) 1.491 (2) 1.394 (2) |
C3–N3–C4
N1–N2–C1 C1–N2–C4 N2–C1–C2 C4–C5–C6 C1–C2–C10 N3–C3–C2 N3–C4–C5 N2–N1–C6 N1–N2–C4 N2–C4–N3 |
116.18 (10)
124.04 (12) 121.41 (12) 116.52 (13) 105.52 (13) 119.58 (13) 123.90 (14) 132.56 (14) 104.27 (11) 114.50(11) 123.02 (13) |
A proposed mechanism to account for the formation of products 6 is illustrated in Scheme 2. The base-catalyzed reaction of cyclic 1,3-diketones 2 with DMFDMA 3 gave the enaminone 7, which subsequently reacted with 5-aminopyrazole 1 at the exocyclic amino function, followed by cyclization through water loss to give 6 (route A). Formation of isomeric product 4, which would be formed by route B, was ruled out based on spectral and X-ray diffraction data.
![[1860-5397-8-3-i2]](/bjoc/content/inline/1860-5397-8-3-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: A proposed mechanism to account for the formation of products 6. The factors that determine the nature of the end product are, however, at present unclear.
Scheme 2: A proposed mechanism to account for the formation of products 6. The factors that determine the nat...
From the data of the X-ray crystal structure it can be concluded that the bridged head nitrogen has bond angles closer to those of sp3 nitrogen. One may thus conclude that the lone pair on this nitrogen atom does not contribute much to the actual state of the molecule and that charge-separated ions also do not contribute significantly; although, the pyrazolo[5,1-b]quinazolin ring is almost planar.
Conclusion
In summary, we can reveal that the reaction of substituted 5-aminopyrazoles, cyclic 1,3-diketones and dimethyformamide dimethylacetal (DMFDMA, 3) proceeds by initial attack of the exocyclic amino function. Although an attack by the ring nitrogen has been proposed for the reaction of 5-aminopyrazoles with acrylonitrile [29], here steric factors hinder such an attack and the reaction occurs exclusively, in every case studied, at the amino function.
Experimental
General information. All the reactions were carried out in a Milestone START Microwave Labstation (temperature control by IR sensor). 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were measured on a Bruker DPX instrument by using DMSO-d6 as solvent and TMS as internal standard. Chemical shifts are expressed as δ in ppm. Coupling constants (J) are given in Hertz (Hz). The melting points were measured in a Gallenkamp melting-point apparatus and are not corrected. Mass spectra were measured by using VG Autospec Q MS 30 and MS 9 (AEI) spectrometer with the EI (70 eV) mode.
General procedure for the synthesis of pyrazoloquinazolinones (6a–g)
A solution of 5-aminopyrazole derivative 1a–f (1 mmol), cyclic 1,3-diketones (2a,b) (1 mmol) and dimethylformamide dimethylacetal (DMFDMA, 3) (1 mmol) in DMF (10 mL) was heated under reflux in a Milestone Microwave Labstation at 150 °C for 15 min. After concentration and cooling to room temperature, the resulting solid product so formed was collected by filtration, washed well with EtOH, dried and recrystallized from EtOH.
2,8,8-Trimethyl-8,9-dihydropyrazolo[5,1-b]quinazolin-6(7H)-one (6a): Greenish yellow plates, 201 mg (88% yield); mp 134–135 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.12 (s, 6H, 2CH3), 2.48 (s, 3H, CH3), 2.56 (s, 2H, CH2 at C-9), 3.32 (s, 2H, CH2 at C-7), 6.70 (s, 1H, CH at C-3), 8.75 (s, 1H, CH at C-5); 13C NMR (100 MHz, DMSO-d6) δ 14.55, 27.89, 32.36, 36.46, 38.87, 50.08, 98.04, 112.39, 146.03, 149.34, 152.21, 157.52, 194.82; EIMS m/z: 229.1 (M+), 214, 173, calcd. for C13H15N3O 229.28; Anal. calcd for C13H15N3O: C, 68.1; H, 6.59; N, 18.33; found: C, 68.22; H, 6.62; N, 18.35%.
2-Methyl-8,9-dihydropyrazolo[5,1-b]quinazolin-6(7H)-one (6b): Yellow plates, 170 mg (85% yield); mp 154–155 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.21–2.27 (m, 2H, CH2 at C-8), 2.66 (t, J = 6.8 Hz, 2H, CH2 at C-9), 3.40 (t, J = 6.4 Hz, 2H, CH2 at C-7), 6.71 (s,1H, CH at C-3), 8.77 (s, 1H, CH at C-5); 13C NMR (100 MHz, DMSO-d6) δ 14.53, 19.95, 33.37, 36.54, 97.91, 113.3, 146.3, 149.0, 153.9, 157.42, 194.81; EIMS m/z 201.12 (M+), calcd for C11H11N3O 201.22; Anal. calcd for C11H11N3O: C, 65.66; H, 5.51; N, 20.88; found: C, 65.68; H, 5.49; N, 20.67%.
Ethyl 2-amino-6-oxo-6,7,8,9-tetrahydropyrazolo[5,1-b]quinazolin-3-carboxylate (6c): Yellow crystals, 243 mg (89% yield); mp 184–185 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.31 (t, J = 7.2 Hz, 3H, CH3), 2.10–2.20 (m, 2H, CH2 at C-8), 2.63 (t, J = 6.8 Hz, 2H, CH2 at C-9), 3.25 (t, J = 6.8 Hz, 2H, CH2 at C-7), 4.31 (q, J = 6.8 Hz, 2H, CH2), 6.7 (br s, 2H, NH2), 8.82 (s, 1H, CH at C-5); EIMS m/z 274.1 (M+), 228, 174.1, calcd for C13H14N4O3 274.28; Anal. calcd for C13H14N4O3: C, 56.93; H, 5.14; 20.43; found: C, 57.12; H, 5.23; N, 20.45%
2,8,8-Trimethyl-3-phenyl-8,9-dihydropyrazolo[5,1-b]quinazolin-6(7H)-one (6d): Pale yellow crystals, 253 mg (83% yield); mp 279–280 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.15 (s, 6H, 2 CH3), 2.49 (s, 2H, CH2 at C-9), 2.58 (s, 3H, CH3 at C-2), 2.63 (s, 2H, CH2 at C-7), 7.13–7.55 (m, 5H, Ph-H), 8.83 (s, 1H, CH at C-5); 13C NMR (100 MHz, DMSO-d6) δ 14.41, 24.42, 27.90, 36.42, 38.87, 50.15, 112.99, 119.22, 125.88, 126.67, 128.30, 129.20, 132.43, 140.64, 144.52, 159.05, 194.70; EIMS m/z 305.2 (M+), 299, 179.1, calcd for C19H19N3O 305.37; Anal. calcd for C19H19N3O: C, 74.73; H, 6.27; N, 13.76; found: C, 74.66; H, 6.35, N, 13.82%.
2-Phenyl-8,9-dihydropyrazolo[1,5-a]quinazolin-6(7H)-one (6e): Pale yellow crystals, 215 mg (82% yield); mp 197–198 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.25 (m, 2H, CH2 at C-8), 2.64 (t, J = 5.6 Hz, 2H, CH2 at C-9), 3.41 (t, J = 5.6 Hz, 2H, CH2 at C-7), 7.39 (br s, 1H, CH at C-3), 7.48 (m, 3H, Ph-H), 8.08 (d, J = 7.2 Hz, 2H, Ph-H), 8.78 (s, 1H, CH at C-5); 13C NMR (100 MHz, DMSO-d6) δ 19.97, 23.46, 36.63, 79.19, 95.49, 114.10, 126.44, 129.0, 129.69, 131.85, 146.77, 149.69, 154.39, 157.60, 162.32, 194.84; EIMS m/z 263.1 (M+), 235.1, 152.1, calcd. for C16H13N3O 263.11; Anal. calcd for C16H13N3O: C, 72.99; H, 4.98; N, 15.96; found: C, 72.94; H, 5.18; N, 16.32%.
8,8-Dimethyl-2-phenyl-8,9-dihydropyrazolo[1,5-a]quinazolin-6(7H)-one (6f): Pale yellow crystals, 242 mg (83% yield); mp 244–245 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.18 (s, 6H, 2 CH3), 2.59 (s, 2H, CH2 at C-9), 3.44 (s, 2H, CH2 at C-7), 7.34 (s, 1H, CH at C-3), 7.50 (m, 3H, Ph-H), 8.09 (m, 2H, Ph-H), 8.81 (s, 1H, CH at C-5); 13C NMR (100 MHz, DMSO-d6) δ 28.47, 32.73, 37.17, 50.86, 95.94, 113.79, 127.02, 129.29, 129.97, 132.53, 146.90, 150.61, 152.87, 158.37, 194.85; Anal. calcd for C18H17N3O: C, 74.20; H, 5.88; N, 14.42; found: C, 74.32; H, 5.91; N, 14.44%.
2-Hydroxy-8,8-dimethyl-3-(phenyldiazenyl)-8,9-dihydropyrazolo[1,5-a]quina-zolin-6(7H)-one (6g): Orange crystals, 295 mg (88% yield); mp 254–255 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.14 (s, 6H, 2 CH3), 2.66 (s, 2H, CH2 at C-9), 3.26 (s, 2H, CH2 at C-7), 7.45 (t, J = 7.2 Hz, 1H, Ph-H), 7.55 (t, J = 7.6 Hz, 2H, Ph-H), 7.85 (d, J = 7.6 Hz, 2H, Ph-H), 8.95 (s, 1H, CH at C-5); 13C NMR (100 MHz, DMSO-d6) δ 27.96, 32.25, 36.44, 50.14, 79.20, 115.14, 115.74, 121.33, 129.34, 129.80, 144.26, 148.99, 151.95, 152.61, 162.10, 194.3; EIMS m/z 335.1 (M+), 307.1, 258.1, calcd for C18H17N5O2 335.14; Anal. calcd for C18H17N5O2: C, 64.47; 5.11; 20.88; found: C, 64.43; 5.33; 20.95%.
Synthesis of (Z)-5,5-dimethyl-3-[(3-methyl-1H-pyrazol-5-yl)amino]cyclohexanone (5)
A solution of 1a (1 mmol) and 2a (1 mmol) in DMF (10 mL) was heated under reflux in a Milestone Microwave Labstation at 150 °C for 10 min. After concentration and cooling to room temperature, the resulting solid product so formed was collected by filtration, washed well with EtOH, dried and recrystallized from EtOH to afford a pure sample of compound 5 as yellow crystals, 186 mg (85% yield); mp 233–235 °C.
Synthesis of 4a: A solution of 1a (1 mmol) and 2a (1 mmol) in DMF (10 mL) was heated under reflux in a Milestone Microwave Labstation at 150 °C for 10 min. After concentration and cooling to room temperature, the resulting solid product so formed was collected by filtration, washed well with EtOH, dried and recrystallized from EtOH to afford a pure sample of (Z)-3,3-dimethyl-5-(3-methyl-1H-pyrazol-5-ylimino)cyclo-hexanone (5) as yellow crystals, 186 mg (85% yield); mp 233–235 °C.
Reaction of 5 with dimethylformamide dimethylacetal (DMFDMA, 3): A solution of 5 (1 mmol) and DMFDMA (3) (1 mmol) in DMF (10 mL) was heated under reflux in a Milestone Microwave Labstation at 150 °C for 10 min. After evaporation to dryness under reduced pressure, the resulting solid product was collected by filtration, washed well with EtOH, dried and recrystallized from EtOH to give 4a.
Alternative synthesis of 6g: Synthesis of 2-((3-hydroxy-4-(phenyldiazenyl)-1H-pyrazol-5-ylamino)methylene)-5,5-dimethylcyclohexane-1,3-dione (8): A solution of 1f (1 mmol), enaminone 7 (1 mmol) in DMF (10 mL) was heated under reflux in a Milestone Microwave Labstation at 150 °C for 2 min. After concentration and cooling to room temperature, the precipitated product was collected by filtration, washed well with EtOH, dried and recrystallized from EtOH to give a pure sample of 8 as orange crystals, 303 mg (88% yield); mp 255–256 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.01 (s, 6H, 2 CH3), 2.40 (s, 2H, CH2), 3.26 (s, 2H, CH2), 7.24–7.85 (m, 6H, 5 Ph-H and CH-NH), 11.76 (s, 1H, NH), 12.59 (s, 1H, pyrazole NH); 13C NMR (100 MHz, DMSO-d6) δ 27.95, 30.70, 50.12, 109.66, 115.16, 115.74, 121.31, 126.16, 129.32, 129.64, 129.80, 144.34, 148.97, 152.57, 158.40, 194.23, 195.33; EIMS m/z 353.2 (M+), 335.1, 242.1, calcd. for C18H19N5O3 353.15; Anal. calcd for C18H19N5O3: C, 61.18; H, 5.42; N, 19.82; found: C, 61.23; H, 5.45; N, 19.92%.
Cyclization of 8. A solution of 8 (1 mmol) in DMF (10 ml) was heated under reflux in a Milestone Microwave Labstation at 150 °C for 13 min. The reaction mixture was evaporated to dryness in vacuo. The precipitated solid product was filtered off, washed with a small amount of EtOH, dried and recrystallized from EtOH to give an analytical pure sample of 6g (identical with an authentic sample, MS, 1H NMR and 13C NMR).
Acknowledgements
K. U. Sadek is grateful to the Alexander von Humboldt Foundation for donation of a Milestone START Microwave Labstation, which was of great help in finishing this work. M. H. Elnagdi and Moustafa Sherief Moustafa are grateful to Kuwait University Research Administration for the financial support of project SC1/10, and the analytical facilities provided by SAF projects No. GS 03/08 (Single crystal X-ray crystallography-Rigaku Rapid II) & GS 01/01 & GS 01/03 & GS 01/05 are greatly appreciated.
References
-
Kessler, M.; Baudry, M.; Lynch, G. Brain Res. 1989, 489, 377. doi:10.1016/0006-8993(89)90875-5
Return to citation in text: [1] -
McQuaid, L. A.; Smith, E. C. R.; South, K. K.; Mitch, C. H.; Schoepp, D. D.; True, R. A.; Calligaro, D. O.; O’Malley, P. J.; Lodge, D.; Ornstein, P. L. J. Med. Chem. 1992, 35, 3319. doi:10.1021/jm00096a002
Return to citation in text: [1] -
Orvieto, F.; Branca, D.; Giomini, C.; Jones, P.; Koch, U.; Ontoria, J. M.; Palumbi, M. C.; Rowley, M.; Toniatti, C.; Muraglia, E. Bioorg. Med. Chem. Lett. 2009, 19, 4196. doi:10.1016/j.bmcl.2009.05.113
Return to citation in text: [1] -
Watson, C. Y.; Whish, W. J. D.; Threadgill, M. D. Bioorg. Med. Chem. 1998, 6, 721. doi:10.1016/S0968-0896(98)00029-7
Return to citation in text: [1] -
Weber, L.; Illgen, K.; Almstetter, M. Synlett 1999, 366. doi:10.1055/s-1999-2612
Return to citation in text: [1] -
Kantevari, S.; Vuppalapati, S. V. N.; Nagarapu, L. Catal. Commun. 2007, 8, 1857. doi:10.1016/j.catcom.2007.02.022
Return to citation in text: [1] -
Sami, S.; Nandi, G. C.; Kumar, R.; Singh, M. S. Tetrahedron Lett. 2009, 50, 7096. doi:10.1016/j.tetlet.2009.10.022
Return to citation in text: [1] -
Zhu, J.; Bienaymé, H., Eds. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005.
Return to citation in text: [1] -
Dömling, A.; Ugi, I. Angew. Chem., Int. Ed. 2000, 39, 3168. doi:10.1002/1521-3773(20000915)39:18<3168::AID-ANIE3168>3.0.CO;2-U
Return to citation in text: [1] [2] -
Simon, C.; Constantieux, T.; Rodriguez, J. Eur. J. Org. Chem. 2004, 4957. doi:10.1002/ejoc.200400511
Return to citation in text: [1] -
Carey, F. A.; Sundberg, R. J. Advanced Organic Chemistry, Part A, 5th ed.; Springer: New York, 2007.
Return to citation in text: [1] -
Erkkilä, A.; Majander, I.; Pihko, P. M. Chem. Rev. 2007, 107, 5416. doi:10.1021/cr068388p
Return to citation in text: [1] -
Laschat, S.; Becheanu, A.; Bell, T.; Baro, A. Synlett 2005, 2547. doi:10.1055/s-2005-918922
Return to citation in text: [1] -
Kappe, C. O. Angew. Chem., Int. Ed. 2004, 43, 6250. doi:10.1002/anie.200400655
Return to citation in text: [1] -
de la Hoz, A.; Díaz-Ortiz, Á.; Moreno, A. Chem. Soc. Rev. 2005, 34, 164. doi:10.1039/b411438h
Return to citation in text: [1] -
Bonrath, W.; Paz Schmidt, R. A. Ultrasound in Synthetic Organic Chemistry. In Advances in Organic Synthesis; Atta-ur-Rahman, Ed.; Bentham Sciences: Oak Park, IL, 2005; Vol. 1, pp 81–117.
Return to citation in text: [1] -
Gravotto, G.; Cintas, P. J. Chem. Soc. Rev. 2006, 35, 180. doi:10.1039/b503848k
Return to citation in text: [1] -
Quiroga, J.; Mejía, D.; Insuasty, B.; Abonía, R.; Nogueras, M.; Sánchez, A.; Cobo, J.; Low, J. N. Tetrahedron 2001, 57, 6947. doi:10.1016/S0040-4020(01)00649-4
Return to citation in text: [1] -
Chebanov, V. A.; Saraev, V. E.; Desenko, S. M.; Chernenko, V. N.; Shishkina, S. V.; Shishkin, O. V.; Kobzar, K. M.; Kappe, C. O. Org. Lett. 2007, 9, 1691. doi:10.1021/ol070411l
Return to citation in text: [1] -
Chebanov, V. A.; Saraev, V. E.; Desenko, S. M.; Chernenko, V. N.; Knyazeva, I. V.; Groth, U.; Glasnov, T. N.; Kappe, C. O. J. Org. Chem. 2008, 73, 5110. doi:10.1021/jo800825c
Return to citation in text: [1] [2] [3] [4] -
Ghotekar, B. K.; Jachak, M. N.; Toche, R. B. J. Heterocycl. Chem. 2009, 46, 708. doi:10.1002/jhet.128
Return to citation in text: [1] -
Sadek, K. U.; Shaker, R. M.; Abd Elrady, M.; Elnagdi, M. H. Tetrahedron Lett. 2010, 51, 6319. doi:10.1016/j.tetlet.2010.09.114
Return to citation in text: [1] -
Abd El Latif, F. M.; Barsy, M. A.; Aref, A. M.; Sadek, K. U. Green Chem. 2002, 4, 196. doi:10.1039/b110723m
Return to citation in text: [1] -
Mekheimer, R. A.; Sadek, K. U. J. Heterocycl. Chem. 2009, 46, 149. doi:10.1002/jhet.13
Return to citation in text: [1] -
CCDC 825123 contains the supplementary crystallographic data for compound 6a. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk
Return to citation in text: [1] -
CCDC 833076 contains the supplementary crystallographic data for compound 5. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk
Return to citation in text: [1] -
CCDC 827653 contains the supplementary crystallographic data for compound 6e. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk
Return to citation in text: [1] -
CCDC 826742 contains the supplementary crystallographic data for compound 6g. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk
Return to citation in text: [1] -
Elnagdi, M. H. Tetrahedron 1974, 30, 2791. doi:10.1016/S0040-4020(01)97447-2
Return to citation in text: [1]
| 27. | CCDC 827653 contains the supplementary crystallographic data for compound 6e. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk |
| 28. | CCDC 826742 contains the supplementary crystallographic data for compound 6g. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk |
| 1. | Kessler, M.; Baudry, M.; Lynch, G. Brain Res. 1989, 489, 377. doi:10.1016/0006-8993(89)90875-5 |
| 2. | McQuaid, L. A.; Smith, E. C. R.; South, K. K.; Mitch, C. H.; Schoepp, D. D.; True, R. A.; Calligaro, D. O.; O’Malley, P. J.; Lodge, D.; Ornstein, P. L. J. Med. Chem. 1992, 35, 3319. doi:10.1021/jm00096a002 |
| 11. | Carey, F. A.; Sundberg, R. J. Advanced Organic Chemistry, Part A, 5th ed.; Springer: New York, 2007. |
| 12. | Erkkilä, A.; Majander, I.; Pihko, P. M. Chem. Rev. 2007, 107, 5416. doi:10.1021/cr068388p |
| 13. | Laschat, S.; Becheanu, A.; Bell, T.; Baro, A. Synlett 2005, 2547. doi:10.1055/s-2005-918922 |
| 20. | Chebanov, V. A.; Saraev, V. E.; Desenko, S. M.; Chernenko, V. N.; Knyazeva, I. V.; Groth, U.; Glasnov, T. N.; Kappe, C. O. J. Org. Chem. 2008, 73, 5110. doi:10.1021/jo800825c |
| 8. | Zhu, J.; Bienaymé, H., Eds. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005. |
| 9. | Dömling, A.; Ugi, I. Angew. Chem., Int. Ed. 2000, 39, 3168. doi:10.1002/1521-3773(20000915)39:18<3168::AID-ANIE3168>3.0.CO;2-U |
| 10. | Simon, C.; Constantieux, T.; Rodriguez, J. Eur. J. Org. Chem. 2004, 4957. doi:10.1002/ejoc.200400511 |
| 25. | CCDC 825123 contains the supplementary crystallographic data for compound 6a. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk |
| 26. | CCDC 833076 contains the supplementary crystallographic data for compound 5. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk |
| 5. | Weber, L.; Illgen, K.; Almstetter, M. Synlett 1999, 366. doi:10.1055/s-1999-2612 |
| 6. | Kantevari, S.; Vuppalapati, S. V. N.; Nagarapu, L. Catal. Commun. 2007, 8, 1857. doi:10.1016/j.catcom.2007.02.022 |
| 7. | Sami, S.; Nandi, G. C.; Kumar, R.; Singh, M. S. Tetrahedron Lett. 2009, 50, 7096. doi:10.1016/j.tetlet.2009.10.022 |
| 21. | Ghotekar, B. K.; Jachak, M. N.; Toche, R. B. J. Heterocycl. Chem. 2009, 46, 708. doi:10.1002/jhet.128 |
| 3. | Orvieto, F.; Branca, D.; Giomini, C.; Jones, P.; Koch, U.; Ontoria, J. M.; Palumbi, M. C.; Rowley, M.; Toniatti, C.; Muraglia, E. Bioorg. Med. Chem. Lett. 2009, 19, 4196. doi:10.1016/j.bmcl.2009.05.113 |
| 4. | Watson, C. Y.; Whish, W. J. D.; Threadgill, M. D. Bioorg. Med. Chem. 1998, 6, 721. doi:10.1016/S0968-0896(98)00029-7 |
| 22. | Sadek, K. U.; Shaker, R. M.; Abd Elrady, M.; Elnagdi, M. H. Tetrahedron Lett. 2010, 51, 6319. doi:10.1016/j.tetlet.2010.09.114 |
| 23. | Abd El Latif, F. M.; Barsy, M. A.; Aref, A. M.; Sadek, K. U. Green Chem. 2002, 4, 196. doi:10.1039/b110723m |
| 24. | Mekheimer, R. A.; Sadek, K. U. J. Heterocycl. Chem. 2009, 46, 149. doi:10.1002/jhet.13 |
| 20. | Chebanov, V. A.; Saraev, V. E.; Desenko, S. M.; Chernenko, V. N.; Knyazeva, I. V.; Groth, U.; Glasnov, T. N.; Kappe, C. O. J. Org. Chem. 2008, 73, 5110. doi:10.1021/jo800825c |
| 20. | Chebanov, V. A.; Saraev, V. E.; Desenko, S. M.; Chernenko, V. N.; Knyazeva, I. V.; Groth, U.; Glasnov, T. N.; Kappe, C. O. J. Org. Chem. 2008, 73, 5110. doi:10.1021/jo800825c |
| 19. | Chebanov, V. A.; Saraev, V. E.; Desenko, S. M.; Chernenko, V. N.; Shishkina, S. V.; Shishkin, O. V.; Kobzar, K. M.; Kappe, C. O. Org. Lett. 2007, 9, 1691. doi:10.1021/ol070411l |
| 20. | Chebanov, V. A.; Saraev, V. E.; Desenko, S. M.; Chernenko, V. N.; Knyazeva, I. V.; Groth, U.; Glasnov, T. N.; Kappe, C. O. J. Org. Chem. 2008, 73, 5110. doi:10.1021/jo800825c |
| 18. | Quiroga, J.; Mejía, D.; Insuasty, B.; Abonía, R.; Nogueras, M.; Sánchez, A.; Cobo, J.; Low, J. N. Tetrahedron 2001, 57, 6947. doi:10.1016/S0040-4020(01)00649-4 |
| 14. | Kappe, C. O. Angew. Chem., Int. Ed. 2004, 43, 6250. doi:10.1002/anie.200400655 |
| 15. | de la Hoz, A.; Díaz-Ortiz, Á.; Moreno, A. Chem. Soc. Rev. 2005, 34, 164. doi:10.1039/b411438h |
| 16. | Bonrath, W.; Paz Schmidt, R. A. Ultrasound in Synthetic Organic Chemistry. In Advances in Organic Synthesis; Atta-ur-Rahman, Ed.; Bentham Sciences: Oak Park, IL, 2005; Vol. 1, pp 81–117. |
| 17. | Gravotto, G.; Cintas, P. J. Chem. Soc. Rev. 2006, 35, 180. doi:10.1039/b503848k |
| 9. | Dömling, A.; Ugi, I. Angew. Chem., Int. Ed. 2000, 39, 3168. doi:10.1002/1521-3773(20000915)39:18<3168::AID-ANIE3168>3.0.CO;2-U |
© 2012 Sadek et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)