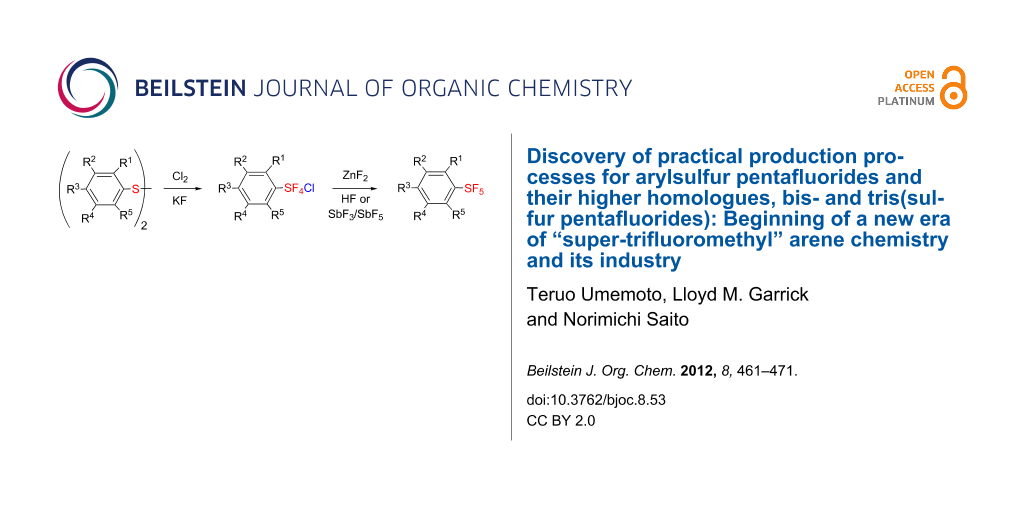
Abstract
Various arylsulfur pentafluorides, ArSF5, have long been desired in both academic and industrial areas, and ArSF5 compounds have attracted considerable interest in many areas such as medicines, agrochemicals, and other new materials, since the highly stable SF5 group is considered a “super-trifluoromethyl group” due to its significantly higher electronegativity and lipophilicity. This article describes the first practical method for the production of various arylsulfur pentafluorides and their higher homologues, bis- and tris(sulfur pentafluorides), from the corresponding diaryl disulfides or aryl thiols. The method consists of two steps: (Step 1) treatment of a diaryl disulfide or an aryl thiol with chlorine in the presence of an alkali metal fluoride, and (step 2) treatment of the resulting arylsulfur chlorotetrafluoride with a fluoride source, such as ZnF2, HF, and Sb(III/V) fluorides. The intermediate arylsulfur chlorotetrafluorides were isolated by distillation or recrystallization and characterized. The aspects of these new reactions are revealed and reaction mechanisms are discussed. As the method offers considerable improvement over previous methods in cost, yield, practicality, applicability, and large-scale production, the new processes described here can be employed as the first practical methods for the economical production of various arylsulfur pentafluorides and their higher homologues, which could then open up a new era of “super-trifluoromethyl” arene chemistry and its applications in many areas.

Graphical Abstract
Introduction
Pentafluorosulfanyl (SF5) is considered a “super-trifluoromethyl group” as SF5 has the peculiarity of fluorine beyond a trifluoromethyl (CF3) group [1]. Arylsulfur pentafluorides (ArSF5) are very thermally and chemically stable [2]. Pioneering work by Sheppard half a century ago on the synthesis and properties of arylsulfur pentafluorides revealed that the SF5 group has absolutely unique properties [2,3]. SF5 is more electronegative (Hammet constants σI: +0.55 for SF5; +0.39 for CF3) [2] and more lipophilic than CF3 (Hansch hydrophobicity constants π: 1.51 for SF5; 1.09 for CF3) [4]. The high electronegativity results in high polarity in molecules. Thus, it is of significant note that there is no functional group other than SF5 that has both high electronegativity and high lipophilicity, because the two natures are generally in conflict. In addition, SF5 has high hydrolytic stability, which equals or exceeds CF3 [2]. These extraordinary properties render SF5 compounds highly attractive particularly in medicinal [5-14], agrochemical [15-18], and new material [19-23] chemistry and industry. However, there have been no practical, economical methods for the production of arylsulfur pentafluorides. In comparison, trifluoromethyl arenes (ArCF3) have grown into a significantly large field in chemistry and industry since their practical two-step production method was developed in the 1930s through to the 1940s. The first step was chlorination of ArCH3 to ArCCl3 with Cl2 and the second step was its conversion to ArCF3 with HF or SbF3 [24-26]. A large number of trifluoromethyl arenes are currently produced on a large scale and used in many areas such as medicines, agrochemicals, dyes, materials for electronics, and others [27-30]. Accordingly, many chemists have long desired to have easy access to the “super-trifluoromethyl” arenes; however, there have been no economical methods for their production until now.
In 1961, Sheppard first reported the synthesis of phenylsulfur pentafluoride by stepwise fluorination of diphenyl disulfide with expensive silver difluorides (AgF2) in a fluorocarbon solvent [3]. However, the yield was only 9%. Since then, many substituted phenylsulfur pentafluorides have been prepared by this method, but still with very low yields [3,20,31,32]. In 2000, a new method using molecular fluorine (F2) was reported [33]. Thus, bis(p- or m-nitrophenyl) disulfide was treated with F2 diluted with nitrogen (F2:N2 = 1:9 v/v) at low temperature in acetonitrile to give the nitrophenylsulfur pentafluoride in ca. 40% yield. However, in addition to the low yields, this method requires F2, which is a highly toxic, corrosive and explosive gas, and it applies only for the case of electron-deficient nitrophenylsulfur pentafluorides, due to the extremely high reactivity of F2. These factors significantly limited the scope and application of this method. Another method reported used expensive xenon difluoride to fluorinate diphenyl disulfide giving phenylsulfur pentafluoride, but its yield was only 25% [34].
Multiple-step methods have previously been developed for the preparation of arylsulfur pentafluorides. In 1964, it was reported that the reaction of sparsely available and toxic gaseous SF5Cl with acetylene, followed by bromination, dehydrobromination, and reduction with zinc, giving pentafluorosulfanylacetylene (HC≡CSF5), which was then reacted with butadiene, followed by an aromatization reaction at very high temperature, gave phenylsulfur pentafluoride [35]. Recently, phenylsulfur pentafluoride was prepared by reaction of 1,4-bis(acetoxy)-2-cyclohexene with SF5Br under 250 W sunlamp irradiation, followed by dehydrobromination and then aromatization reactions [36]. A triethylborane-catalyzed reaction of 4,5-dichloro-1-cyclohexene with SF5Cl followed by dehydrochlorination has also been reported [37]. The multistep method has recently been extended to the preparation of 2-naphthylsulfur pentafluoride and heteroarylsulfur pentafluorides [22,38-41].
5-Nitrophenyl-1,3-bis(sulfur pentafluoride) was prepared by reaction of the corresponding polymeric disulfide with AgF2 in 12% yield [3]. Two isomers of phenyl tris(sulfur pentafluorides) were synthesized by many steps starting from the reaction of SF5Cl with acetylene via HC≡CSF5 [42]. Complex Co(CO)4(HC≡CSF5) derived from HC≡CSF5 and Co2(CO)8 was decomposed in the presence of Br2 to give phenyl-1,2,4-tris(sulfur pentafluoride). Photoreaction of HC≡CSF5 in the presence of SF5Cl gave phenyl-1,3,5-tris(sulfur pentafluoride), but in low yield (19%) [42].
All of the previous methods described above suffer from multiple drawbacks of low yields, the necessity of costly and dangerous fluoro-reagents, and the quite limited scope and applicability. In response to these, we now report practical, inexpensive, and widely applicable methods suitable for the large-scale production of arylsulfur pentafluorides and their higher homologues, which have the potential to open up a new era of sulfur pentafluoride chemistry and its industry. These new methods have been described in patents and patent applications [43-48].
Results and Discussion
Recently we synthesized various arylsulfur trifluorides (ArSF3) by treatment of diaryl disulfides with chlorine in the presence of potassium [49,50] or cesium fluoride [50] and thus discovered 4-tert-butyl-2,6-dimethylphenylsulfur trifluoride (Fluolead reagent) as an excellent fluorinating agent with high thermal stability, ease of handling, and wide applicability [50,51]. During the research, we unexpectedly discovered that an arylsulfur chlorotetrafluoride (ArSF4Cl) is formed when a diaryl disulfide is treated with an excess of chlorine in the presence of an excess of the alkaline metal fluoride. Janzen et al. reported that cis- and trans-phenylsulfur chlorotetrafluoride and its p-methyl- and p-nitro-derivatives were formed by reaction of diaryl disulfide with XeF2 and tetraethylammonium chloride [52]. However, the physical properties of the arylsulfur chlorotetrafluorides were not determined since they were not isolated, presumably because the chlorotetrafluorides were considered to be unstable. Instead, their chemical structures were assigned by 19F- and 13C NMR spectroscopy of the reaction solution.
Arylsulfur chlorotetrafluorides were prepared in high yield by the reactions of diaryl disulfides with an excess amount of chlorine (ca. 7 mol or more per mole of the disulfide) in the presence of an excess amount of potassium or cesium fluoride (ca. 16 mol or more per mole of the disulfide) in dry acetonitrile at ice-bath temperature to room temperature. Although conventionally dried and powdered potassium fluoride can be used satisfactorily, spray-dried potassium fluoride, having a large surface area, is preferable. The normal dry potassium fluoride must be used in greater quantities than the spray-dried potassium fluoride. When the reaction is not taken to completion, the distilled product (ArSF4Cl) is contaminated with its intermediate ArSF3. By this method, many arylsulfur chlorotetrafluorides 2a–o having different substituents on the aromatic ring were prepared, as summarized in Scheme 1 and Table 1. The products were trans isomers, except for in the case of polyfluorinated arylsulfur chlorotetrafluorides, which formed a mixture of trans and cis isomers. The products were distilled under reduced pressure or crystallized and then characterized.
![[1860-5397-8-53-i1]](/bjoc/content/inline/1860-5397-8-53-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Table 1: Preparation of arylsulfur chlorotetrafluorides 2a–o.
| runa | ArSSAr or ArSH (mmol)b | Cl2c | MFd | ArSF4Cle | conditionsf | yield (%)g |
|---|---|---|---|---|---|---|
| 1 | 1a (150) | ≈8 | KF (16) | 2a (t) | ice bath, 9.5 h | 88 |
| 2 | C6H5SH (91) | 4.9 | KF (9) | 2a (t) | 6–10 °C, 6.5 h | 83 |
| 3 | 1b (500) | 7.7 | KF (16) | 2b (t) | ice bath, 10.5 h | 73 |
| 4 | p-(t-Bu)C6H4SH (60) | 7.5 | CsF (10) | 2c (t) | 5–10 °C, 3.5 h to rt, 24 h | 84 |
| 5 | 1d (39) | 7.2 | KF (16) | 2d (t) | ice bath, 2.5 h to rt, o.n. | 67 |
| 6 | 1e (39) | 8 | KF (16) | 2e (t) | ice bath, 2.5 h to rt, o.n. | 80 |
| 7 | 1f (87) | 6.6 | KF (17) | 2f (t) | 5–8 °C, 3.5 h | 88 |
| 8 | 1g (100) | 7.2 | KF (16) | 2g (t) | ice bath, 4.5 h to rt, o.n. | 77 |
| 9 | 1h (127) | 6.9 | KF (15.7) | 2h (t) | ice bath, 5.5 h to rt, o.n. | 86 |
| 10 | 1i (100) | 7.2 | KF (16) | 2i (t) | ice bath, 4.5 h to rt, o.n. | 60 |
| 11 | 1j (26) | 27 | KF (40) | 2j (t) | rt, 3 d | 97h |
| 12 | 1k (100) | 10 | CsF (18) | 2k (t/c = 92/8) | ice bath, 5 h to rt, o.n. | 82 |
| 13 | 1l (130) | 16 | KF (22) | 2l (t/c = 89/11) | ice bath, 6 h to rt, o.n. | 80 |
| 14 | 1m (77) | 16 | KF (24) | 2m (t/c = 96/4) | ice bath, 6 h to rt, o.n. | 87 |
| 15 | 1n (70) | 15 | KF (19) | 2n (t/c = 86/14) | ice bath, 7.5 h to rt, o.n. | 83 |
| 16 | 1o (65) | 15 | KF (22) | 2o (t/c = 60/40) | ice bath, 5 h to rt, o.n. | 86 |
aThe experimental procedure is described in the Supporting Information File 1. bThe number in parentheses is the amount (mmol) of ArSSAr or ArSH used. cMolar ratio of Cl2 per mole of ArSSAr or ArSH. dThe number in parentheses is molar ratio of MF per mole of ArSSAr or ArSH. et = trans-isomer, c = cis-isomer. The t/c ratio was determined by 19F NMR of the reaction mixture before post-treatment. frt = room temperature, o.n. = overnight. gIsolated yields. hCrude product.
Arylsulfur chlorotetrafluorides having an electron-donating alkyl group, such as methyl or tert-butyl, or an electron-withdrawing substituent, such as a halogen atom, a nitro-, or a methanesulfonyl group, were prepared in good to high yields from the corresponding diaryl disulfides. 2-Fluoro and 2,6-difluorophenylsulfur chlorotetrafluoride were formed in high yields due to the small steric effect of fluorine atom(s). However, bis(2-bromophenyl) disulfide gave a 11:1 mixture of 2-bromophenylsulfur trifluoride and chlorotetrafluoride. 2-Bromophenylsulfur chlorotetrafluoride was a minor product due to the steric hindrance of the relatively large bromo substituent at the ortho position. Arylsulfur chlorotetrafluorides were also prepared from the corresponding aryl thiols in high yields, as shown in runs 2 and 4, Table 1.
The method using aryl thiols as starting materials was successfully applied to the preparation of aryl bis- and tris(sulfur chlorotetrafluorides) as shown in Scheme 2 and Table 2. The method using the corresponding polymeric disulfides did not work well because of their extremely low solubility.
![[1860-5397-8-53-i2]](/bjoc/content/inline/1860-5397-8-53-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of Ar(SF4Cl)n from Ar(SH)n (n = 2, 3).
Scheme 2: Preparation of Ar(SF4Cl)n from Ar(SH)n (n = 2, 3).
Table 2: Preparation of aryl bis- and tris(sulfur chlorotetrafluorides) 2p'–u'.
| runa | Ar(SH)n (mmol)b | Cl2c | KFd | Ar(SF4Cl)ne | conditionsf | yield (%)g |
|---|---|---|---|---|---|---|
| 1 | 1p (68.7) | 18 | 25 | 2p′ (t) | ice bath, 6 h to rt, 2 d | 56 |
| 2 | 1q (64) | 20 | 47 | 2q′ (t) | ice bath, 6 h to rt, 2 d | 74 |
| 3 | 1r (80) | 23 | 38 | 2r′ (t/c = 89/11) | rt, 2 d | 96h |
| 4 | 1s (63) | 20 | 20 | 2s′ (t/c = 95/5) | ice bath, 7 h to rt, o.n. | 52 |
| 5 | 1t (64) | 20 | 27 | 2t′ (t/c = 58/42) | ice bath, 7 h to rt, o.n. | 79 |
| 6 | 1u (57) | 40 | 60 | 2u′ (t/c = 79/21) | rt, 3 d | 98h |
aThe experimental procedure is described in the Supporting Information File 1. bThe number in parentheses is the amount (mmol) of Ar(SH)n used. cMolar ratio of Cl2 per mole of Ar(SH)n. dMolar ratio of KF per mole of Ar(SH)n. et = trans-configuration, c = cis-configuration. The t/c ratio was determined by 19F NMR of the reaction mixture or crude product. frt = room temperature, o.n. = overnight. gIsolated yields. hCrude product.
The reaction of a diaryl disulfide with Cl2 and KF is given as Equation 1. Per 1 mol of a diaryl disulfide, 5 mol of Cl2, and 8 mol of KF are theoretically consumed.
![[1860-5397-8-53-i10]](/bjoc/content/inline/1860-5397-8-53-i10.svg?max-width=590&scale=1.18182)
Scheme 3 shows a postulated reaction mechanism, which consists of six steps including intermediates 4, 5, 6, 7 and 8. Treatment of p-nitrophenylsulfenyl chloride with Cl2/KF gave p-nitrophenylsulfur chlorotetrafluoride in 76% yield. Treatment of phenylsulfur trifluoride with Cl2/KF gave phenylsulfur chlorotetrafluoride in 84% yield. These results support the hypothesis that arylsulfenyl chloride 4 and trifluoride 7 are intermediates for the reaction of diaryl disulfide giving arylsulfur chlorotetrafluoride.
![[1860-5397-8-53-i3]](/bjoc/content/inline/1860-5397-8-53-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction mechanism for the formation of ArSF4Cl.
Scheme 3: Reaction mechanism for the formation of ArSF4Cl.
In a typical reaction of diphenyl disulfide, an orange color appears immediately as chlorine (Cl2) gas is introduced into a mixture of Ar2S2 and KF in acetonitrile. Cl2 is absorbed as fast as it is introduced until ArSF3 7 is formed, at which point the solution becomes colorless. 19F NMR analysis of the reaction mixture at this moment confirms the formation of 7. After that, the absorption of Cl2 becomes slow. Thus, the sequence of steps 1 to 4 giving 7 is fast, while the sequence of steps 5 and 6 giving the final product 2 is slow. The slow reaction is probably due to an equilibrium reaction (step 5) between 7 and 8. The reaction of aryl thiol as a starting material is similar to that of the disulfide 1 since aryl thiol reacts with Cl2 to form disulfide 1.
The arylsulfur chlorotetrafluorides 2a–j and bis(sulfur chlorotetrafluorides) 2p′ and 2q′ obtained were trans-isomers, while the polyfluorinated 2k–o, bromo and polyfluoro bis(sulfur chlorotetrafluorides) 2r′–t′, and tris(sulfur chlorotetrafluoride) 2u′ were a mixture of trans- and cis-configuration. Since we did not observe any isomerization at room temperature or upon distillation, it is reasonable to conclude that each isomer was formed through each isomeric salt of 8 as shown in Scheme 4. The multifluoro derivatives 2o and 2t′ have the highest ratio of cis-configuration. This suggests that the relative stability of the cis-isomeric salts 8 increases particularly with increased fluorine substitution. The high ratio of cis-configuration of 2u′ suggests the ability of electron-withdrawing groups, such as –SF4Cl, to stabilize the cis-configuration salt form.
![[1860-5397-8-53-i4]](/bjoc/content/inline/1860-5397-8-53-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction mechanism for the formation of trans and cis-ArSF4Cl.
Scheme 4: Reaction mechanism for the formation of trans and cis-ArSF4Cl.
It is noticeable that, except for the polyfluorinated compounds and others discussed above, trans-isomeric ArSF4Cl was exclusively formed by the reactions of Ar2S2 with Cl2/KF, while the reactions of Ar2S2 with XeF2/Et4NCl gave a mixture of cis- and trans-isomers according to Janzen's report [52]. Janzen proposed radical reactions with Cl· species [52]. The Cl2/KF reactions are ionic in nature, in which trans-form salts 8 exclusively form and react with Cl2 to give the trans-isomers.
Arylsulfur chlorotetrafluorides are stable during long periods of storage in a fluoropolymer vessel at room temperature. We did not observe any isomerization between trans- and cis-isomers on standing. Whereas arylsulfur trifluorides are extremely sensitive to moisture (water) [50,53], with the exception of the Fluolead reagent [50], arylsulfur chlorotetrafluorides are relatively insensitive to moisture. The half life time of decomposition of phenylsulfur chlorotetrafluoride (2a) in a CDCl3 solution (≈1.2 mol/L) on direct contact with water was 300 to 500 min at room temperature. The tracing experiment was conducted with a NMR tube. The 19F NMR was measured by using an internal standard (p-chlorobenzotrifluoride) and the NMR tube was shaken between the measurements. Gas chromatography (GC) could not be used because of decomposition of arylsulfur chlorotetrafluorides in the GC column. When a few drops of D2O were added to a solution of 2a in CD3CN (1 mL), 2a decomposed in 1 h and the decomposition product was phenylsulfonyl chloride. Phenylsulfonyl fluoride was detected in a trace amount (≤1%) by GC–mass analysis.
Arylsulfur chlorotetrafluorides 2 have relatively high thermal stability. Thus, 2a did not decompose during 134 hours at 100 °C or 48 h at 150 °C in a Teflon tube. The isomerization of the trans- to the cis-isomer occurred very slowly. A very small amount (3–4%) of the cis-isomer was formed after heating of 2a (trans-isomer) at 150 °C for 48 h. The real thermal decomposition temperatures of 2 could not be determined by a differential scanning calorimeter (DSC), because 2 reacted with the cell materials, i.e., stainless steel and gold, at elevated temperature due to their strongly oxidizing effect on the hexavalent sulfur(VI) element. Many arylsulfur chlorotetrafluorides were measured with DSC and these are discussed in Supporting Information File 1.
We examined reaction conditions for the conversion of trans-PhSF4Cl (2a) to PhSF5 (3a) with various reactive fluorides using an approximately one-gram scale of 2a, as seen in Table 3. Janzen et al. described in the experimental section that bubbling an excess of BF3 into a mixture of cis- and trans-2a in CD2Cl2 at 25 °C led to the gradual disappearance of 2a and the formation of PhSF5 (3a) [52]. We conducted the reaction of trans-2a (isolated) with BF3 in a sealed reactor and found that all the starting material became a solid residue, probably a polymer (run 1, Table 3). The reaction in dichloromethane solvent resulted in only a 28% yield of 3a (run 2). The use of HBF4·OEt2 at room temperature provided a better yield (40%) (run 3). A strong Lewis acid SbF5 led to polymeric product, but SbF3 at 80 °C gave 33% (run 4). A combination of SbF3/SbCl5(cat.) at room temperature provided a better yield (54%) (run 5). At 80 °C, transition-metal fluorides TiF4 and CuF2 afforded 35 and 57%, respectively (runs 7, 8). Finally, we found that inexpensive and easily handled ZnF2 produced 3a in high yield (runs 9, 10).
Table 3: Conversion of phenylsulfur chlorotetrafluoride (2a) to PhSF5 (3a).
| runa | 2a (mmol)b | fluoride (mmol)c | solvent (mL)d | temperature (°C) | time | yield of 3a (%)e |
|---|---|---|---|---|---|---|
| 1 | 4.5 | BF3f | none | rt | 3 d | 0 |
| 2 | 6.4 | BF3f | CH2Cl2 (6.4) | rt | 5 h | 28 |
| 3 | 4.5 | HBF4·OEt2 (5.4) | CH2Cl2 (4.5) | rt | 21 h | 40 |
| 4 | 4.5 | SbF3 (2.2) | none | 80 | 5 h | 33 |
| 5 | 4.5 | SbF3/SbCl5 (2.0/cat.) | hexane (2) | rt | 3 d | 54 |
| 6 | 4.5 | SnF4 (1.4) | none | 80 | 2 h | 34 |
| 7 | 4.5 | TiF4 (1.4) | none | 80 | 16 h | 35 |
| 8 | 4.5 | CuF2 (2.8) | none | 80 | 22 h | 57 |
| 9 | 4.5 | ZnF2 (2.7) | none | 80 | 20 h | 85 |
| 10 | 4.5 | ZnF2 (2.7) | none | 120 | 4 h | 88 |
| 11 | 13.6 | ZnF2/SbCl5 (8.2/1.4) | heptane (5) | rt | 17 h | 53 |
aThe experimental procedure is described in Supporting Information File 1. bThe amount (mmol) of 2a used. cThe number in parentheses is the amount (mmol) of fluoride used. dThe number in parentheses is the amount (mL) of solvent used. eDetermined by 19F NMR. fSee Supporting Information File 1 for the amount of BF3 used.
A 19F NMR tracing experiment of the conversion reaction of trans-PhSF4Cl (trans-2a) provided some information on the reaction mechanism. With HBF4·OEt2, it was observed that the molar ratio of trans-2a:cis-2a:PhSF5 was 156:172:100 in the reaction mixture after 7 h, and 3:6:100 after 21 h. With ZnF2, the ratio observed was 22:117:100 during the reaction. A considerable amount of the cis-isomer was formed as an intermediate. It may thus be suggested that there are two routes, a direct route of the trans-isomers to the SF5 products and an indirect route via cis-isomers. The experiment with HBF4·OEt2 or ZnF2 may be considered to be thermal isomerization of trans-2a to cis-2a with an acid catalyst, suggesting that the cis-isomer is more thermodynamically stable than the trans-isomer. With ZnF2/SbCl5 the cis-isomer was barely detected (run 11, Table 3). The ratio of trans-2a:cis-2a:PhSF5 was 385:0:100 after 10 min; 63:trace:100 after 1.5 h; 34:trace:100 after 3 h; and 18:2:100 after 17 h. Thus, the lack of detectable cis-isomer may suggest that the addition of a strong Lewis acid such as SbCl5 gives priority to the direct route. However, we cannot rule out the possibility that the conditions could very quickly convert the cis-isomer to the product.
The method with ZnF2 was applied to 10–50 gram scale reactions of PhSF4Cl (2a) and its derivatives 2b,d,e–i,k (Scheme 5). A fluoropolymer reactor charged with the reactants (ArSF4Cl and ZnF2) was heated under the pressure of a balloon filled with N2 gas (no flow of N2). The reaction conditions and yields are shown in Table 4. Liquid 2a,b,d–f,h efficiently reacted with solid ZnF2 (powder) under stirring without solvent. The two fluorine atoms of ZnF2 were effectively consumed for the reaction. 2a and monohalogenated 2d–h were converted to the corresponding products 3 in good to high yields at 120 °C (bath temperature). It was observed that the start of the exothermic reaction of halogenated 2f was significantly delayed compared to that of unsubstituted 2a. p-Methyl-2b reacted with ZnF2 at 90 °C (run 2). p-Nitro-2i and 2,6-difluoro-2k required a high temperature of 150 °C or more and their yields were fair to poor (runs 8 and 9). Thus, the electron-donating substituents increase the reactivity of –SF4Cl, while the electron-withdrawing ones decrease it.
![[1860-5397-8-53-i5]](/bjoc/content/inline/1860-5397-8-53-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Preparation of ArSF5 with ZnF2.
Scheme 5: Preparation of ArSF5 with ZnF2.
Table 4: Preparation of various arylsulfur pentafluorides 3 with ZnF2.
| runa | 2 (mmol)b | solvent | temp (°C) | time (h) | product 3 | yield (%)c |
|---|---|---|---|---|---|---|
| 1 | 2a (200) | none | 120 | 20 | 3ad | 75 |
| 2 | 2b (137) | none | 90 | overnight | 3be | 71 |
| 3 | 2d (42) | none | 120 | 16 | 3df | 62 |
| 4 | 2e (42) | none | 120 | 15 | 3e | 59 |
| 5 | 2f (175) | none | 120 | 16 | 3fd,g | 73 |
| 6 | 2g (100) | heptane (20 mL) | reflux | 17 | 3gd | 79 |
| 7 | 2h (33) | none | 120 | 15 | 3hd | 78 |
| 8 | 2i (100) | none | 150 | 72 | 3id | 36 |
| 9 | 2k (160) | none | 130→180 | 4→6 | 3k | 52 |
aThe amount of ZnF2 used was 0.6 mol per 1 mol of 2 in runs 1–4 and 6–8, 0.53 mol in run 5, and 1.06 mol in run 9. The experimental procedure is described in Supporting Information File 1. bThe number in parentheses is the amount (mmol) of 2 used. cIsolated yields. dSee [2]. eSee [52]. fSee [54]. gProduct 3f (purity 97%) obtained after distillation was contaminated with 3% of p-dichlorobenzene (major) and trichlorobenzene (minor). Purity was determined by GC.
It was found that the product p-chloro-3f obtained after distillation (run 5, Table 4) was contaminated (3%) with p-dichlorobenzene (major) and trichlorobenzene (minor), which were formed by cleavage of the C–S bond, in the case of the major byproduct, and further chlorination, in the case of the minor one, during the reaction. The complete removal of the byproducts from 3f was difficult due to similar boiling points. We found that the byproducts were suppressed by the addition of a strong Lewis acid AlCl3 and that the addition of ZnCl2 modified the exothermic reaction of 2f with ZnF2. The addition of AlCl3 decreased the reaction temperature and the addition of ZnCl2 probably helped to form reactive “ZnFCl” species. Finally the byproduct was restricted to less than 1% with ZnF2/ZnCl2/AlCl3 (molar ratio 100:10:5). The detailed experimental procedure is described in Supporting Information File 1.
The reaction of PhSF4Cl (2a) with ZnF2 in Table 4 was conducted under nonflowing N2 gas (under the pressure of a N2 balloon). When the reaction of 2a was conducted under a flow of N2 gas, the reaction rate became low. We then examined the reaction conditions for 2a in more detail and found that the reaction was dependent upon the atmosphere of the reaction mixture. While the reaction conducted under a N2 balloon (no flow of N2) was completed in 4 h in 88% yield, the reaction conducted under a flow of N2 (through a reactor) was not completed in 5 h and its yield was down to 67%. With a faster flow of N2, it became slower and the yield decreased. Apparently, a small amount of gas was generated at the beginning of the reaction. The gas was not analyzed because of experimental difficulty. Therefore, it was most likely that removal of the gas by the flow of N2 gas made the reaction slow. Surprisingly, when the reactor was filled with Cl2 gas, the reaction was completed in a short time (1.7 h) and its yield was very high (92%). Thus, the presence of Cl2 significantly accelerated the reaction rate and increased the yield. We assumed that one of the effects of the Cl2 atmosphere may be to inhibit a possible disproportionation reaction [2PhSF4Cl (2a) → PhSF5 + PhSF3 + Cl2 ↑], as the disproportionation leads to the formation of Cl2. Although it remains unclear as to why Cl2 accelerated the reaction, Cl2 may also activate ZnF2 or intermediate “ZnFCl” species.
Interestingly, this Cl2 atmosphere was effective for the reaction of the p-methyl derivative 2b with ZnF2, but not for the p-chloro derivative 2f. The reaction of 2f with ZnF2 was not affected by N2 flow. As it is known that sulfur-related disproportionation, for instance arylsulfinic acid giving arylsulfonic acid and S-aryl arylthiosulfonate ester, is retarded by an electron-withdrawing group [55], it may thus be suggested that an electron-withdrawing substituent limits the disproportionation. Possibly the electron-withdrawing substituent lowers the disproportionation rate or increases the temperature necessary for disproportionation such that it is greater than that required for the replacement reaction of –SF4Cl to –SF5.
The method with easily handled and inexpensive ZnF2 under a Cl2 atmosphere was successfully applied to a large-scale production (≈0.5 kg) of 3a from 2a, in which the addition method was adopted and a small amount of product 3a was used as the reaction solvent. This procedure is described in Supporting Information File 1.
In contrast, anhydrous hydrogen fluoride (HF) is not easy to handle under normal laboratory conditions due to its high toxicity. However, in industry, in addition to its availability as a cheap fluorine source, the gaseous or liquid nature of HF (bp 19 °C) is quite suitable for large-scale industrial processes due to its ease in transfer, recovery and recycling.
2a satisfactorily reacted with HF at less than its boiling point to produce 3a along with the evolution of hydrogen chloride (Scheme 6). As seen in Table 5, this method has successfully been applied to various substituted arylsulfur chlorotetrafluorides. The products obtained by the method were of high purity (≥99%) except for the cases of methyl derivative 2b (runs 4–6). When the reaction of 2a was conducted with the addition of KHF2 (KF:HF = 1:1), the yield improved (run 3). KF suppressed the formation of impurities, such as polymeric residue and chlorinated byproducts, because basic KF neutralizes the strong acid HCl formed in the reaction.
![[1860-5397-8-53-i6]](/bjoc/content/inline/1860-5397-8-53-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Preparation of PhSF5 with anhydrous HF.
Scheme 6: Preparation of PhSF5 with anhydrous HF.
Table 5: Preparation of arylsulfur pentafluorides 3 with anhydrous hydrogen fluoride.
| runa | ArSF4Cl 2 (mmol)b | molar ratio 2/HF/additive | additivec | temperature (°C) | time (h) | ArSF5 3 | yield (%)d |
|---|---|---|---|---|---|---|---|
| 1 | 2a (87.1) | 1/29/– | none | 15 | 20 | 3a | 62 |
| 2 | 2a (152) | 1/24/– | none | −10 | 20 | 3a | 66 |
| 3 | 2a (96.2) | 1/25/1.1 | KHF2 | 15 | 18 | 3a | 73 |
| 4 | 2b (144) | 1/22/– | none | 15 | 19 | 3b | 73e |
| 5 | 2b (89.8) | 1/22/1.2 | KHF2 | 15 | 20 | 3b | 79f |
| 6 | 2b (80.8) | 1/30/0.37 | PhH | 15 | 79 | 3b | 57g |
| 7 | 2b (91.8) | 1/23/1.2/0.33i | KHF2, PhH | 15 | 17 | 3b | 56h |
| 8 | 2d (90.2) | 1/28/- | none | 15 | 21 | 3d | 67 |
| 9 | 2ej (292) | 1/23/- | none | 19 | 22 | 3e | 76h |
| 10 | 2f (146) | 1/23/- | none | 15 | 20 | 3f | 71 |
| 11 | 2g (250) | 1/32/- | none | 20 | 2 d | 3g | 77 |
aThe experimental procedure is described in Supporting Information File 1. bThe number in parentheses is the amount (mmol) of 2 used. cKHF2 = potassium hydrogen difluoride. PhH = benzene. dIsolated yields after distillation. Purities of the products were >99% except for the cases labeled with superscripts e, f, g, h. Purity was determined by GC. ePurity was 91%. fPurity was 97%. gPurity was 90%. hPurity was 99%. iMolar ratio: 1/23/1.2/0.33 = 2b/HF/KHF2/PhH. j2e (71 wt %) in CH3CN was used, which was obtained by concentration (with a vacuum pump) of the filtrate of the reaction mixture after completion of reaction of Scheme 1.
In run 4, the purity of product 3b was 91%, which was contaminated with 8% of 3-chloro-4-methylphenylsulfur pentafluoride (3b·Cl) as the main byproduct. Compound 3b·Cl was tentatively assigned by GC–mass analysis. When the reaction was conducted with the addition of KHF2, the purity greatly increased to 97% (run 5). The occurrence of an intermolecular side chlorination reaction was clearly demonstrated by the experiment of run 6 in which benzene was used as an additive. The distilled product 3b (purity 90%) was contaminated with p-dichlorobenzene (6%) and o-dichlorobenzene (3%) in addition to the byproduct 3b·Cl (1%). The dichlorobenzenes were formed by chlorination of the added benzene. Chlorobenzene formed in this reaction was contained in an initial distillation fraction, which was separated. The method of run 7 using both KHF2 and benzene as additives provided product 3b with 99% purity, as the formation of impurity 3b·Cl was completely suppressed. It is likely that the SF4Cl part of 2b acts as a chlorinating agent toward 3b, or another 2b and benzene, under the strong acidic conditions that are formed from the HCl generated in the anhydrous HF.
As seen in runs 8–11, halogenated arylsulfur pentafluorides 3d–g of high purity (≥99%) were obtained in good isolated yields without any additive. The side reactions such as polymerization and chlorination are restrained, as the aromatic nuclei are deactivated by the electron-withdrawing effect of the halogen atoms.
It was also found that when PhSF4Cl (2a) was treated with a 70:30 w/w mixture of HF–pyridine at 55 °C, it gave PhSF5 (3a) in 63% isolated yield (Scheme 7). The HF–pyridine reagent is a nonvolatile and easily handled chemical in the laboratory.
![[1860-5397-8-53-i7]](/bjoc/content/inline/1860-5397-8-53-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Preparation of 3a with HF–pyridine.
Scheme 7: Preparation of 3a with HF–pyridine.
Polyfluorinated arylsulfur chlorotetrafluorides 2k–o were smoothly converted to the corresponding sulfur pentafluorides 3k–o in good yields by treatment with a combination of SbF3 and a strong Lewis acid, such as SbF5 or SbCl5, as shown in Scheme 8 and Table 6.
![[1860-5397-8-53-i8]](/bjoc/content/inline/1860-5397-8-53-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Preparation of polyfluorinated ArSF5.
Scheme 8: Preparation of polyfluorinated ArSF5.
Table 6: Preparation of polyfluorinated arylsulfur pentafluorides with Sb(III)/(V) fluorides.
| runa | 2 (mmol)b | Sb(III) (mmol)c | Sb(V) (mmol)c | molar ratio Sb(III)/Sb(V) | solvent (mL)d | temperature (°C) | time (h) | 3 | yield (%)e |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2k (3.9) | SbF3 (5.7) | SbCl5 (0.4) | 14/1 | FC-72 (8) | rt | 1 | 3k | 71 |
| 2 | 2l (36) | SbF3 (39) | SbF5 (9) | 4.3/1 | FC-72 (40) | −60 → rt | 5 | 3l | 70 |
| 3 | 2m (106) | SbF3 (121) | SbF5 (26.7) | 4.5/1 | FC-72 (110) | −60 → rt | ≈5 | 3m | 77 |
| 4 | 2n (118) | SbF3 (145) | SbF5 (22.3) | 6.5/1 | FC-72 (200) | rt | 4.5 | 3n | 61 |
| 5 | 2o (30.3) | SbF3 (32) | SbF5 (30.4) | 1.05/1 | FC-72 (40) | rt | 1 | 3o | 71 |
aThe experimental procedure is described in Supporting Information File 1. bThe number in parentheses is the amount (mmol) of 2 used. cThe number in parentheses is the amount (mmol) of the Sb halide used. dThe number of parentheses is the amount (mL) of the solvent used. FC-72 is a perfluorocarbon with bp 56 °C (3M Fluorinert™ Electronic Liquid FC-72, 3M Specialty Materials, St. Paul, MN, USA). eIsolated yields.
While treatment of 2,4,6-trifluoro-2m with a mixture of SbF3/SbF5 (4.5/1) gave 77% of product 3m (run 3, Table 6), treatment of 2m with SbF5 alone gave a byproduct (20%) in addition to 3m (60%). The byproduct was tentatively assigned as 3-chloro-2,4,6-trifluorophenylsulfur pentafluoride by 1H and 19F NMR and GC–mass analysis. The chlorination as a side reaction may occur from the action of a strong Lewis acid SbF5 on the fluorine atoms of the SF4Cl group of ArSF4Cl 2m, forming a [ArSF3Cl]+ [SbF6]− species (Ar = 2,4,6-trifluorophenyl), which may act as a strong chlorinating agent (Cl+) toward 3m or another molecule of 2m. Pentafluoro-2o was converted to 3o in good yield with a high molar ratio of SbF5 (run 5) as the reactivity of 2o was considerably decreased by the five fluorine atoms.
Phenyl bis(sulfur chlorotetrafluorides) 2p′ and 2q′, bromo derivative 2r′, and fluoro derivatives 2s′ and 2t′ were smoothly converted to the corresponding bis(sulfur pentafluorides) 3p″–t″ in fair to good yields with SbF5 alone, as shown in Scheme 9 and Table 7 (runs 1–5). Phenyl tris(sulfur chlorotetrafluoride) 2u′ was converted to phenyl tris(sulfur pentafluoride) 3u″ in 55% yield under similar conditions (run 6, Table 7).
![[1860-5397-8-53-i9]](/bjoc/content/inline/1860-5397-8-53-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Preparation of aryl bis- and tris(sulfur pentafluorides), Ar(SF5)n (n = 2,3).
Scheme 9: Preparation of aryl bis- and tris(sulfur pentafluorides), Ar(SF5)n (n = 2,3).
Table 7: Preparation of aryl bis- and tris(sulfur pentafluorides) with SbF5.
| runa | 2 (mmol)b | SbF5 (mmol)c | solvent (mL)d | temperature (°C) | time (h)e | product 3 | yield (%)f |
|---|---|---|---|---|---|---|---|
| 1 | 2p′ (10.8) | 21.4 | CH2Cl2 (45) | −85 → −25 | 2.5 | 3p″ | 57 |
| 2 | 2q′ (11) | 24 | CH2Cl2 (80) | −85 → −15 | 1.5 | 3q″ | 71 |
| 3 | 2r′ (77) | 130 | CH2Cl2 (450) | ca. −85 → −15 | 5 | 3r″ | 66 |
| 4 | 2s′ (27.8) | 30.7 | FC-72 (390) | −80 → rt, then rt | 8, then o.n. | 3s″ | 52 |
| 5 | 2t′ (12.4) | 14.4 | FC-72 (70) | rt | o.n. | 3t″ | 67 |
| 6 | 2u′ (32) | 99 | CH2Cl2/FC-72 (160/68) | −95 → +7 | 6 | 3u″ | 55 |
aThe experimental procedure is described in Supporting Information File 1. bThe number in parentheses is the amount (mmol) of 2 used. cThe number is the amount (mmol) of SbF5 used. dThe number in parentheses is the amount (mL) of the solvent used. FC-72 is a perfluorocarbon having bp 56 °C (3M Fluorinert™ Electronic Liquid FC-72, 3M Specialty Materials, St. Paul, MN, USA). eo.n. = overnight. fIsolated yields.
Conclusion
We have developed the first practical and economical method for the production of various arylsulfur pentafluorides and their higher homologues, which consists of the treatment of diaryl disulfides or aryl thiols with chlorine in the presence of potassium or cesium fluoride, followed by treatment of the resulting arylsulfur chlorotetrafluorides with a fluoride, such as ZnF2, HF, and Sb(III/V) fluorides. The important characteristics of these new reactions were revealed and some reactions were modified to provide the products in high purity and in high yields. Since these methods are superior to the previous methods using AgF2, F2, or XeF2, or multiple-step methods starting from SF5Br or SF5Cl, in terms of cost, applicability, and scalability of production, the processes developed here can be used as the first practical and economical methods for the production of many kinds of arylsulfur pentafluorides. Thus, it is expected that this will lead to new and rapid advances in “super-trifluoromethyl” arene chemistry and associated industries in many areas.
Supporting Information
| Supporting Information File 1: Experimental details and copies of 1H-, 19F-, and 13C NMR spectra of new products. | ||
| Format: PDF | Size: 7.8 MB | Download |
References
-
Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2004; p 151. doi:10.1002/352760393X
Return to citation in text: [1] -
Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3072–3076. doi:10.1021/ja00875a007
Return to citation in text: [1] [2] [3] [4] [5] -
Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3064–3072. doi:10.1021/ja00875a006
Return to citation in text: [1] [2] [3] [4] -
Hansch, C.; Muir, R. M.; Fujita, T.; Maloney, P. P.; Geiger, F.; Streich, M. J. Am. Chem. Soc. 1963, 85, 2817–2824. doi:10.1021/ja00901a033
Return to citation in text: [1] -
Stump, B.; Eberle, C.; Schweizer, W. B.; Kaiser, M.; Brun, R.; Krauth-Siegel, R. L.; Lentz, D.; Diederich, F. ChemBioChem 2009, 10, 79–83. doi:10.1002/cbic.200800565
Return to citation in text: [1] -
Mo, T.; Mi, X.; Milner, E. E.; Dow, G. S.; Wipf, P. Tetrahedron Lett. 2010, 51, 5137–5140. doi:10.1016/j.tetlet.2010.07.113
Return to citation in text: [1] -
Gujjar, R.; El Mazouni, F.; White, K. L.; White, J.; Creason, S.; Shackleford, D. M.; Deng, X.; Charman, W. N.; Bathurst, I.; Burrows, J.; Floyd, D. M.; Matthews, D.; Buckner, F. S.; Charman, S. A.; Phillips, M. A.; Rathod, P. K. J. Med. Chem. 2011, 54, 3935–3949. doi:10.1021/jm200265b
Return to citation in text: [1] -
Coteron, J. M.; Marco, M.; Esquivias, J.; Deng, X.; White, K. L.; White, J.; Koltun, M.; El Mazouni, F.; Kokkonda, S.; Katneni, K.; Bhamidipati, R.; Shackleford, D. M.; Angulo-Barturen, I.; Ferrer, S. B.; Jiménez-Díaz, M. B.; Gamo, F.-J.; Goldsmith, E. J.; Charman, W. N.; Bathurst, I.; Floyd, D.; Matthews, D.; Burrows, J. N.; Rathod, P. K.; Charman, S. A.; Phillips, M. A. J. Med. Chem. 2011, 54, 5540–5561. doi:10.1021/jm200592f
Return to citation in text: [1] -
Bossemaier, B.; Friebe, W.-G.; Georges, G.; Rueth, M.; Voss, E. Novel pentafluorosulfanyl compounds. U.S. Patent Appl. 0,197,370, Sept 8, 2005.
Return to citation in text: [1] -
Andeotti, D.; Checchia, A.; Hamprecht, D.; Micheli, F. 3-triazolylthioalkyl-3-azabicyclo(3.1.0)hexanes and their use as dopamine D3 receptor ligands. WO Patent WO/2006/108700, Oct 19, 2006.
Return to citation in text: [1] -
Frank, R.; Sundermann, B.; Schick, H. Pentafluorosulphanyl-substituted compound and its use for producing medicaments. WO Patent WO/2006/122773, Nov 23, 2006.
Return to citation in text: [1] -
Billen, D.; Boyle, J.; Critcher, D. J.; Gethin, D. M.; Hall, K. T.; Kyne, G. M. Substituted arylpyrazoles. US Patent Appl. 176,865, July 24, 2008.
Return to citation in text: [1] -
Kleeman, H.-W. Pentafluorosulfanylphenyl-substituted benzoylguanidines, method for the production thereof, their use as a medicament or diagnostic agent, and a medicament containing these compounds. U.S. Patent 7,446,225, Nov 4, 2008.
Return to citation in text: [1] -
Stamford, A. W.; Cumming, J. N. Pentafluorosulfurimino heterocyclic compounds as BACE-1 inhibitors, compositions and their use. WO Patent WO/2011/044184, April 11, 2011.
Return to citation in text: [1] -
Crowley, P. J.; Mitchell, G.; Salmon, R.; Worthington, P. A. Chimia 2004, 58, 138–142.
Return to citation in text: [1] -
Lim, D. S.; Choi, J. S.; Pak, C. S.; Welch, J. T. J. Pestic. Sci. 2007, 32, 255–259. doi:10.1584/jpestics.G06-50
Return to citation in text: [1] -
Chern, R. T.; Zingerman, J. R.; Clark, J. N.; Drag, M. D. Sulfurpentafluorophenyl pyrazoles for controlling ectoparasitic infestations. WO Patent WO/1999/047139, Sept 23, 1999.
Return to citation in text: [1] -
Matsuzaki, Y.; Morimoto, M.; Fujioka, S.; Tohnishi, M. Phtalimide derivative, agricultural or horticultural insecticide, and method of use thereof. WO Patent WO/2003/093228, Nov 13, 2003.
Return to citation in text: [1] -
Nixon, P. G.; Winter, R.; Castner, D. G.; Holcomb, N. R.; Grainger, D. W.; Gard, G. L. Chem. Mater. 2000, 12, 3108–3112. doi:10.1021/cm000339k
Return to citation in text: [1] -
Kirsch, P.; Hahn, A. Eur. J. Org. Chem. 2005, 3095–3100. doi:10.1002/ejoc.200500125
Return to citation in text: [1] [2] -
Gao, H.; Ye, C.; Winter, R. W.; Gard, G. L.; Sitzmann, M. E.; Shreeve, J. M. Eur. J. Inorg. Chem. 2006, 3221–3226. doi:10.1002/ejic.200600098
Return to citation in text: [1] -
Ye, C.; Gard, G. L.; Winter, R. W.; Syvret, R. G.; Twamley, B.; Shreeve, J. M. Org. Lett. 2007, 9, 3841–3844. doi:10.1021/ol701602a
Return to citation in text: [1] [2] -
Zahn, S.; Nordquist, A. F.; Minnich, K. E.; Lal, G. S.; Burgoyne, W. F., Jr.; Klauck-Jacobs, A. Pentafluorosulfanyl-substituted thienothiophene monomers and conducting polymers. U.S. Patent 7,060,846, June 13, 2006.
Return to citation in text: [1] -
Simons, J. H.; Lewis, C. J. J. Am. Chem. Soc. 1938, 60, 492. doi:10.1021/ja01269a507
Return to citation in text: [1] -
Pouterman, E.; Girardet, A. Helv. Chim. Acta 1947, 30, 107–112. doi:10.1002/hlca.19470300114
Return to citation in text: [1] -
McBee, E. T.; Hass, H. B.; Weiner, P. E.; Rothrock, G. M.; Burt, W. E.; Robb, R. M.; Van Dyken, A. R. Ind. Eng. Chem. 1947, 39, 298–301. doi:10.1021/ie50447a613
Return to citation in text: [1] -
Banks, R. E., Ed. Organofluorine Chemicals and Their Industrial Applications; Ellis Horwood Ltd.: Chichester, 1979.
Return to citation in text: [1] -
Banks, R. E.; Smart, B. E.; Tatlow, J. C., Eds. Organofluorine Chemistry, Principles and Commercial Applications; Plenum Press: New York, 1994.
Return to citation in text: [1] -
Hiyama, T. In Organofluorine Compounds, Chemsitry and Applications; Yamamoto, H., Ed.; Springer: Heidelberg, Germany, 2000.
Return to citation in text: [1] -
Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/352760393X
Return to citation in text: [1] -
Sipyagin, A. M.; Bateman, C. P.; Tan, Y.-T.; Thrasher, J. S. J. Fluorine Chem. 2001, 112, 287–295. doi:10.1016/S0022-1139(01)00514-0
Return to citation in text: [1] -
Sipyagin, A. M.; Enshov, V. S.; Kashtanov, S. A.; Bateman, C. P.; Mullen, B. D.; Tan, Y.-T.; Thrasher, J. S. J. Fluorine Chem. 2004, 125, 1305–1316. doi:10.1016/j.jfluchem.2004.03.008
Return to citation in text: [1] -
Bowden, R. D.; Comina, P. J.; Greenhall, M. P.; Kariuki, B. M.; Loveday, A.; Philp, D. Tetrahedron 2000, 56, 3399–3408. doi:10.1016/S0040-4020(00)00184-8
Return to citation in text: [1] -
Ou, X.; Janzen, A. F. J. Fluorine Chem. 2000, 101, 279–283. doi:10.1016/S0022-1139(99)00171-2
Return to citation in text: [1] -
Hoover, F. W.; Coffman, D. D. J. Org. Chem. 1964, 29, 3567–3570. doi:10.1021/jo01035a030
Return to citation in text: [1] -
Winter, R. W.; Gard, G. L. J. Fluorine Chem. 2004, 125, 549–552. doi:10.1016/j.jfluchem.2003.11.028
Return to citation in text: [1] -
Sergeeva, T. A.; Dolbier, W. R., Jr. Org. Lett. 2004, 6, 2417–2419. doi:10.1021/ol0491991
Return to citation in text: [1] -
Dolbier, W. R., Jr.; Mitani, A.; Warren, R. D. Tetrahedron Lett. 2007, 48, 1325–1326. doi:10.1016/j.tetlet.2006.12.123
Return to citation in text: [1] -
Dolbier, W. R., Jr.; Mitani, A.; Xu, W.; Ghiviriga, I. Org. Lett. 2006, 8, 5573–5575. doi:10.1021/ol0622662
Return to citation in text: [1] -
Dolbier, W. R., Jr.; Zheng, Z. J. Fluorine Chem. 2011, 132, 389–393. doi:10.1016/j.jfluchem.2011.03.017
Return to citation in text: [1] -
Dolbier, W. R., Jr.; Zheng, Z. J. Org. Chem. 2009, 74, 5626–5628. doi:10.1021/jo9007699
Return to citation in text: [1] -
Wessel, J.; Hartl, H.; Seppelt, K. Chem. Ber. 1986, 119, 453–463. doi:10.1002/cber.19861190208
Return to citation in text: [1] [2] -
Umemoto, T. Process for producing arylsulfur pentafluorides. WO Patent WO/2008/118787, Oct 2, 2008.
Return to citation in text: [1] -
Umemoto, T. Process for producing arylsulfur pentafluorides. U.S. Patent 7,592,491, Sept 22, 2009.
Return to citation in text: [1] -
Umemoto, T. Process for producing arylsulfur pentafluorides. U.S. Patent 7,820,864, Oct 26, 2010.
Return to citation in text: [1] -
Umemoto, T. Process for producing arylsulfur pentafluorides. U.S. Patent 7,851,646, Dec 14, 2010.
Return to citation in text: [1] -
Umemoto, T. Method for producing fluorinated phenylsulfur pentafluorides. U.S. Patent Appl. 2010/0,130,790, May 27, 2010.
Return to citation in text: [1] -
Umemoto, T. Processes for preparing poly(pentafluorosulfanyl)aromatic compounds. WO Patent WO/2010/033930, March 25, 2010.
Return to citation in text: [1] -
Pashinnik, V. E.; Martyniuk, E. G.; Tabachuk, M. R.; Shermolovich, Yu. G.; Yagupolskii, L. M. Synth. Commun. 2003, 33, 2505–2509. doi:10.1081/SCC-120021841
Return to citation in text: [1] -
Umemoto, T.; Singh, R. P.; Xu, Y.; Saito, N. J. Am. Chem. Soc. 2010, 132, 18199–18205. doi:10.1021/ja106343h
Return to citation in text: [1] [2] [3] [4] [5] -
Xu, W.; Martinez, H.; Dolbier, W. R., Jr. J. Fluorine Chem. 2011, 132, 482–488. doi:10.1016/j.jfluchem.2011.05.001
(Discussing the use of bromine (Br2) instead of chlorine (Cl2) for the preparation of arylsulfur trifluorides.)
Return to citation in text: [1] -
Ou, X.; Bernard, G. M.; Janzen, A. F. Can. J. Chem. 1997, 75, 1878–1884. doi:10.1139/v97-621
Return to citation in text: [1] [2] [3] [4] [5] -
Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3058–3063. doi:10.1021/ja00875a005
It was described that the hydrolysis of PhSF3 to benzenesulfinic acid occurs with almost explosive violence.
Return to citation in text: [1] -
Taft, R. W.; Price, E.; Fox, I. R.; Lewis, I. C.; Anderson, K. K.; Davis, G. T. J. Am. Chem. Soc. 1963, 85, 3146–3155. doi:10.1021/ja00903a022
Return to citation in text: [1] -
Kice, J. L.; Hampton, D. C.; Fitzgerald, A. J. Org. Chem. 1965, 30, 882–885. doi:10.1021/jo01014a053
Return to citation in text: [1]
| 52. | Ou, X.; Bernard, G. M.; Janzen, A. F. Can. J. Chem. 1997, 75, 1878–1884. doi:10.1139/v97-621 |
| 52. | Ou, X.; Bernard, G. M.; Janzen, A. F. Can. J. Chem. 1997, 75, 1878–1884. doi:10.1139/v97-621 |
| 50. | Umemoto, T.; Singh, R. P.; Xu, Y.; Saito, N. J. Am. Chem. Soc. 2010, 132, 18199–18205. doi:10.1021/ja106343h |
| 53. |
Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3058–3063. doi:10.1021/ja00875a005
It was described that the hydrolysis of PhSF3 to benzenesulfinic acid occurs with almost explosive violence. |
| 1. | Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2004; p 151. doi:10.1002/352760393X |
| 4. | Hansch, C.; Muir, R. M.; Fujita, T.; Maloney, P. P.; Geiger, F.; Streich, M. J. Am. Chem. Soc. 1963, 85, 2817–2824. doi:10.1021/ja00901a033 |
| 34. | Ou, X.; Janzen, A. F. J. Fluorine Chem. 2000, 101, 279–283. doi:10.1016/S0022-1139(99)00171-2 |
| 35. | Hoover, F. W.; Coffman, D. D. J. Org. Chem. 1964, 29, 3567–3570. doi:10.1021/jo01035a030 |
| 2. | Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3072–3076. doi:10.1021/ja00875a007 |
| 3. | Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3064–3072. doi:10.1021/ja00875a006 |
| 3. | Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3064–3072. doi:10.1021/ja00875a006 |
| 20. | Kirsch, P.; Hahn, A. Eur. J. Org. Chem. 2005, 3095–3100. doi:10.1002/ejoc.200500125 |
| 31. | Sipyagin, A. M.; Bateman, C. P.; Tan, Y.-T.; Thrasher, J. S. J. Fluorine Chem. 2001, 112, 287–295. doi:10.1016/S0022-1139(01)00514-0 |
| 32. | Sipyagin, A. M.; Enshov, V. S.; Kashtanov, S. A.; Bateman, C. P.; Mullen, B. D.; Tan, Y.-T.; Thrasher, J. S. J. Fluorine Chem. 2004, 125, 1305–1316. doi:10.1016/j.jfluchem.2004.03.008 |
| 54. | Taft, R. W.; Price, E.; Fox, I. R.; Lewis, I. C.; Anderson, K. K.; Davis, G. T. J. Am. Chem. Soc. 1963, 85, 3146–3155. doi:10.1021/ja00903a022 |
| 33. | Bowden, R. D.; Comina, P. J.; Greenhall, M. P.; Kariuki, B. M.; Loveday, A.; Philp, D. Tetrahedron 2000, 56, 3399–3408. doi:10.1016/S0040-4020(00)00184-8 |
| 55. | Kice, J. L.; Hampton, D. C.; Fitzgerald, A. J. Org. Chem. 1965, 30, 882–885. doi:10.1021/jo01014a053 |
| 19. | Nixon, P. G.; Winter, R.; Castner, D. G.; Holcomb, N. R.; Grainger, D. W.; Gard, G. L. Chem. Mater. 2000, 12, 3108–3112. doi:10.1021/cm000339k |
| 20. | Kirsch, P.; Hahn, A. Eur. J. Org. Chem. 2005, 3095–3100. doi:10.1002/ejoc.200500125 |
| 21. | Gao, H.; Ye, C.; Winter, R. W.; Gard, G. L.; Sitzmann, M. E.; Shreeve, J. M. Eur. J. Inorg. Chem. 2006, 3221–3226. doi:10.1002/ejic.200600098 |
| 22. | Ye, C.; Gard, G. L.; Winter, R. W.; Syvret, R. G.; Twamley, B.; Shreeve, J. M. Org. Lett. 2007, 9, 3841–3844. doi:10.1021/ol701602a |
| 23. | Zahn, S.; Nordquist, A. F.; Minnich, K. E.; Lal, G. S.; Burgoyne, W. F., Jr.; Klauck-Jacobs, A. Pentafluorosulfanyl-substituted thienothiophene monomers and conducting polymers. U.S. Patent 7,060,846, June 13, 2006. |
| 27. | Banks, R. E., Ed. Organofluorine Chemicals and Their Industrial Applications; Ellis Horwood Ltd.: Chichester, 1979. |
| 28. | Banks, R. E.; Smart, B. E.; Tatlow, J. C., Eds. Organofluorine Chemistry, Principles and Commercial Applications; Plenum Press: New York, 1994. |
| 29. | Hiyama, T. In Organofluorine Compounds, Chemsitry and Applications; Yamamoto, H., Ed.; Springer: Heidelberg, Germany, 2000. |
| 30. | Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/352760393X |
| 15. | Crowley, P. J.; Mitchell, G.; Salmon, R.; Worthington, P. A. Chimia 2004, 58, 138–142. |
| 16. | Lim, D. S.; Choi, J. S.; Pak, C. S.; Welch, J. T. J. Pestic. Sci. 2007, 32, 255–259. doi:10.1584/jpestics.G06-50 |
| 17. | Chern, R. T.; Zingerman, J. R.; Clark, J. N.; Drag, M. D. Sulfurpentafluorophenyl pyrazoles for controlling ectoparasitic infestations. WO Patent WO/1999/047139, Sept 23, 1999. |
| 18. | Matsuzaki, Y.; Morimoto, M.; Fujioka, S.; Tohnishi, M. Phtalimide derivative, agricultural or horticultural insecticide, and method of use thereof. WO Patent WO/2003/093228, Nov 13, 2003. |
| 52. | Ou, X.; Bernard, G. M.; Janzen, A. F. Can. J. Chem. 1997, 75, 1878–1884. doi:10.1139/v97-621 |
| 5. | Stump, B.; Eberle, C.; Schweizer, W. B.; Kaiser, M.; Brun, R.; Krauth-Siegel, R. L.; Lentz, D.; Diederich, F. ChemBioChem 2009, 10, 79–83. doi:10.1002/cbic.200800565 |
| 6. | Mo, T.; Mi, X.; Milner, E. E.; Dow, G. S.; Wipf, P. Tetrahedron Lett. 2010, 51, 5137–5140. doi:10.1016/j.tetlet.2010.07.113 |
| 7. | Gujjar, R.; El Mazouni, F.; White, K. L.; White, J.; Creason, S.; Shackleford, D. M.; Deng, X.; Charman, W. N.; Bathurst, I.; Burrows, J.; Floyd, D. M.; Matthews, D.; Buckner, F. S.; Charman, S. A.; Phillips, M. A.; Rathod, P. K. J. Med. Chem. 2011, 54, 3935–3949. doi:10.1021/jm200265b |
| 8. | Coteron, J. M.; Marco, M.; Esquivias, J.; Deng, X.; White, K. L.; White, J.; Koltun, M.; El Mazouni, F.; Kokkonda, S.; Katneni, K.; Bhamidipati, R.; Shackleford, D. M.; Angulo-Barturen, I.; Ferrer, S. B.; Jiménez-Díaz, M. B.; Gamo, F.-J.; Goldsmith, E. J.; Charman, W. N.; Bathurst, I.; Floyd, D.; Matthews, D.; Burrows, J. N.; Rathod, P. K.; Charman, S. A.; Phillips, M. A. J. Med. Chem. 2011, 54, 5540–5561. doi:10.1021/jm200592f |
| 9. | Bossemaier, B.; Friebe, W.-G.; Georges, G.; Rueth, M.; Voss, E. Novel pentafluorosulfanyl compounds. U.S. Patent Appl. 0,197,370, Sept 8, 2005. |
| 10. | Andeotti, D.; Checchia, A.; Hamprecht, D.; Micheli, F. 3-triazolylthioalkyl-3-azabicyclo(3.1.0)hexanes and their use as dopamine D3 receptor ligands. WO Patent WO/2006/108700, Oct 19, 2006. |
| 11. | Frank, R.; Sundermann, B.; Schick, H. Pentafluorosulphanyl-substituted compound and its use for producing medicaments. WO Patent WO/2006/122773, Nov 23, 2006. |
| 12. | Billen, D.; Boyle, J.; Critcher, D. J.; Gethin, D. M.; Hall, K. T.; Kyne, G. M. Substituted arylpyrazoles. US Patent Appl. 176,865, July 24, 2008. |
| 13. | Kleeman, H.-W. Pentafluorosulfanylphenyl-substituted benzoylguanidines, method for the production thereof, their use as a medicament or diagnostic agent, and a medicament containing these compounds. U.S. Patent 7,446,225, Nov 4, 2008. |
| 14. | Stamford, A. W.; Cumming, J. N. Pentafluorosulfurimino heterocyclic compounds as BACE-1 inhibitors, compositions and their use. WO Patent WO/2011/044184, April 11, 2011. |
| 50. | Umemoto, T.; Singh, R. P.; Xu, Y.; Saito, N. J. Am. Chem. Soc. 2010, 132, 18199–18205. doi:10.1021/ja106343h |
| 24. | Simons, J. H.; Lewis, C. J. J. Am. Chem. Soc. 1938, 60, 492. doi:10.1021/ja01269a507 |
| 25. | Pouterman, E.; Girardet, A. Helv. Chim. Acta 1947, 30, 107–112. doi:10.1002/hlca.19470300114 |
| 26. | McBee, E. T.; Hass, H. B.; Weiner, P. E.; Rothrock, G. M.; Burt, W. E.; Robb, R. M.; Van Dyken, A. R. Ind. Eng. Chem. 1947, 39, 298–301. doi:10.1021/ie50447a613 |
| 52. | Ou, X.; Bernard, G. M.; Janzen, A. F. Can. J. Chem. 1997, 75, 1878–1884. doi:10.1139/v97-621 |
| 22. | Ye, C.; Gard, G. L.; Winter, R. W.; Syvret, R. G.; Twamley, B.; Shreeve, J. M. Org. Lett. 2007, 9, 3841–3844. doi:10.1021/ol701602a |
| 38. | Dolbier, W. R., Jr.; Mitani, A.; Warren, R. D. Tetrahedron Lett. 2007, 48, 1325–1326. doi:10.1016/j.tetlet.2006.12.123 |
| 39. | Dolbier, W. R., Jr.; Mitani, A.; Xu, W.; Ghiviriga, I. Org. Lett. 2006, 8, 5573–5575. doi:10.1021/ol0622662 |
| 40. | Dolbier, W. R., Jr.; Zheng, Z. J. Fluorine Chem. 2011, 132, 389–393. doi:10.1016/j.jfluchem.2011.03.017 |
| 41. | Dolbier, W. R., Jr.; Zheng, Z. J. Org. Chem. 2009, 74, 5626–5628. doi:10.1021/jo9007699 |
| 36. | Winter, R. W.; Gard, G. L. J. Fluorine Chem. 2004, 125, 549–552. doi:10.1016/j.jfluchem.2003.11.028 |
| 37. | Sergeeva, T. A.; Dolbier, W. R., Jr. Org. Lett. 2004, 6, 2417–2419. doi:10.1021/ol0491991 |
| 50. | Umemoto, T.; Singh, R. P.; Xu, Y.; Saito, N. J. Am. Chem. Soc. 2010, 132, 18199–18205. doi:10.1021/ja106343h |
| 51. |
Xu, W.; Martinez, H.; Dolbier, W. R., Jr. J. Fluorine Chem. 2011, 132, 482–488. doi:10.1016/j.jfluchem.2011.05.001
(Discussing the use of bromine (Br2) instead of chlorine (Cl2) for the preparation of arylsulfur trifluorides.) |
| 52. | Ou, X.; Bernard, G. M.; Janzen, A. F. Can. J. Chem. 1997, 75, 1878–1884. doi:10.1139/v97-621 |
| 49. | Pashinnik, V. E.; Martyniuk, E. G.; Tabachuk, M. R.; Shermolovich, Yu. G.; Yagupolskii, L. M. Synth. Commun. 2003, 33, 2505–2509. doi:10.1081/SCC-120021841 |
| 50. | Umemoto, T.; Singh, R. P.; Xu, Y.; Saito, N. J. Am. Chem. Soc. 2010, 132, 18199–18205. doi:10.1021/ja106343h |
| 50. | Umemoto, T.; Singh, R. P.; Xu, Y.; Saito, N. J. Am. Chem. Soc. 2010, 132, 18199–18205. doi:10.1021/ja106343h |
| 42. | Wessel, J.; Hartl, H.; Seppelt, K. Chem. Ber. 1986, 119, 453–463. doi:10.1002/cber.19861190208 |
| 43. | Umemoto, T. Process for producing arylsulfur pentafluorides. WO Patent WO/2008/118787, Oct 2, 2008. |
| 44. | Umemoto, T. Process for producing arylsulfur pentafluorides. U.S. Patent 7,592,491, Sept 22, 2009. |
| 45. | Umemoto, T. Process for producing arylsulfur pentafluorides. U.S. Patent 7,820,864, Oct 26, 2010. |
| 46. | Umemoto, T. Process for producing arylsulfur pentafluorides. U.S. Patent 7,851,646, Dec 14, 2010. |
| 47. | Umemoto, T. Method for producing fluorinated phenylsulfur pentafluorides. U.S. Patent Appl. 2010/0,130,790, May 27, 2010. |
| 48. | Umemoto, T. Processes for preparing poly(pentafluorosulfanyl)aromatic compounds. WO Patent WO/2010/033930, March 25, 2010. |
| 42. | Wessel, J.; Hartl, H.; Seppelt, K. Chem. Ber. 1986, 119, 453–463. doi:10.1002/cber.19861190208 |
© 2012 Umemoto et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)