Abstract
Water-soluble dendritic iridium catalysts were synthesized by attaching a reactive metal complex to DAB-Am dendrimers via an adapted asymmetric bipyridine ligand. These dendritic catalysts were applied in the aqueous reductive amination of valine while contained in a dialysis bag. Comparable conversions were observed as for the noncompartmentalized counterparts, albeit with somewhat longer reaction times. These results clearly show that the encapsulated catalyst system is suitable to successfully drive a complex reaction mixture with various equilibrium reactions to completion.
Graphical Abstract
Introduction
Cascade catalysis, a bioinspired strategy to conduct multiple consecutive catalytic steps in one pot, is attracting the attention of an increasing number of chemists [1-9]. Advantages of cascade processes include a reduction in the number of workup and purification steps, but also the fact that unstable intermediates can be immediately further reacted, or that unfavorable equilibria can be driven to the desired product. An obvious drawback of such a strategy may be the often intrinsic incompatibility of the catalysts and/or enzymes involved, which will lead to incomplete conversions. Incompatible catalysts can be physically separated in various ways, e.g., by applying biphasic reaction conditions [10], membrane reactors [11] or sol–gels [12]. Another way to circumvent incompatibility problems is to achieve compartmentalization by attaching the actual catalyst to larger particles and contain them in an environment that is accessible for the substrate molecules, but impermeable to the macromolecular catalyst. Such “pseudo-homogeneous catalysts” can, amongst other methods, be created by immobilizing soluble metal complexes on nanoparticles, either nonmagnetic [13,14] or magnetic ones [15,16], which can be contained in semipermeable membranes and hence be physically separated from other catalysts. Nanoparticles, however, may be unstable, and stabilizers, which may affect the catalyst behavior, are often necessary to prevent aggregation [17,18].
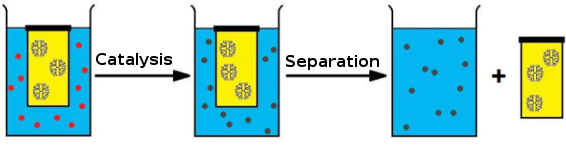
Dendrimers can also be used as scaffolds for creating macromolecular catalysts [19], which generally show only a limited loss in activity compared to their “nonexpanded” congeners [20,21]. Such dendritic catalysts are kept in a compartment due to the macromolecular nature of these polymers, and have been utilized for purification purposes after the reaction [22] and in continuous-flow reactors during the reaction [23]. Dendritic catalysts have also been applied while enclosed in commercially available dialysis bags [24-26]. The latter examples, however, were conducted in organic, environmentally unfriendly solvents. As part of a research program, in which we focus on conducting cascade reactions catalyzed by the joint action of organometallic catalysts and enzymes, we studied possibilities to compartmentalize metal catalysts in an aqueous environment. Inspired by the aforementioned dendrimer examples, we decided to design a metallodendrimer that would show catalytic activity in a cascade process while compartmentalized under aqueous conditions. To this end, an iridium catalyst was selected that was known to be suitable for reductive amination in an aqueous environment. We showed that by applying a dendritic analogue in a dialysis device we were able to successfully drive the aqueous reductive amination of an unprotected amino acid to completion, despite the unfavorable equilibria for iminium ion formation in water. Since the dendrimer remains inside the dialysis bag due to its size, the catalyst can also be easily removed from the reaction mixture after the reaction has been completed (Figure 1).
![[1860-5397-9-110-1]](/bjoc/content/figures/1860-5397-9-110-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: General approach for the use of dendritic catalysts in a dialysis bag.
Figure 1: General approach for the use of dendritic catalysts in a dialysis bag.
Results and Discussion
To study this approach, we selected iridium catalyst 3, which has been previously successfully applied in aqueous reductive aminations [27]. The catalyst is based on related iridium complexes that are capable of performing transfer hydrogenation reactions, as has been documented by Fukuzumi et al. [28-30]. The water-soluble iridium catalyst 3 was prepared according to a procedure of Francis in high yield (Scheme 1) [27]. The starting material is [Cp*IrCl2]2 which readily coordinates to bipyridine 1 to form iridium complex 2. Subsequent abstraction of the chloride ion in the presence of Ag2SO4 afforded the active species 3 in excellent yield.
![[1860-5397-9-110-i1]](/bjoc/content/inline/1860-5397-9-110-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of water-soluble iridium catalyst 3.
Scheme 1: Synthesis of water-soluble iridium catalyst 3.
To attach catalyst 3 to a dendrimer, an amine functionality was introduced on the bipyridine ligand in three steps (Scheme 2). The first step involved demethylation of 4,4’-dimethoxy-2,2’-bipyridine (1) in 92% yield [31]. Monofunctionalization of the ligand will lead to the highest possible catalyst loading per dendrimer and also prevents cross-linking between dendrimers. Therefore, a 1:1 mixture of isopropyl mesylate (5) and tetraethylene glycol azido mesylate 6 was reacted with diol 4, affording a mixture of the desired asymmetric bipyridine 7 and the two corresponding symmetric bipyridines. This mixture was separated by column chromatography resulting in 32% isolated yield of pure azide 7. Subsequent Staudinger reduction and purification by acid–base extraction afforded the amine-containing ligand 8 in 96% yield.
![[1860-5397-9-110-i2]](/bjoc/content/inline/1860-5397-9-110-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the desymmetrized bipyridine 8.
Scheme 2: Synthesis of the desymmetrized bipyridine 8.
Ligand 8 was connected to water-soluble DAB-Am dendrimers via a protocol described by Peerlings and Meijer (Scheme 3) [32]. First, a multi-isocyanate was prepared in situ by using di-tert-butyl tricarbonate (11). After the addition of pyridine to quench the excess of tricarbonate 11, ligand 8 was added, leading to the formation of dendrimers 12 and 13 in good yields. The products were completely characterized by 1H and 13C NMR, thereby confirming that all amines had been reacted. Furthermore, IR spectroscopy showed signals at 1580 and 1640 cm−1 derived from the newly formed urea functionalities. The next reaction, coordination of iridium to the dendritic ligands in methanol, afforded the corresponding iridium complexes 14 and 15 in quantitative yield. In the final step, the complex was converted into the water-soluble dendritic catalysts 16 and 17 by overnight treatment with Ag2SO4 in 46 and 47% yield, respectively. During the removal of AgCl by centrifugation, some dendritic material was probably lost in the pellet, explaining the rather moderate yield of the final step. Dendrimers 16 and 17 were purified by aqueous dialysis. Unfortunately, the NMR spectra of dendrimers 14–17 could not be completely resolved due to the broad peaks observed as well as the low intensity of the resonances in the 13C NMR measurements. However, each transformation was accompanied by a clear and complete change in chemical shift of the aromatic bipyridine peaks.
![[1860-5397-9-110-i3]](/bjoc/content/inline/1860-5397-9-110-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Attachment of the adapted ligand 8 to the dendrimers via a multi-isocyanate coupling, followed by iridium coordination.
Scheme 3: Attachment of the adapted ligand 8 to the dendrimers via a multi-isocyanate coupling, followed by i...
The reactivity of macromolecular catalysts 16 and 17 was examined in the aqueous reductive amination of valine (18, Scheme 4). This reaction is in fact a multistep process, in which the equilibria unfavorable in water for the formation of hemiaminal 20 and iminium ion 21 are compensated by the iridium-catalyzed reduction to form the benzylated amino acid 19. Iridium complexes 3, 16 and 17 were capable of reducing the intermediate imine in the presence of HCO2K, which acted as the hydride source. The reaction was monitored by taking aliquots of the reaction mixture and by HPLC analysis. An excess of benzaldehyde was used, because it was also partially reduced to benzyl alcohol in a side reaction. The optimal pH appeared to be 5, comparable with a similar catalyst published by Fukuzumi [29,30].
![[1860-5397-9-110-i4]](/bjoc/content/inline/1860-5397-9-110-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Catalytic reductive amination of valine (18) via unfavorable equilibrium reactions in water.
Scheme 4: Catalytic reductive amination of valine (18) via unfavorable equilibrium reactions in water.
In the presence of the nondendritic catalyst 3, formation of the N-alkylated amino acid 19 proceeded considerably faster than with the dendrimer-supported reductive aminations (Figure 2). However, after longer reaction times the yields were only slightly lower for the dendritic catalysts.
![[1860-5397-9-110-2]](/bjoc/content/figures/1860-5397-9-110-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Formation of 19 catalyzed by the three iridium catalysts.
Figure 2: Formation of 19 catalyzed by the three iridium catalysts.
In order to study the same reaction with the dendritic catalysts 16 and 17 in a dialysis bag, a different setup was used based on commercially available materials. This consisted of a cup which could be closed and contained a dialysis membrane with a molecular-weight cut-off value of 2,000. We assumed that all reaction components except the catalyst would be able to pass through the membrane, turning the dialysis tube into a compartment suitable for catalysis. To verify this hypothesis, catalyst 16 was injected in the dialysis device and the reductive amination of valine was performed under otherwise identical conditions (Figure 3). HPLC analysis showed that the reaction rate and conversion were fairly similar to the noncompartmentalized aqueous reaction (Figure 4), demonstrating that mass transport of the small substrate and product molecules through the membrane is hardly rate-limiting.
![[1860-5397-9-110-3]](/bjoc/content/figures/1860-5397-9-110-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Reaction setup to perform compartmentalized catalysis.
Figure 3: Reaction setup to perform compartmentalized catalysis.
![[1860-5397-9-110-4]](/bjoc/content/figures/1860-5397-9-110-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: G3 catalyst 16 activity in dialysis device.
Figure 4: G3 catalyst 16 activity in dialysis device.
After the appropriate reaction time, the dialysis tube was removed from the reaction mixture, dialyzed in buffer to remove all the other reaction contents and added to a fresh reaction mixture. In the second run, the recovered G3 dendrimer 16 still showed catalytic activity (Figure 4), albeit that a distinct decrease in reaction rate was observed. This phenomenon may be attributed to leakage of the catalyst through the dialysis membrane.
Similar experiments were conducted with the G4 dendrimer 17 (Figure 5). The figure shows that the reaction rate is somewhat lower, but that the conversion is fairly similar to that for the free dendritic catalyst. In contrast to the smaller G3 dendrimer 16, however, the G4 catalyst 17 was equally active in a second run, clearly demonstrating the viability of containment of the catalyst in the dialysis bag. This result also suggests that the immobilized catalyst should be applicable in cascade reactions with reagents that are normally incompatible with the iridium complex. Since our process was successfully conducted in water, combinations with enzymes should also be possible.
![[1860-5397-9-110-5]](/bjoc/content/figures/1860-5397-9-110-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: G4 catalyst 17 activity in subsequent runs.
Figure 5: G4 catalyst 17 activity in subsequent runs.
Conclusion
Dendritic catalysts were successfully synthesized by connecting Ir-bipyridine complexes to third and fourth generation DAB-Am dendrimers. The dendritic catalysts showed good activity in the multistep reductive amination of a free amino acid in water, albeit that prolonged reaction times as compared to the free catalyst were required. Due to the size of the macromolecular catalysts, it was possible to employ them in a dialysis device for conducting reductive aminations of free amino acids in water. The G4 catalyst showed the same reaction rate in a second run, which validates the concept of maintaining the macromolecular catalyst in the compartment. The work described here could be more widely applicable for compartmentalization of catalytic systems in aqueous media, where the iridium system may be used either in combination with other catalysts, or with enzymes. These topics are currently under investigation in our group.
References
-
Bruggink, A.; Schoevaart, R.; Kieboom, T. Org. Process Res. Dev. 2003, 7, 622–640. doi:10.1021/op0340311
Return to citation in text: [1] -
Tietze, L. F. Chem. Rev. 1996, 96, 115–136. doi:10.1021/cr950027e
Return to citation in text: [1] -
Zhou, J. Chem.–Asian J. 2010, 5, 422–434. doi:10.1002/asia.200900458
Return to citation in text: [1] -
Ricca, E.; Brucher, B.; Schrittwieser, J. H. Adv. Synth. Catal. 2011, 353, 2239–2262. doi:10.1002/adsc.201100256
Return to citation in text: [1] -
Enders, D.; Hüttl, M. R. M.; Grondal, C.; Raabe, G. Nature 2006, 441, 861–863. doi:10.1038/nature04820
Return to citation in text: [1] -
Rueping, M.; Lin, M.-Y. Chem.–Eur. J. 2010, 16, 4169–4172. doi:10.1002/chem.201000203
Return to citation in text: [1] -
Carlone, A.; Cabrera, S.; Marigo, M.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2007, 46, 1101–1104. doi:10.1002/anie.200604479
Return to citation in text: [1] -
Babich, L.; van Hemert, L. J. C.; Bury, A.; Hartog, A. F.; Falcicchio, P.; van der Oost, J.; van Herk, T.; Wever, R.; Rutjes, F. P. J. T. Green Chem. 2011, 13, 2895–2900. doi:10.1039/c1gc15429j
Return to citation in text: [1] -
Babich, L.; Hartog, A. F.; van Hemert, L. J. C.; Rutjes, F. P. J. T.; Wever, R. ChemSusChem 2012, 5, 2348–2353. doi:10.1002/cssc.201200468
Return to citation in text: [1] -
Karmee, S. K.; Roosen, C.; Kohlmann, C.; Lütz, S.; Greiner, L.; Leitner, W. Green Chem. 2009, 11, 1052–1055. doi:10.1039/b820606f
Return to citation in text: [1] -
Cantarella, L.; Gallifuoco, A.; Malandra, A.; Martínková, L.; Spera, A.; Cantarella, M. Enzyme Microb. Technol. 2011, 48, 345–350. doi:10.1016/j.enzmictec.2010.12.010
Return to citation in text: [1] -
Gelman, F.; Blum, J.; Avnir, D. J. Am. Chem. Soc. 2000, 122, 11999–12000. doi:10.1021/ja003029b
Return to citation in text: [1] -
Astruc, D.; Lu, F.; Aranzaes, J. R. Angew. Chem., Int. Ed. 2005, 44, 7852–7872. doi:10.1002/anie.200500766
Return to citation in text: [1] -
Wang, S.; Zhang, M.; Zhong, L.; Zhang, W. J. Mol. Catal. A: Chem. 2010, 327, 92–100. doi:10.1016/j.molcata.2010.05.018
Return to citation in text: [1] -
Jin, M.-J.; Lee, D.-H. Angew. Chem., Int. Ed. 2010, 49, 1119–1122. doi:10.1002/anie.200905626
Return to citation in text: [1] -
Stevens, P. D.; Li, G.; Fan, J.; Yenb, M.; Gao, Y. Chem. Commun. 2005, 4435–4437. doi:10.1039/b505424a
Return to citation in text: [1] -
Hayek, K.; Kramer, R.; Paál, Z. Appl. Catal., A 1997, 162, 1–15. doi:10.1016/S0926-860X(97)00243-3
Return to citation in text: [1] -
Schmidt, E.; Kleist, W.; Krumeich, F.; Mallat, T.; Baiker, A. Chem.–Eur. J. 2010, 16, 2181–2192. doi:10.1002/chem.200902517
Return to citation in text: [1] -
Grayson, S. M.; Fréchet, J. M. J. Chem. Rev. 2001, 101, 3819–3867. doi:10.1021/cr990116h
Return to citation in text: [1] -
Méry, D.; Astruc, D. Coord. Chem. Rev. 2006, 250, 1965–1979. doi:10.1016/j.ccr.2005.11.012
Return to citation in text: [1] -
Knapen, J. W. J.; van der Made, A. W.; de Wilde, J. C.; van Leeuwen, P. W. N. M.; Wijkens, P.; Grove, D. M.; van Koten, G. Nature 1994, 372, 659–663. doi:10.1038/372659a0
Return to citation in text: [1] -
Köllner, C.; Pugin, B.; Togni, A. J. Am. Chem. Soc. 1998, 120, 10274–10275. doi:10.1021/ja981137g
Return to citation in text: [1] -
Brinkmann, N.; Giebel, D.; Lohmer, G.; Reetz, M. T.; Kragl, U. J. Catal. 1999, 183, 163–168. doi:10.1006/jcat.1999.2462
Return to citation in text: [1] -
Pijnenburg, N. J. M.; Dijkstra, H. P.; van Koten, G.; Klein Gebbink, R. J. M. Dalton Trans. 2011, 40, 8896–8905. doi:10.1039/c1dt10502g
Return to citation in text: [1] -
Arink, A. M.; van de Coevering, R.; Wieczorek, B.; Firet, J.; Jastrzebski, J. T. B. H.; Klein Gebbink, R. J. M.; van Koten, G. J. Organomet. Chem. 2004, 689, 3813–3819. doi:10.1016/j.jorganchem.2004.07.023
Return to citation in text: [1] -
Gaab, M.; Bellemin-Laponnaz, S.; Gade, L. H. Chem.–Eur. J. 2009, 15, 5450–5462. doi:10.1002/chem.200900504
Return to citation in text: [1] -
McFarland, J. M.; Francis, M. B. J. Am. Chem. Soc. 2005, 127, 13490–13491. doi:10.1021/ja054686c
Return to citation in text: [1] [2] -
Abura, T.; Ogo, S.; Watanabe, Y.; Fukuzumi, S. J. Am. Chem. Soc. 2003, 125, 4149–4154. doi:10.1021/ja0288237
Return to citation in text: [1] -
Ogo, S.; Uehara, K.; Abura, T.; Fukuzumi, S. J. Am. Chem. Soc. 2004, 126, 3020–3021. doi:10.1021/ja031633r
Return to citation in text: [1] [2] -
Ogo, S.; Uehara, K.; Abura, T.; Watanabe, Y.; Fukuzumi, S. J. Am. Chem. Soc. 2004, 126, 16520–16527. doi:10.1021/ja0473541
Return to citation in text: [1] [2] -
Hong, Y.-R.; Gorman, C. B. J. Org. Chem. 2003, 68, 9019–9025. doi:10.1021/jo0351116
Return to citation in text: [1] -
Peerlings, H. W. I.; Meijer, E. W. Tetrahedron Lett. 1999, 40, 1021–1024. doi:10.1016/S0040-4039(98)02515-5
Return to citation in text: [1]
| 32. | Peerlings, H. W. I.; Meijer, E. W. Tetrahedron Lett. 1999, 40, 1021–1024. doi:10.1016/S0040-4039(98)02515-5 |
| 29. | Ogo, S.; Uehara, K.; Abura, T.; Fukuzumi, S. J. Am. Chem. Soc. 2004, 126, 3020–3021. doi:10.1021/ja031633r |
| 30. | Ogo, S.; Uehara, K.; Abura, T.; Watanabe, Y.; Fukuzumi, S. J. Am. Chem. Soc. 2004, 126, 16520–16527. doi:10.1021/ja0473541 |
| 1. | Bruggink, A.; Schoevaart, R.; Kieboom, T. Org. Process Res. Dev. 2003, 7, 622–640. doi:10.1021/op0340311 |
| 2. | Tietze, L. F. Chem. Rev. 1996, 96, 115–136. doi:10.1021/cr950027e |
| 3. | Zhou, J. Chem.–Asian J. 2010, 5, 422–434. doi:10.1002/asia.200900458 |
| 4. | Ricca, E.; Brucher, B.; Schrittwieser, J. H. Adv. Synth. Catal. 2011, 353, 2239–2262. doi:10.1002/adsc.201100256 |
| 5. | Enders, D.; Hüttl, M. R. M.; Grondal, C.; Raabe, G. Nature 2006, 441, 861–863. doi:10.1038/nature04820 |
| 6. | Rueping, M.; Lin, M.-Y. Chem.–Eur. J. 2010, 16, 4169–4172. doi:10.1002/chem.201000203 |
| 7. | Carlone, A.; Cabrera, S.; Marigo, M.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2007, 46, 1101–1104. doi:10.1002/anie.200604479 |
| 8. | Babich, L.; van Hemert, L. J. C.; Bury, A.; Hartog, A. F.; Falcicchio, P.; van der Oost, J.; van Herk, T.; Wever, R.; Rutjes, F. P. J. T. Green Chem. 2011, 13, 2895–2900. doi:10.1039/c1gc15429j |
| 9. | Babich, L.; Hartog, A. F.; van Hemert, L. J. C.; Rutjes, F. P. J. T.; Wever, R. ChemSusChem 2012, 5, 2348–2353. doi:10.1002/cssc.201200468 |
| 13. | Astruc, D.; Lu, F.; Aranzaes, J. R. Angew. Chem., Int. Ed. 2005, 44, 7852–7872. doi:10.1002/anie.200500766 |
| 14. | Wang, S.; Zhang, M.; Zhong, L.; Zhang, W. J. Mol. Catal. A: Chem. 2010, 327, 92–100. doi:10.1016/j.molcata.2010.05.018 |
| 27. | McFarland, J. M.; Francis, M. B. J. Am. Chem. Soc. 2005, 127, 13490–13491. doi:10.1021/ja054686c |
| 12. | Gelman, F.; Blum, J.; Avnir, D. J. Am. Chem. Soc. 2000, 122, 11999–12000. doi:10.1021/ja003029b |
| 31. | Hong, Y.-R.; Gorman, C. B. J. Org. Chem. 2003, 68, 9019–9025. doi:10.1021/jo0351116 |
| 11. | Cantarella, L.; Gallifuoco, A.; Malandra, A.; Martínková, L.; Spera, A.; Cantarella, M. Enzyme Microb. Technol. 2011, 48, 345–350. doi:10.1016/j.enzmictec.2010.12.010 |
| 27. | McFarland, J. M.; Francis, M. B. J. Am. Chem. Soc. 2005, 127, 13490–13491. doi:10.1021/ja054686c |
| 10. | Karmee, S. K.; Roosen, C.; Kohlmann, C.; Lütz, S.; Greiner, L.; Leitner, W. Green Chem. 2009, 11, 1052–1055. doi:10.1039/b820606f |
| 28. | Abura, T.; Ogo, S.; Watanabe, Y.; Fukuzumi, S. J. Am. Chem. Soc. 2003, 125, 4149–4154. doi:10.1021/ja0288237 |
| 29. | Ogo, S.; Uehara, K.; Abura, T.; Fukuzumi, S. J. Am. Chem. Soc. 2004, 126, 3020–3021. doi:10.1021/ja031633r |
| 30. | Ogo, S.; Uehara, K.; Abura, T.; Watanabe, Y.; Fukuzumi, S. J. Am. Chem. Soc. 2004, 126, 16520–16527. doi:10.1021/ja0473541 |
| 20. | Méry, D.; Astruc, D. Coord. Chem. Rev. 2006, 250, 1965–1979. doi:10.1016/j.ccr.2005.11.012 |
| 21. | Knapen, J. W. J.; van der Made, A. W.; de Wilde, J. C.; van Leeuwen, P. W. N. M.; Wijkens, P.; Grove, D. M.; van Koten, G. Nature 1994, 372, 659–663. doi:10.1038/372659a0 |
| 23. | Brinkmann, N.; Giebel, D.; Lohmer, G.; Reetz, M. T.; Kragl, U. J. Catal. 1999, 183, 163–168. doi:10.1006/jcat.1999.2462 |
| 19. | Grayson, S. M.; Fréchet, J. M. J. Chem. Rev. 2001, 101, 3819–3867. doi:10.1021/cr990116h |
| 24. | Pijnenburg, N. J. M.; Dijkstra, H. P.; van Koten, G.; Klein Gebbink, R. J. M. Dalton Trans. 2011, 40, 8896–8905. doi:10.1039/c1dt10502g |
| 25. | Arink, A. M.; van de Coevering, R.; Wieczorek, B.; Firet, J.; Jastrzebski, J. T. B. H.; Klein Gebbink, R. J. M.; van Koten, G. J. Organomet. Chem. 2004, 689, 3813–3819. doi:10.1016/j.jorganchem.2004.07.023 |
| 26. | Gaab, M.; Bellemin-Laponnaz, S.; Gade, L. H. Chem.–Eur. J. 2009, 15, 5450–5462. doi:10.1002/chem.200900504 |
| 17. | Hayek, K.; Kramer, R.; Paál, Z. Appl. Catal., A 1997, 162, 1–15. doi:10.1016/S0926-860X(97)00243-3 |
| 18. | Schmidt, E.; Kleist, W.; Krumeich, F.; Mallat, T.; Baiker, A. Chem.–Eur. J. 2010, 16, 2181–2192. doi:10.1002/chem.200902517 |
| 15. | Jin, M.-J.; Lee, D.-H. Angew. Chem., Int. Ed. 2010, 49, 1119–1122. doi:10.1002/anie.200905626 |
| 16. | Stevens, P. D.; Li, G.; Fan, J.; Yenb, M.; Gao, Y. Chem. Commun. 2005, 4435–4437. doi:10.1039/b505424a |
| 22. | Köllner, C.; Pugin, B.; Togni, A. J. Am. Chem. Soc. 1998, 120, 10274–10275. doi:10.1021/ja981137g |
© 2013 Willemsen et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)