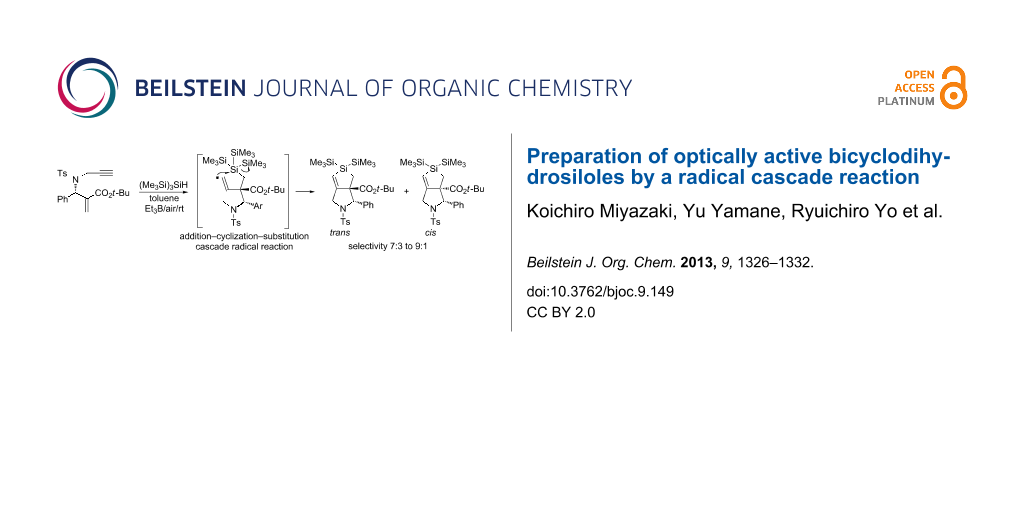
Abstract
Bicyclodihydrosiloles were readily prepared from optically active enyne compounds by a radical cascade reaction triggered by tris(trimethylsilyl)silane ((Me3Si)3SiH). The reaction was initiated by the addition of a silyl radical to an α,β-unsaturated ester, forming an α-carbonyl radical that underwent radical cyclization to a terminal alkyne unit. The resulting vinyl radical attacked the silicon atom in an SHi manner to give dihydrosilole. The reaction preferentially formed trans isomers of bicyclosiloles with an approximately 7:3 to 9:1 selectivity.

Graphical Abstract
Introduction
Radical cyclization occupies a unique position in organic synthesis because it is a useful reaction for the construction of cyclic molecules [1-10]. The radical cascade cyclization process is also an interesting synthetic reaction that often provides an efficient method [11-13]. Recently, we reported a new type of higher-order radical cascade reaction between chiral enyne compounds and Bu3SnH, which is recognized as a useful reagent in radical reactions [14]. In this reaction, radical addition–cyclization cascade followed by intramolecular radical substitution (SHi) occurred in one-pot to give optically active bicyclostannolanes in good yields [15]. We are interested in whether such a cascade SHi process might occur with other radical species. We have found that a methylthiyl radical also undergoes such a radical cascade reaction to stereoselectively give bicyclic dihydrothiophenes [16]. We expected that tris(trimethylsilyl)silane (Me3Si)3SiH [17], which is a well-known alternative to Bu3SnH in radical reactions [18-22], would be a good promoter of a similar cascade SHi reaction, because there were several reports so far that show such SHi reaction on silicon atoms progressing efficiently [23-28]. In this paper, we report a new synthesis of chiral bicyclodihydrosiloles through an addition–cyclization–SHi cascade reaction in one-pot treatment of chiral enyne compounds. A good trans-selectivity was observed in the reaction.
Results and Discussion
We examined the cascade process using optically active enyne precursor 1a, which was prepared by a Michael/aldol domino reaction to chiral sulfinimines followed by thermal elimination and N-propargylation [29,30]. We first optimized the reaction conditions. The results are summarized in Table 1.
Table 1: Radical cascade reaction under various reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-149-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | Initiator (equiv) | Temp (°C) | 2a; Yield (%)a | trans/cisb |
|---|---|---|---|---|
| 1 | AIBN (0.1) | 110 | 14 | n/a |
| 2 | AIBN (1.0) | 110 | 39 | 69/31 |
| 3 | Et3B (3.0) | 25 | 58 | 80/20 (95)c |
| 4 | Et3B (3.0) | 0 | 48 | 86/14 |
aIsolated yield. bDetermined by HPLC analyses. cEnantiomeric excess for trans-2a. Determined by chiral HPLC analysis using ChiralPak ID.
Treatment of 1 with (Me3Si)3SiH in the presence of catalytic amounts of AIBN at 110 °C resulted in the formation of the desired bicyclodihydrosilole 2a in 14% yield (Table 1, entry 1). The use of one equivalent of AIBN improved the yield of 2a to 39% (Table 1, entry 2). These results suggest that the radical chain reaction insufficiently progressed during the reaction initiated by AIBN. The product contained two diastereomers, which were separated by chromatography. The use of Et3B/air as an initiator enhanced the yield of 2a to 58% (Table 1, entry 3). The enantiomeric excess of trans-2a was estimated to be 95% by HPLC analysis, which was the same ee level of precursor 1a. Thus, no epimerization at the C3 chiral center occurred during the reaction. The stereoselectivity was improved to 8:2. The stereoselectivity was sensitive to the reaction temperature, and an 86/14 mixture of trans-2a and cis-2a was obtained when the reaction was performed at 0 °C, although the yield was less than that obtained when the reaction was performed at room temperature (Table 1, entry 4).
Having determined the optimized reaction conditions, we examined the generality of the reaction. The results are summarized in Table 2.
Table 2: Preparation of pyrrolidinodihydrosiloles 2.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-149-i5.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Ar | Product | Yielda (%) | trans/cisb | ee for trans-2c |
|---|---|---|---|---|---|
| 1 | 2-MeC6H4 | 2b | 60 | 84/16 | ndd |
| 2 | 4-MeC6H4 | 2c | 53 | 91/9 | ndd |
| 3 | 4-MeOC6H4 | 2d | 42 | 86/14 | 97 |
| 4 | 3-ClC6H4 | 2e | 42 | 71/29 | ndd |
| 5 | 4-ClC6H4 | 2f | 51 | 81/19 | 90 |
| 6 | 4-FC6H4 | 2g | 61 | 80/20 | 97 |
| 7 | 4-CF3C6H4 | 2h | 61 | 80/20 | 68 |
| 8 | 2-thienyl | 2i | 48 | 75/25 | 98 |
| 9 | 2-naphthyl | 2j | 51 | 81/19 | 99 |
aIsolated yield. bDetermined by HPLC analyses. cDetermined by HPLC analyses with a Chiral-Pak-ID. dNot determined owing to insufficient separation by chiral HPLC analyses with ChiralPak ID and IC.
For example, the reaction of 1b smoothly occurred, giving bicyclic dihydrosilole 2b in 60% yield. HPLC analysis of the reaction mixture revealed that the diastereomeric ratio of 2b was 84/16. Dihydrosiloles 2c–2j were isolated in good yields from other precursors in a trans-selective manner (Table 2, entries 2–9). Their diastereomeric ratios ranged from 9/1 to 7/3. Although we could not determine the enantiomeric excesses for some compounds of 2 because of insufficient separation by chiral HPLC analyses using ChiralPak ID and IC (Table 2, entries 1, 2, and 4), the enantiomeric excesses of most of products 2 were high, and their original values were maintained (Table 2, entries 3, 5, 6, 8, and 9). Interestingly, significant epimerization occurred during the reaction of 1h; the enantiomeric excess of 2h was only 68% ee (Table 2, entry 7).
The configuration of 2 was determined in the following manner: The major isomer of 2a was highly crystalline, which allowed the performance of X-ray crystallography. The observed data clearly showed a trans- 2a structure [31]. The ORTEP structure of major 2a, which unambiguously indicates a trans configuration, is shown in Figure 1. The 1H NMR spectra of trans-2a and other major 2 showed similar trends, and trans configurations for other major 2 were determined unambiguously.
![[1860-5397-9-149-1]](/bjoc/content/figures/1860-5397-9-149-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Unfortunately, none of the minor 2 formed suitable crystals, which precluded X-ray analysis of the minor isomers. However, their 1H NMR spectra showed several diagnostic points. For example, the tert-butyl group in the ester at the C3a position in minor 2a appeared at 1.17 ppm; this peak was substantially shifted toward higher field than trans-2a. Compared with X-ray data for the sulfur analogue of cis-2a, the tert-butyl ester group is located above the aromatic ring at C3, and expected to introduce an anisotropic effect that subsequently causes a high-field shift for the tert-butyl protons [28]. Other typical differences between the 1H NMR spectra of minor 2a and major 2a (= trans-2a) included the following: The peaks of the CH2Si group at the C4 position in minor 2a appeared at 0.92 and 2.00 ppm, whereas the corresponding peaks of trans-2a were observed at 0.49 and 1.14 ppm. In addition, we found that H6 and H3 appeared at 5.51 and 4.46 ppm, respectively, in the spectrum of minor 2a. The corresponding protons in trans-2a appeared at substantially lower-field positions at 5.86 and 5.53 ppm. We assumed that this shift was caused by another anisotropic effect of the Ts group at N2. These trends in the 1H NMR spectra were also observed in the sulfur analogues of cis-2. Thus, we concluded that the minor isomer of 2 exhibited cis configuration.
To explore the reaction mechanism, we examined the reaction of 1a without additional solvents (Scheme 1).
![[1860-5397-9-149-i1]](/bjoc/content/inline/1860-5397-9-149-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Formation of bicyclic dihydrosilole 2a under high concentration conditions.
Scheme 1: Formation of bicyclic dihydrosilole 2a under high concentration conditions.
The treatment of 1a, (Me3Si)3SiH, and Et3B in hexane under an air atmosphere gave 2a in 72%. To our surprise, this yield was better than that of the reaction performed under the usual conditions. We expected that exo-methylenepyrrolidine 4 would be a side product under these conditions, and we indeed detected an exo-methylene compound in 5% yield in the reaction mixture. However, NMR spectra and HRMS results indicated that the isolated product was compound 3, which contained a Me3SiCH2- group instead of a (Me3Si)3SiCH2- group. These results suggest that Me3Si radicals were generated during the cascade reaction, and that a small part of the radical was subsequently trapped by 1 under such conditions.
We believe this process progressed in a similar manner to our previously investigated reaction that involved tributyltin radicals [15]. A plausible reaction mechanism is depicted in Scheme 2.
![[1860-5397-9-149-i2]](/bjoc/content/inline/1860-5397-9-149-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
The (Me3Si)3Si radical attacks the β-carbon of the α,β-unsaturated ester in 1, and α-carbonyl radical A is generated. Intermediate A undergoes radical cyclization in a 5-exo-dig mode giving vinyl radical intermediate B, which is potentially reactive for attacking the silyl group in an SHi manner to give a Me3Si radical and 2. The process from B to 2 should be very rapid. Giese and co-workers have reported that the reaction rate for a similar SHi process reaches 2.4 × 105 s−1 at 80 °C [25]. Although most of the Me3Si radicals undergo hydrogen abstraction from (Me3Si)3SiH to yield a new (Me3Si)3Si radical and Me3SiH, a small fraction of the Me3Si radicals compete to attack 1; a similar cascade reaction progresses consequently, and compound 3 is formed in 5% yield under very high concentration conditions. We assume that compound 4 was not detected in the reaction product under such conditions for two reasons: first, as previously mentioned, the SHi process from intermediate B to 2 is very rapid, and the process occurs faster than intermolecular hydrogen abstraction from (Me3Si)3SiH, even under high concentration conditions. Second, the addition rate of (Me3Si)3Si radicals to alkenes should be relatively slow; the rate competes with the addition rate of Me3Si radicals to alkenes. This reason is supported by the results that indicated the yield of 2a to be much improved under high (Me3Si)3SiH concentration conditions because the addition rate should be accelerated as the concentration of (Me3Si)3SiH increased.
We examined whether a germyl radical might undergo a similar reaction with 1. Treatment of 1 with Et3GeH in the presence of Et3B, however, failed in the formation of the corresponding compound 5. This failure was probably because a carbon–germanium bond, which is supposed to be stronger than a Si–Si bond, was never cleaved under these conditions (Scheme 3). Another possibility of this failure might be that the addition rate of a triethylgermyl radical to enyne 1a was slow and less efficient.
![[1860-5397-9-149-i3]](/bjoc/content/inline/1860-5397-9-149-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, we have successfully converted chiral enyne compounds 1, which were readily available from an asymmetric aza-Morita–Baylis–Hillman equivalent reaction, into bicyclic pyrolidinodihydrosiloles 2 in good yields. These reactions progressed in a highly stereoselective manner. Further application of the present silole synthesis is now underway in our laboratory.
Experimental
General methods: All 1H and 13C NMR spectra were recorded on a JEOL JNM-ECA500 Delta2 (500 MHz for 1H, 126 MHz for 13C) spectrometer. All the reactions in this study were performed under nitrogen atmosphere unless otherwise noted. CH2Cl2 was dried over CaH2, and distilled under nitrogen before use. High-resolution mass spectra (HRMS) were measured at the Tokiwa Instrumentation Analysis Center, Yamaguchi University.
Preparation of (3S)-tert-butyl 3-phenyl-2-tosyl-5,5-bis(trimethylsilyl)-1,2,3,3a,4,5-hexahydrosilolo[3,4-c]pyrrole-3a-carboxylate (2a). A solution of 1a (85 mg, 0.201 mmol, 95% ee), (Me3Si)3SiH (0.06 mL, 0.195 mmol), and Et3B (1.0 M in hexane, 0.60 mL, 0.60 mmol) in toluene (20 mL) was stirred at room temperature under air for 15 min. The reaction mixture was concentrated in vacuo, and the residue was purified by flash chromatography (silica gel/hexane–EtOAc 15/1 to 10/1, v/v) to give 2a in 58% yield (70.2 mg, 0.117 mmol). The two diastereomers, trans-2a and cis-2a, were separated by further careful chromatography.
(3S,3aS)-tert-Butyl 3-phenyl-2-tosyl-5,5-bis(trimethylsilyl)-1,2,3,3a,4,5-hexahydrosilolo[3,4-c]pyrrole-3a-carboxylate (trans-2a). White solid; mp 144–145 °C; [α]D −31.8 (c 0.68, CHCl3); the enantiomeric purity was determined by HPLC analysis, tR 10.0 min (major), tR 11.5 min (minor) [CHIRALPAK ID (0.46 cm × 25 cm), hexane/iPrOH, 95/5, 40 °C, 1.0 mL/min] to be 95% ee; 1H NMR (500 MHz, CHCl3) δ 7.32 (d, J = 8.2 Hz, 2H), 7.26 (s, 3H), 7.24–7.07 (m, 2H), 7.03 (d, J = 7.8 Hz, 2H), 5.86 (s, 1H), 5.23 (s, 1H), 4.42 (d, J = 13.0 Hz, 1H), 3.95 (d, J = 13.0 Hz, 1H), 2.32 (s, 3H), 1.51 (s, 9H), 1.15 (d, J = 14.9 Hz, 1H), 0.50 (d, J = 14.8 Hz, 1H), 0.07 (s, 9H), −0.20 (s, 9H); 13C NMR (126 MHz, CHCl3) δ 173.9, 157.6, 142.6, 138.6, 137.1, 129.1 (2C), 128.3 (br, 4C), 127.5, 127.0 (2C), 124.2, 82.3, 71.1, 69.7, 50.5, 28.0 (3C), 21.5, 12.2, −0.3 (3C), −0.9 (3C); HRMS–ESI (positive mode; M + Na) m/z 622.2282, calcd for C30H45NNaO4SSi3, 622.2275.
(3S,3aR)-tert-Butyl 3-phenyl-2-tosyl-5,5-bis(trimethylsilyl)-1,2,3,3a,4,5-hexahydrosilolo[3,4-c]pyrrole-3a-carboxylate (cis-2a). Pale yellow oil; [α]D +97.3 (c 0.27, CHCl3); 1H NMR (500 MHz, CHCl3) δ 7.63 (d, J = 7.8 Hz, 2H), 7.57–7.50 (m, 2H), 7.33–7.22 (m, 5H), 5.51 (s, 1H), 4.60 (d, J = 14.3 Hz, 1H), 4.23 (s, 1H), 4.11 (dd, J = 14.3, 1.6 Hz, 1H), 2.39 (s, 3H), 2.00 (d, J = 12.8 Hz, 1H), 1.17 (s, 9H), 0.92 (d, J = 15.0 Hz, 1H), 0.04 (s, 9H), −0.11 (s, 9H); 13C NMR (126 MHz, CHCl3) δ 169.5, 157.8, 143.8, 138.1, 133.1, 129.9 (2C), 128.0 (2C), 127.7 (2C), 127.7, 127.1 (br, 2C), 122.8, 82.1, 75.2, 72.5, 53.7, 27.9 (3C), 21.6, 17.4, 0.3 (3C), −1.4 (3C); HRMS–ESI (positive mode; M + Na) m/z 622.2292, calcd for C30H45NNaO4SSi3, 622.2275.
Preparation of 2a under no solvent conditions (Scheme 3, neat condition). A solution of 1a (85 mg, 0.201 mmol), (Me3Si)3SiH (0.07 mL, 0.228 mmol), and Et3B (1.0 M in hexane, 0.60 mL, 0.60 mmol) was stirred at room temperature for 15 min under air. The reaction mixture was concentrated in vacuo, and the yellow residue was purified by flash chromatography (silica gel/hexane–EtOAc 30/1 to 20/1 v/v) to give 2a in 72% yield (85.6 mg, 0.143 mmol). The trans-2a/cis-2a ratio was determined to be 84/16. Careful separation of these two diastereomers gave pure trans-2a and minor isomers that contained cis-2a and 3 in a 74/26 ratio. The separation of 3 was achieved using a recycling GPC apparatus, giving pure 3 in 5% yield (5.1 mg, 0.011 mmol).
(2S,3S)-tert-Butyl 3-((trimethylsilyl)methyl)-4-methylene-2-phenyl-1-tosylpyrrolidine-3-carboxylate (3). Pale yellow oil; [α]D +3.0 (c 0.01, CHCl3); 1H NMR (500 MHz, CHCl3) δ 7.20–7.09 (m, 5H), 7.00–6.94 (m, 4H), 5.36 (s, 1H), 5.21 (t, J = 1.8 Hz, 1H), 5.14 (dd, J = 2.7, 1.5 Hz, 1H), 4.36 (dt, J = 13.0, 2.5 Hz, 1H), 3.90 (dt, J = 13.0, 1.5 Hz, 1H), 2.29 (s, 3H), 1.50 (s, 9H), 0.90 (d, J = 14.6 Hz, 1H), 0.48 (d, J = 14.7 Hz, 1H), −0.13 (s, 9H); 13C NMR (126 MHz, CHCl3) δ 172.4, 148.8, 142.4, 138.2, 136.7, 129.0 (4C), 128.1 (2C), 127.8, 127.0 (2C), 110.0, 82.4, 70.2, 61.0, 51.5, 27.9 (3C), 21.5, 19.6, 0.7 (3C); HRMS–ESI (positive mode; M + Na) m/z 522.2108, calcd for C27H37NNaO4SSi, 522.2110.
References
-
Zimmerman, J.; Halloway, A.; Sibi, M. P. Free Radical Cyclization Reactions. In Handbook of Cyclization Reactions; Ma, S., Ed.; Wiley-VCH: Weinheim, Germany, 2010; Vol. 2, pp 1099–1148.
Return to citation in text: [1] -
Majumdar, K. C.; Mukhopadhyay, P. P.; Basu, P. K. Heterocycles 2004, 63, 1903. doi:10.3987/REV-04-577
Return to citation in text: [1] -
Majumdar, K. C.; Basu, P. K.; Mukhopadhyay, P. P. Tetrahedron 2004, 60, 6239. doi:10.1016/j.tet.2004.05.001
Return to citation in text: [1] -
Majumdar, K. C.; Basu, P. K.; Mukhopadhyay, P. P. Tetrahedron 2005, 61, 10603. doi:10.1016/j.tet.2005.07.079
Return to citation in text: [1] -
Ishibashi, H. Chem. Rec. 2006, 6, 23–31. doi:10.1002/tcr.20069
Return to citation in text: [1] -
Barrero, A. F.; Quilez del Moral, J. F.; Sanchez, E. M.; Arteaga, J. F. Eur. J. Org. Chem. 2006, 1627. doi:10.1002/ejoc.200500849
Return to citation in text: [1] -
Majumdar, K. C.; Basu, P. K.; Chattopadhyay, S. K. Tetrahedron 2007, 63, 793. doi:10.1016/j.tet.2006.09.049
Return to citation in text: [1] -
Yoshioka, E.; Kohtani, S.; Miyabe, H. Heterocycles 2009, 79, 229. doi:10.3987/REV-08-SR(D)8
Return to citation in text: [1] -
Rowlands, G. J. Tetrahedron 2010, 66, 1593. doi:10.1016/j.tet.2009.12.023
Return to citation in text: [1] -
Galli, C. Chem. Rev. 1988, 88, 765. doi:10.1021/cr00087a004
Return to citation in text: [1] -
Curran, D. P.; Kim, D.; Liu, H. T.; Shen, W. J. Am. Chem. Soc. 1988, 110, 5900. doi:10.1021/ja00225a052
Return to citation in text: [1] -
Snieckus, V.; Cuevas, J.-C.; Sloan, C. P.; Liu, H.; Curran, D. P. J. Am. Chem. Soc. 1990, 112, 896. doi:10.1021/ja00158a075
Return to citation in text: [1] -
Dènés, F.; Beaufils, F.; Renaud, P. Synlett 2008, 2389. doi:10.1055/s-2008-1078016
Return to citation in text: [1] -
Chatgilialoglu, C.; Newcomb, M. Adv. Organomet. Chem. 1999, 44, 67. doi:10.1016/S0065-3055(08)60620-6
See for a review om tin hydride and related hydride reagents.
Return to citation in text: [1] -
Kamimura, A.; Ishikawa, S.; Noguchi, F.; Moriyama, T.; So, M.; Murafuji, T.; Uno, H. Chem. Commun. 2012, 48, 6592. doi:10.1039/c2cc31753b
Return to citation in text: [1] [2] -
Kamimura, A.; Miyazaki, K.; Yamane, Y.; Yo, R.; Ishikawa, S.; Uno, H. submitted.
Return to citation in text: [1] -
Gilman, H.; Atwell, W. H.; Sen, P. K.; Smith, C. L. J. Organomet. Chem. 1965, 4, 163. doi:10.1016/S0022-328X(00)84384-3
Return to citation in text: [1] -
Chatgilialoglu, C. Chem.–Eur. J. 2008, 14, 2310. doi:10.1002/chem.200701415
Return to citation in text: [1] -
Kanabus-Kaminska, J. M.; Hawari, J. A.; Griller, D.; Chatgilialoglu, C. J. Am. Chem. Soc. 1987, 109, 5267. doi:10.1021/ja00251a035
Return to citation in text: [1] -
Chatgilialoglu, C.; Griller, D.; Lesage, M. J. Org. Chem. 1988, 53, 3641. doi:10.1021/jo00250a051
Return to citation in text: [1] -
Chatgilialoglu, C. Acc. Chem. Res. 1992, 25, 188. doi:10.1021/ar00016a003
Return to citation in text: [1] -
Chatgilialoglu, C. Organosilanes in Radical Chemistry; Wiley: Chichester (UK), 2004. doi:10.1002/0470024755
Return to citation in text: [1] -
Miura, K.; Oshima, K.; Utimoto, K. Chem. Lett. 1992, 21, 2477. doi:10.1246/cl.1992.2477
Return to citation in text: [1] -
Miura, K.; Oshimaa, K.; Utimoto, K. Bull. Chem. Soc. Jpn. 1993, 66, 2348. doi:10.1246/bcsj.66.2348
Return to citation in text: [1] -
Kulicke, K.; Chatgilialoglu, C. S.; Kopping, B.; Giese, B. Helv. Chim. Acta 1992, 75, 935. doi:10.1002/hlca.19920750327
Return to citation in text: [1] [2] -
Studer, A. Angew. Chem., Int. Ed. 1998, 37, 462. doi:10.1002/(SICI)1521-3773(19980302)37:4<462::AID-ANIE462>3.0.CO;2-M
Return to citation in text: [1] -
Studer, A.; Steen, H. Chem.–Eur. J. 1999, 5, 759. doi:10.1002/(SICI)1521-3765(19990201)5:2<759::AID-CHEM759>3.0.CO;2-V
Return to citation in text: [1] -
Rouquet, G.; Robert, F.; Méreau, R.; Castet, F.; Renaud, P.; Landais, Y. Chem.–Eur. J. 2012, 18, 940. doi:10.1002/chem.201102318
Return to citation in text: [1] [2] -
Kamimura, A.; Okawa, H.; Morisaki, Y.; Ishikawa, S.; Uno, H. J. Org. Chem. 2007, 72, 3569. doi:10.1021/jo062251h
Return to citation in text: [1] -
Ishikawa, S.; Noguchi, F.; Kamimura, A. J. Org. Chem. 2010, 75, 3578. doi:10.1021/jo100315j
Return to citation in text: [1] -
Crystallographic data (excluding structure factors) for the structures of trans 2a have been deposited with the Cambridge Crystallographic Data Centre under supplementary publication numbers CCDC 931894. Copies of the data can be obtained, free of charge, upon request from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: +44(0)-1223-336033 or email: deposit@ccdc.cam.ac.uk].
Return to citation in text: [1]
| 1. | Zimmerman, J.; Halloway, A.; Sibi, M. P. Free Radical Cyclization Reactions. In Handbook of Cyclization Reactions; Ma, S., Ed.; Wiley-VCH: Weinheim, Germany, 2010; Vol. 2, pp 1099–1148. |
| 2. | Majumdar, K. C.; Mukhopadhyay, P. P.; Basu, P. K. Heterocycles 2004, 63, 1903. doi:10.3987/REV-04-577 |
| 3. | Majumdar, K. C.; Basu, P. K.; Mukhopadhyay, P. P. Tetrahedron 2004, 60, 6239. doi:10.1016/j.tet.2004.05.001 |
| 4. | Majumdar, K. C.; Basu, P. K.; Mukhopadhyay, P. P. Tetrahedron 2005, 61, 10603. doi:10.1016/j.tet.2005.07.079 |
| 5. | Ishibashi, H. Chem. Rec. 2006, 6, 23–31. doi:10.1002/tcr.20069 |
| 6. | Barrero, A. F.; Quilez del Moral, J. F.; Sanchez, E. M.; Arteaga, J. F. Eur. J. Org. Chem. 2006, 1627. doi:10.1002/ejoc.200500849 |
| 7. | Majumdar, K. C.; Basu, P. K.; Chattopadhyay, S. K. Tetrahedron 2007, 63, 793. doi:10.1016/j.tet.2006.09.049 |
| 8. | Yoshioka, E.; Kohtani, S.; Miyabe, H. Heterocycles 2009, 79, 229. doi:10.3987/REV-08-SR(D)8 |
| 9. | Rowlands, G. J. Tetrahedron 2010, 66, 1593. doi:10.1016/j.tet.2009.12.023 |
| 10. | Galli, C. Chem. Rev. 1988, 88, 765. doi:10.1021/cr00087a004 |
| 16. | Kamimura, A.; Miyazaki, K.; Yamane, Y.; Yo, R.; Ishikawa, S.; Uno, H. submitted. |
| 15. | Kamimura, A.; Ishikawa, S.; Noguchi, F.; Moriyama, T.; So, M.; Murafuji, T.; Uno, H. Chem. Commun. 2012, 48, 6592. doi:10.1039/c2cc31753b |
| 14. |
Chatgilialoglu, C.; Newcomb, M. Adv. Organomet. Chem. 1999, 44, 67. doi:10.1016/S0065-3055(08)60620-6
See for a review om tin hydride and related hydride reagents. |
| 25. | Kulicke, K.; Chatgilialoglu, C. S.; Kopping, B.; Giese, B. Helv. Chim. Acta 1992, 75, 935. doi:10.1002/hlca.19920750327 |
| 11. | Curran, D. P.; Kim, D.; Liu, H. T.; Shen, W. J. Am. Chem. Soc. 1988, 110, 5900. doi:10.1021/ja00225a052 |
| 12. | Snieckus, V.; Cuevas, J.-C.; Sloan, C. P.; Liu, H.; Curran, D. P. J. Am. Chem. Soc. 1990, 112, 896. doi:10.1021/ja00158a075 |
| 13. | Dènés, F.; Beaufils, F.; Renaud, P. Synlett 2008, 2389. doi:10.1055/s-2008-1078016 |
| 29. | Kamimura, A.; Okawa, H.; Morisaki, Y.; Ishikawa, S.; Uno, H. J. Org. Chem. 2007, 72, 3569. doi:10.1021/jo062251h |
| 30. | Ishikawa, S.; Noguchi, F.; Kamimura, A. J. Org. Chem. 2010, 75, 3578. doi:10.1021/jo100315j |
| 28. | Rouquet, G.; Robert, F.; Méreau, R.; Castet, F.; Renaud, P.; Landais, Y. Chem.–Eur. J. 2012, 18, 940. doi:10.1002/chem.201102318 |
| 23. | Miura, K.; Oshima, K.; Utimoto, K. Chem. Lett. 1992, 21, 2477. doi:10.1246/cl.1992.2477 |
| 24. | Miura, K.; Oshimaa, K.; Utimoto, K. Bull. Chem. Soc. Jpn. 1993, 66, 2348. doi:10.1246/bcsj.66.2348 |
| 25. | Kulicke, K.; Chatgilialoglu, C. S.; Kopping, B.; Giese, B. Helv. Chim. Acta 1992, 75, 935. doi:10.1002/hlca.19920750327 |
| 26. | Studer, A. Angew. Chem., Int. Ed. 1998, 37, 462. doi:10.1002/(SICI)1521-3773(19980302)37:4<462::AID-ANIE462>3.0.CO;2-M |
| 27. | Studer, A.; Steen, H. Chem.–Eur. J. 1999, 5, 759. doi:10.1002/(SICI)1521-3765(19990201)5:2<759::AID-CHEM759>3.0.CO;2-V |
| 28. | Rouquet, G.; Robert, F.; Méreau, R.; Castet, F.; Renaud, P.; Landais, Y. Chem.–Eur. J. 2012, 18, 940. doi:10.1002/chem.201102318 |
| 15. | Kamimura, A.; Ishikawa, S.; Noguchi, F.; Moriyama, T.; So, M.; Murafuji, T.; Uno, H. Chem. Commun. 2012, 48, 6592. doi:10.1039/c2cc31753b |
| 18. | Chatgilialoglu, C. Chem.–Eur. J. 2008, 14, 2310. doi:10.1002/chem.200701415 |
| 19. | Kanabus-Kaminska, J. M.; Hawari, J. A.; Griller, D.; Chatgilialoglu, C. J. Am. Chem. Soc. 1987, 109, 5267. doi:10.1021/ja00251a035 |
| 20. | Chatgilialoglu, C.; Griller, D.; Lesage, M. J. Org. Chem. 1988, 53, 3641. doi:10.1021/jo00250a051 |
| 21. | Chatgilialoglu, C. Acc. Chem. Res. 1992, 25, 188. doi:10.1021/ar00016a003 |
| 22. | Chatgilialoglu, C. Organosilanes in Radical Chemistry; Wiley: Chichester (UK), 2004. doi:10.1002/0470024755 |
| 17. | Gilman, H.; Atwell, W. H.; Sen, P. K.; Smith, C. L. J. Organomet. Chem. 1965, 4, 163. doi:10.1016/S0022-328X(00)84384-3 |
| 31. | Crystallographic data (excluding structure factors) for the structures of trans 2a have been deposited with the Cambridge Crystallographic Data Centre under supplementary publication numbers CCDC 931894. Copies of the data can be obtained, free of charge, upon request from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: +44(0)-1223-336033 or email: deposit@ccdc.cam.ac.uk]. |
© 2013 Miyazaki et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)