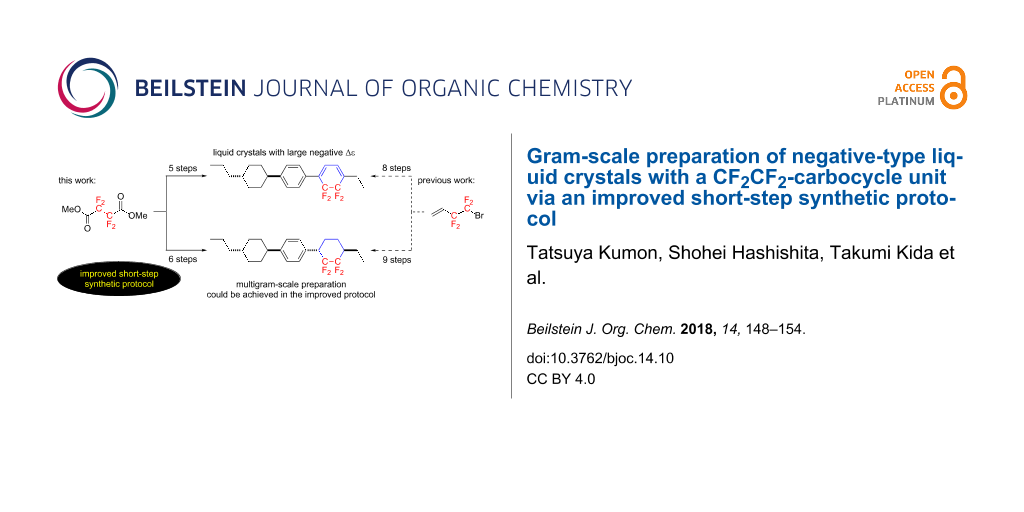
Abstract
Herein, we demonstrate an improved short-step protocol for the synthesis of multicyclic molecules having a CF2CF2-containing cyclohexadiene or cyclohexane framework in a mesogenic structure. These molecules are promising candidates for vertical alignment (VA)-mode liquid crystal (LC) display devices owing to their large negative dielectric constant. The tetrafluorinated multicyclic molecules were successfully obtained in only five or six reaction steps without the need for special handling techniques, as is generally required for thermally unstable organometallic species, representing a reduction of three reaction steps. The improved short-step synthetic protocol was also amenable to the multigram preparation of these promising molecules, which may contribute significantly to the development of novel negative-type LC molecules containing CF2CF2 carbocycles.

Graphical Abstract
Introduction
Fluorine-containing organic compounds have attracted much attention in various areas, such as the medicinal, agrochemical, and materials science fields [1-3], due to the unique characteristics of the fluorine atom [4-6]. It is well known that fluorine atoms incorporated into organic substances very often lead to intriguing physical as well as chemical properties. Therefore, considerable attention has been devoted to the development of efficient synthetic protocols for fluorine-containing organic compounds.
Owing to the fascinating molecular properties of organofluorine compounds exerted by the fluorine atom, our research group has devoted sustained effort to the development of novel biologically active fluorinated substances and high-functional fluorinated materials thus far [7-9]. Our recent interest inspired by the discovery of fluorinated liquid-crystalline (LC) molecules [10,11] led to the rational molecular design and synthesis of a family of novel fluorinated LC molecules that possess large negative dielectric anisotropy (Δε). In fact, as shown in Figure 1, tricyclic molecules containing a CF2CF2 carbocycle, e.g., the 5,5,6,6-tetrafluorocyclohexa-1,3-diene [12] or the 1,1,2,2-tetrafluorocyclohexane motif [13], were successfully synthesized and found to exhibit a large negative Δε value (−7.3 for 1c and −9.4 for trans-2c) [14]. This indicated that these compounds are promising candidates for vertical alignment (VA)-type display materials.
![[1860-5397-14-10-1]](/bjoc/content/figures/1860-5397-14-10-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Typical examples of previously reported negative-type liquid crystals containing a CF2CF2-carbocycle.
Figure 1: Typical examples of previously reported negative-type liquid crystals containing a CF2CF2-carbocycl...
In spite of their valuable utility, synthetic procedures for generating the aforementioned CF2CF2-containing LC molecules inevitably require a multistep protocol, viz. eight steps for 1 and nine steps for 2, which is a substantial drawback for the practical application of these compounds. Therefore, for practical use of fluorine-containing LC molecules, the development of more efficient synthetic protocols is highly necessary. Herein, an improved short-step synthetic protocol for obtaining promising LC molecules containing the CF2CF2 fragment is demonstrated, where the improved methodology enables us to prepare the CF2CF2-containing cyclohexadiene 1a and the corresponding cyclohexane 2c, as selected examples, on the multigram scale.
Results and Discussion
Improved synthetic design
In order to establish an improved synthetic protocol, we initially designed a method for the multicyclic mesogens 1 and 2 containing a CF2CF2 carbocycle which is shown in Scheme 1.
![[1860-5397-14-10-i1]](/bjoc/content/inline/1860-5397-14-10-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Improved short-step synthetic protocol for multicyclic mesogens 1 and 2.
Scheme 1: Improved short-step synthetic protocol for multicyclic mesogens 1 and 2.
The desired multicyclic molecules 1 with a tetrafluorocyclohexadiene or 2 with a tetrafluorocyclohexane moiety could be prepared starting from the same precursor, e.g., tetrafluorocyclohexane-1,4-diol 3: through dehydration in the case of 1 or radical reduction through the corresponding bisxanthate derivative in the case of 2. The required diol 3 could be obtained through a simultaneous hydrogenation of both, the cyclohexene and vinyl moieties of 1-aryl-4-vinyl-5,5,6,6-tetrafluorocyclohex-2-ene-1,4-diol 4. The latter could be constructed through ring-closing metathesis of the corresponding precursor, e.g., 4,4,5,5-tetrafluoroocta-1,7-diene 5, using a Grubbs' catalyst. The octa-1,7-diene 5 could be obtained through a nucleophilic addition of a vinylic Grignard reagent to the γ-keto ester 6. Lastly, the γ-keto ester 6 could be prepared by an addition–elimination reaction of commercially available tetrafluorosuccinic acid diester 7.
The designed reaction protocol enabled the construction of the target multicyclic molecules in only five or six steps, which is a more efficient protocol with three reaction steps less than the previous method. In addition, the present synthetic protocol involves several standard organic transformations, such as hydrogenation and dehydration, which are advantageous for a large-scale synthesis of the target compounds. Thus, we attempted a detailed examination of the short-step manipulations for obtaining the CF2CF2-containing multicyclic molecules 1 and 2.
Scope and limitation
The synthetic route was initiated by the sequential addition–elimination reaction of 4-n-propylphenylmagnesium bromide (4-n-PrC6H4MgBr) with commercially available dimethyl tetrafluorosuccinate (7, Scheme 2) and the results are summarized in Table 1.
![[1860-5397-14-10-i2]](/bjoc/content/inline/1860-5397-14-10-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Short-step approach to CF2CF2-containing carbocycles.
Scheme 2: Short-step approach to CF2CF2-containing carbocycles.
Table 1: Yields of all reaction steps in Scheme 2.
| Isolated yield [%] | ||||||
| 6 | 5/8 | 4 | 3 | 1 | 2 (trans/cis)a | |
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-10-i6.svg?max-width=637&scale=1.0)
|
85 | 46/42 | 75 | 99 | 82 | 47 (79/21) |
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-10-i7.svg?max-width=637&scale=1.0)
|
67 | 43/47 | 59 | 96 | 96 | 59b (72/28b→100/0c)d |
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-10-i8.svg?max-width=637&scale=1.0)
|
86 | 38/46 | 71 | 100 | 74 | 59b (87/13b→100/0c)d |
aDetermined by 19F NMR. bBefore recrystallization. cAfter recrystallization. dPreviously reported in [13].
Thus, the treatment of 1.0 equiv of 7 with 2.0 equiv of 4-n-PrC6H4MgBr in THF at −78 °C overnight gave the corresponding γ-keto ester 6a in 85% isolated yield. Interestingly, although using an excess amount of Grignard reagent in this reaction, no adducts by over-reactions, e.g., A, B, C, etc. (Figure 2a), were observed.
![[1860-5397-14-10-2]](/bjoc/content/figures/1860-5397-14-10-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: (a) Expected products of over-reaction in the Grignard reaction of dimethyl tetrafluorosuccinate (7) with 4-n-propylmagnesium bromide (reaction step 7→6). (b) Mechanism for the 7→6 reaction step.
Figure 2: (a) Expected products of over-reaction in the Grignard reaction of dimethyl tetrafluorosuccinate (7...
The suppression of the formation of the over-reacted products A–C may be brought about by the following possible reaction pathway (Figure 2b): (i) the nucleophilic attack of the Grignard reagent on the ester carbonyl functionality leads to the formation of the corresponding magnesium acetal Int-D, (ii) the alkoxide attacks another ester carbonyl moiety in the molecule to form the corresponding 5-membered ring acetal intermediate (Int-E) [15-17], after which immediate hydrolysis leads to the exclusive formation of the corresponding monosubstituted product 6. The intermediate Int-E may be quite stable at −78 °C and in equilibrium with Int-D at the stated temperature because of the high electrophilicity of the carbonyl moiety derived from the strong electron-withdrawing effect of the perfluoroalkylene fragment [18,19]. Accordingly, the over-reactions did not occur, and the γ-keto ester 6a was exclusively obtained after acid treatment of the reaction mixture.
As shown in Scheme 2, the isolated γ-keto ester 6a was treated with a large excess (3.6 equiv) of the vinyl Grignard reagent in diethyl ether at reflux overnight to afford the corresponding octa-1,7-diene 5a in only 46% yield. In this case, the 5-membered lactol derivative 8a was also obtained in 42% yield as a side-product. A proposed reaction mechanism for the formation of 5a and 8a is shown in Scheme 3.
![[1860-5397-14-10-i3]](/bjoc/content/inline/1860-5397-14-10-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Mechanism for the reaction of γ-keto ester 6 with vinyl Grignard reagents.
Scheme 3: Mechanism for the reaction of γ-keto ester 6 with vinyl Grignard reagents.
Thus, the nucleophilic addition reactions of vinylmagnesium chloride with the γ-keto ester 6 furnishes the corresponding magnesium alkoxide Int-F, which can be easily converted to the 5-membered ring intermediate, Int-G, as already discussed in the reaction of 7→6 (Figure 2b). However, the reaction at a higher temperature may lead to the elimination of MeOMgCl, generating the lactone Int-H. Accordingly, Int-H could undergo a second Grignard reaction with vinylmagnesium chloride present in the reaction mixture, resulting in the formation of Int-I. The latter intermediate, Int-I which is also in equilibrium with Int-J at reflux temperature, can react with the vinylic Grignard reagent through two possible pathways. As indicated by the blue arrow in Scheme 3, when vinylmagnesium chloride attacks the carbonyl carbon of Int-I, the corresponding adduct Int-K is formed in situ; this adduct can be smoothly converted into the desired octa-1,7-diene 5a after acid hydrolysis. On the other hand, the β-carbon at the α,β-unsaturated carbonyl moiety of Int-I may be susceptible to attack by the vinylic Grignard reagent, as shown by the purple arrow in Scheme 3, because of the high electrophilicity and lack of steric hindrance. As a consequence, Int-I also undergoes conjugate addition reaction, a Michael addition reaction, giving rise to the corresponding magnesium enolate Int-L. The subsequent hydrolysis of Int-L then leads to the γ-hydroxyketone 8a', which easily tautomerizes into the more stable 5-membered hemiacetal 8a.
From the above insight into the reaction mechanism, if the 1,2-addition reaction of Int-I proceeds in preference to the conjugate addition reaction, the desired octa-1,7-diene 5a should be produced in higher yield. However, no significant improvement was observed after various attempts such as employing a more nucleophilic lithium reagent instead of the Grignard reagent [20] or the addition of a Lewis acid to the reaction mixture [21,22]. Fortunately, 5a and 8a are easily separable by silica gel column chromatography, and the obtained octa-1,7-diene 5a was employed in the ensuing reaction without further attempts to improve its yield.
The subsequent ring-closing metathesis [23-25] of 5a under the influence of less than 10 mol % of a Grubbs 1st generation catalyst did not proceed to completion, resulting in recovery of the starting material along with the desired adduct 4a. Increasing the reaction temperature did not lead to satisfactory results. However, performing the reaction in the presence of 10 mol % of the catalyst for 40 h drove the reaction to completion. Intriguingly, the ring-closing metathesis proceeded in a highly diastereoselective manner and produced the corresponding cyclized adduct 4a as a single isomer [26].
Finally, with compound 4a in hand, the successive hydrogenation in the presence of 20 mol % of Pd/C in methanol was performed for 1 d and generated the corresponding tetrafluorinated cyclohexane-1,4-diol 3a in quantitative yield. Compound 3a could be converted to the cyclohexadiene 1a in 82% isolated yield under the influence of a large amount of phosphorus oxychloride in pyridine at 90 °C for 1 d according to the previous literature [12]. On the other hand, 4a could also be transformed into the corresponding bisxanthate derivative 9a according to the literature procedure [13]. Without further purification, 9a was treated with 4.0 equiv of n-Bu3SnH and 2.0 equiv of Et3B in dichloromethane at room temperature for 1 d to afford the desired reduction products 2a as an inseparable diastereomeric mixture in a trans/cis ratio of ca. 80:20 in 47% isolated yield [27].
With the established short-step protocol, we subsequently synthesized 1b, 1c, 2b, and 2c using the corresponding Grignard reagents, e.g., (4-n-PrC6H4)C6H4MgBr or 4-(trans-4-n-Pr-c-C6H10)C6H4MgBr, instead of 4-n-PrC6H4MgBr. As shown in Table 1, all reactions proceeded well to afford the corresponding adducts in acceptable to excellent yields. Tricyclic cyclohexanes containing the CF2CF2 fragment, e.g., 2b and 2c, were also obtained as a mixture of trans/cis diastereomers, but careful recrystallization enabled the isolation of the trans isomers in a pure form. A noteworthy advantage of the current synthetic protocol is that starting from the commercially available fluorine-containing substance 7, multigram-scale preparation of the promising negative-type LC molecules could be firstly achieved and ca. 2.0 g each of tetrafluorocyclohexadiene 1a and the cyclohexane derivative 2c (Scheme 4) were obtained. This achievement may lead to a significant contribution to the LC display industry.
![[1860-5397-14-10-i4]](/bjoc/content/inline/1860-5397-14-10-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: First multigram-scale preparation of CF2CF2-containing multicyclic mesogens.
Scheme 4: First multigram-scale preparation of CF2CF2-containing multicyclic mesogens.
The stereochemistry in the ring-closing metathesis step 5→4 was determined as follows. As depicted in Table 2, it has been generally recognized that the melting points of trans-1,4-disubstituted cyclohexane-1,4-diols are much higher than those of the corresponding cis-counterparts [28].
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-10-i9.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-14-10-i10.svg?max-width=637&scale=1.0)
As shown in Scheme 5, diastereomeric mixtures of trans- and cis-1-ethyl-2,2,3,3-tetrafluoro-4-[4-(4-n-propylphenyl)phenyl]cyclohexane-1,4-diol (3b) or 1-ethyl-2,2,3,3-tetrafluoro-4-[4-(trans-4-n-propylcyclohexyl)phenyl]cyclohexane-1,4-diol (3c) were obtained through an alternative procedure, starting from commercially available 4-bromo-3,3,4,4-tetrafluorobut-1-ene [12,13]. Fortunately, the trans/cis diastereomers 3b and 3c could be separated from each other through silica gel column chromatography. The careful thermal analyses of the diastereomers 3b and 3c [12] revealed that the melting points of the less polar products were approximately 20–50 °C higher than those of the more polar products, indicating that the former and the latter could be successfully assigned as the trans- and cis-isomer, respectively. On the other hand, the products 3b and 3c obtained in the present study were found to be identical to the more polar adducts based on comparison of various physical data, such as the 1H, 13C, 19F nuclear magnetic resonance (NMR) signals, retardation factors (Rf values), melting points, etc. From these analyses, 3b and 3c obtained by the present protocol were eventually determined to be the cis-isomer in both cases.
![[1860-5397-14-10-i5]](/bjoc/content/inline/1860-5397-14-10-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Stereochemical assignment of the ring-closing metathesis products.
Scheme 5: Stereochemical assignment of the ring-closing metathesis products.
Conclusion
In summary, we demonstrated the efficient short-step, gram-scale preparation of tetrafluorinated cyclohexadiene or cyclohexane derivatives, which are very promising negative-type LC molecules, starting from commercially available dimethyl 2,2,3,3-tetrafluorosuccinate (7). A total of only five or six steps were required for the synthesis of the cyclohexadiene or cyclohexane derivatives, respectively. It should also be noted that the present reaction pathways did not require specific techniques or sensitive reagents, and gram-scale preparation was successfully achieved. The present synthetic protocol is promising for the development of a wide range of negative-type LC molecules containing CF2CF2 carbocycles by the selection of the starting Grignard reagent and should contribute to further evolution of VA-type LC display molecules.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, and copies of 1H, 13C and 19F NMR spectra. | ||
| Format: PDF | Size: 7.1 MB | Download |
References
-
Charpentier, J.; Früh, N.; Togni, A. Chem. Rev. 2015, 115, 650–682. doi:10.1021/cr500223h
Return to citation in text: [1] -
Campbell, M. G.; Ritter, T. Chem. Rev. 2015, 115, 612–633. doi:10.1021/cr500366b
Return to citation in text: [1] -
Ni, C.; Hu, M.; Hu, J. Chem. Rev. 2015, 115, 765–825. doi:10.1021/cr5002386
Return to citation in text: [1] -
Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392
Return to citation in text: [1] -
Ni, C.; Hu, J. Chem. Soc. Rev. 2016, 45, 5441–5454. doi:10.1039/C6CS00351F
Return to citation in text: [1] -
O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/B711844A
Return to citation in text: [1] -
Konno, T. Synlett 2014, 25, 1350–1370. doi:10.1055/s-0033-1340867
Return to citation in text: [1] -
Konno, T.; Ishihara, T. In Advances in Organic Synthesis, Modern Organofluorine Chemistry–Synthetic Aspects; Rahman, A.-U.; Laali, K. K., Eds.; Bentham Science Publishers, Ltd.: The Netherlands, 2006; Vol. 2, pp 491–522.
Return to citation in text: [1] -
Konno, T.; Ishiahra, T. A New Aspect of Fluoroalkylated Acetylenes: Synthesis and Applications—Hydrometallation and Carbometallation. In Fluorine-Containing Synthons; Soloshonok, V. A., Ed.; ACS Publication Division and Oxford University Press: Washington DC, 2005; Vol. 911, pp 190–203. doi:10.1021/bk-2005-0911.ch009
Return to citation in text: [1] -
Kirsch, P. J. Fluorine Chem. 2015, 177, 29–36. doi:10.1016/j.jfluchem.2015.01.007
Return to citation in text: [1] -
Hird, M. Chem. Soc. Rev. 2007, 36, 2070–2095. doi:10.1039/b610738a
Return to citation in text: [1] -
Yamada, S.; Hashishita, S.; Asai, T.; Ishihara, T.; Konno, T. Org. Biomol. Chem. 2017, 15, 1495–1509. doi:10.1039/C6OB02431A
Return to citation in text: [1] [2] [3] [4] -
Yamada, S.; Hashishita, S.; Konishi, H.; Nishi, Y.; Kubota, T.; Asai, T.; Ishihara, T.; Konno, T. J. Fluorine Chem. 2017, 200, 47–58. doi:10.1016/j.jfluchem.2017.05.013
Return to citation in text: [1] [2] [3] [4] -
The Δε values were obtained in a binary mixture of the guest fluorinated molecule (10%) and host LC molecule (90%, MLC-6608 (Merck)) by means of extrapolation technique. The experimentally details were described in previous reports; see references [12,13].
Return to citation in text: [1] -
Bonnac, L.; Lee, S. E.; Giuffredi, G. T.; Elphick, L. M.; Anderson, A. A.; Child, E. S.; Mann, D. J.; Gouverneur, V. Org. Biomol. Chem. 2010, 8, 1445–1454. doi:10.1039/b922442d
Return to citation in text: [1] -
Timofte, R. S.; Linclau, B. Org. Lett. 2008, 10, 3673–3676. doi:10.1021/ol801272e
Return to citation in text: [1] -
Boydell, A. J.; Vinader, V.; Linclau, B. Angew. Chem., Int. Ed. 2004, 43, 5677–5679. doi:10.1002/anie.200460746
Return to citation in text: [1] -
Kaneko, S.; Yamazaki, T.; Kitazume, T. J. Org. Chem. 1993, 58, 2302–2312. doi:10.1021/jo00060a055
Return to citation in text: [1] -
Pierce, O. R.; Kane, T. G. J. Am. Chem. Soc. 1954, 76, 300–301. doi:10.1021/ja01630a097
Return to citation in text: [1] -
Amoah, E.; Dieter, R. K. J. Org. Chem. 2017, 82, 2870–2888. doi:10.1021/acs.joc.6b02769
Return to citation in text: [1] -
Pace, V.; Castoldi, L.; Hoyos, P.; Sinisterra, J. V.; Pregnolato, M.; Sánchez-Montero, J. M. Tetrahedron 2011, 67, 2670–2675. doi:10.1016/j.tet.2011.01.067
Return to citation in text: [1] -
Liu, H.-J.; Shia, K.-S.; Shang, X.; Zhu, B.-Y. Tetrahedron 1999, 55, 3803–3830. doi:10.1016/S0040-4020(99)00114-3
Return to citation in text: [1] -
Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746–1787. doi:10.1021/cr9002424
Return to citation in text: [1] -
Nolan, S. P.; Clavier, H. Chem. Soc. Rev. 2010, 39, 3305–3316. doi:10.1039/b912410c
Return to citation in text: [1] -
Coquerel, Y.; Rodriguez, J. Eur. J. Org. Chem. 2008, 1125–1132. doi:10.1002/ejoc.200700696
Return to citation in text: [1] -
Wallace, D. J. Tetrahedron Lett. 2003, 44, 2145–2148. doi:10.1016/S0040-4039(03)00161-8
Return to citation in text: [1] -
Our previous report cited in [13] reported that a catalytic hydrogenation of tetrafluorocyclohexa-1,3-diene provided the corresponding cyclohexane moiety in a cis-selective manner. After careful investigations, the present methodology was successful to obtain the desired tetrafluorocyclohexane unit with a trans-selective manner.
Return to citation in text: [1] -
Courtot, P.; Kinastowski, S.; Lumbroso, H. Bull. Soc. Chim. Fr. 1964, 489–494.
Return to citation in text: [1]
| 12. | Yamada, S.; Hashishita, S.; Asai, T.; Ishihara, T.; Konno, T. Org. Biomol. Chem. 2017, 15, 1495–1509. doi:10.1039/C6OB02431A |
| 13. | Yamada, S.; Hashishita, S.; Konishi, H.; Nishi, Y.; Kubota, T.; Asai, T.; Ishihara, T.; Konno, T. J. Fluorine Chem. 2017, 200, 47–58. doi:10.1016/j.jfluchem.2017.05.013 |
| 27. | Our previous report cited in [13] reported that a catalytic hydrogenation of tetrafluorocyclohexa-1,3-diene provided the corresponding cyclohexane moiety in a cis-selective manner. After careful investigations, the present methodology was successful to obtain the desired tetrafluorocyclohexane unit with a trans-selective manner. |
| 28. | Courtot, P.; Kinastowski, S.; Lumbroso, H. Bull. Soc. Chim. Fr. 1964, 489–494. |
| 1. | Charpentier, J.; Früh, N.; Togni, A. Chem. Rev. 2015, 115, 650–682. doi:10.1021/cr500223h |
| 2. | Campbell, M. G.; Ritter, T. Chem. Rev. 2015, 115, 612–633. doi:10.1021/cr500366b |
| 3. | Ni, C.; Hu, M.; Hu, J. Chem. Rev. 2015, 115, 765–825. doi:10.1021/cr5002386 |
| 12. | Yamada, S.; Hashishita, S.; Asai, T.; Ishihara, T.; Konno, T. Org. Biomol. Chem. 2017, 15, 1495–1509. doi:10.1039/C6OB02431A |
| 12. | Yamada, S.; Hashishita, S.; Asai, T.; Ishihara, T.; Konno, T. Org. Biomol. Chem. 2017, 15, 1495–1509. doi:10.1039/C6OB02431A |
| 10. | Kirsch, P. J. Fluorine Chem. 2015, 177, 29–36. doi:10.1016/j.jfluchem.2015.01.007 |
| 11. | Hird, M. Chem. Soc. Rev. 2007, 36, 2070–2095. doi:10.1039/b610738a |
| 13. | Yamada, S.; Hashishita, S.; Konishi, H.; Nishi, Y.; Kubota, T.; Asai, T.; Ishihara, T.; Konno, T. J. Fluorine Chem. 2017, 200, 47–58. doi:10.1016/j.jfluchem.2017.05.013 |
| 7. | Konno, T. Synlett 2014, 25, 1350–1370. doi:10.1055/s-0033-1340867 |
| 8. | Konno, T.; Ishihara, T. In Advances in Organic Synthesis, Modern Organofluorine Chemistry–Synthetic Aspects; Rahman, A.-U.; Laali, K. K., Eds.; Bentham Science Publishers, Ltd.: The Netherlands, 2006; Vol. 2, pp 491–522. |
| 9. | Konno, T.; Ishiahra, T. A New Aspect of Fluoroalkylated Acetylenes: Synthesis and Applications—Hydrometallation and Carbometallation. In Fluorine-Containing Synthons; Soloshonok, V. A., Ed.; ACS Publication Division and Oxford University Press: Washington DC, 2005; Vol. 911, pp 190–203. doi:10.1021/bk-2005-0911.ch009 |
| 23. | Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746–1787. doi:10.1021/cr9002424 |
| 24. | Nolan, S. P.; Clavier, H. Chem. Soc. Rev. 2010, 39, 3305–3316. doi:10.1039/b912410c |
| 25. | Coquerel, Y.; Rodriguez, J. Eur. J. Org. Chem. 2008, 1125–1132. doi:10.1002/ejoc.200700696 |
| 4. | Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392 |
| 5. | Ni, C.; Hu, J. Chem. Soc. Rev. 2016, 45, 5441–5454. doi:10.1039/C6CS00351F |
| 6. | O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/B711844A |
| 26. | Wallace, D. J. Tetrahedron Lett. 2003, 44, 2145–2148. doi:10.1016/S0040-4039(03)00161-8 |
| 15. | Bonnac, L.; Lee, S. E.; Giuffredi, G. T.; Elphick, L. M.; Anderson, A. A.; Child, E. S.; Mann, D. J.; Gouverneur, V. Org. Biomol. Chem. 2010, 8, 1445–1454. doi:10.1039/b922442d |
| 16. | Timofte, R. S.; Linclau, B. Org. Lett. 2008, 10, 3673–3676. doi:10.1021/ol801272e |
| 17. | Boydell, A. J.; Vinader, V.; Linclau, B. Angew. Chem., Int. Ed. 2004, 43, 5677–5679. doi:10.1002/anie.200460746 |
| 20. | Amoah, E.; Dieter, R. K. J. Org. Chem. 2017, 82, 2870–2888. doi:10.1021/acs.joc.6b02769 |
| 13. | Yamada, S.; Hashishita, S.; Konishi, H.; Nishi, Y.; Kubota, T.; Asai, T.; Ishihara, T.; Konno, T. J. Fluorine Chem. 2017, 200, 47–58. doi:10.1016/j.jfluchem.2017.05.013 |
| 13. | Yamada, S.; Hashishita, S.; Konishi, H.; Nishi, Y.; Kubota, T.; Asai, T.; Ishihara, T.; Konno, T. J. Fluorine Chem. 2017, 200, 47–58. doi:10.1016/j.jfluchem.2017.05.013 |
| 21. | Pace, V.; Castoldi, L.; Hoyos, P.; Sinisterra, J. V.; Pregnolato, M.; Sánchez-Montero, J. M. Tetrahedron 2011, 67, 2670–2675. doi:10.1016/j.tet.2011.01.067 |
| 22. | Liu, H.-J.; Shia, K.-S.; Shang, X.; Zhu, B.-Y. Tetrahedron 1999, 55, 3803–3830. doi:10.1016/S0040-4020(99)00114-3 |
| 14. | The Δε values were obtained in a binary mixture of the guest fluorinated molecule (10%) and host LC molecule (90%, MLC-6608 (Merck)) by means of extrapolation technique. The experimentally details were described in previous reports; see references [12,13]. |
| 12. | Yamada, S.; Hashishita, S.; Asai, T.; Ishihara, T.; Konno, T. Org. Biomol. Chem. 2017, 15, 1495–1509. doi:10.1039/C6OB02431A |
| 13. | Yamada, S.; Hashishita, S.; Konishi, H.; Nishi, Y.; Kubota, T.; Asai, T.; Ishihara, T.; Konno, T. J. Fluorine Chem. 2017, 200, 47–58. doi:10.1016/j.jfluchem.2017.05.013 |
| 18. | Kaneko, S.; Yamazaki, T.; Kitazume, T. J. Org. Chem. 1993, 58, 2302–2312. doi:10.1021/jo00060a055 |
| 19. | Pierce, O. R.; Kane, T. G. J. Am. Chem. Soc. 1954, 76, 300–301. doi:10.1021/ja01630a097 |
| 12. | Yamada, S.; Hashishita, S.; Asai, T.; Ishihara, T.; Konno, T. Org. Biomol. Chem. 2017, 15, 1495–1509. doi:10.1039/C6OB02431A |
| 13. | Yamada, S.; Hashishita, S.; Konishi, H.; Nishi, Y.; Kubota, T.; Asai, T.; Ishihara, T.; Konno, T. J. Fluorine Chem. 2017, 200, 47–58. doi:10.1016/j.jfluchem.2017.05.013 |
© 2018 Kumon et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)