Abstract
Copper(II)-salt-promoted oxidative ring-opening reactions of bicyclic cyclopropanol derivatives were investigated. The regioselectivities of these processes were found to be influenced by the structure of cyclopropanols as well as the counter anion of the copper(II) salts. A mechanism involving rearrangement reactions of radical intermediates and their competitive trapping by copper ions is proposed.

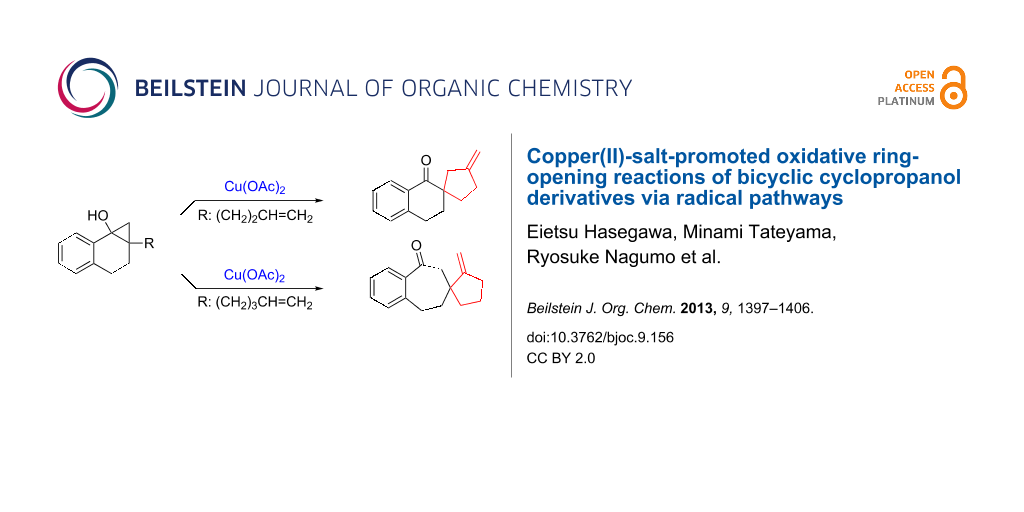
Graphical Abstract
Introduction
Radical ions are key intermediates in electron-transfer (ET) reactions of organic molecules [1-5] and they often undergo fragmentations to yield free radicals and ions [6-10]. The ensuing reaction pathways followed by the resulting radicals are governed not only by their intrinsic nature but also by the nature of co-existing redox reagents. In principle, radical intermediates in ET-promoted reactions have a tendency to participate in further ET processes to generate ionic species when stoichiometric amounts of redox reagents are used (Scheme 1) [1-10]. In contrast, radical intermediates formed by a photoinduced ET (PET) are less likely to undergo these secondary reactions, because steady-state concentrations of PET-generated redox reagents are low [11-19]. When radical intermediates and ions derived from their precursor radical ions undergo different rearrangement reactions, it is often possible to distinguish respective reaction pathways of radicals and ions by examining the product distributions of the reactions of substrates that contain appropriate probe moieties (Scheme 2).
![[1860-5397-9-156-i1]](/bjoc/content/inline/1860-5397-9-156-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Comparison of fragmentation reaction pathways of organic radical ions generated under the redox-reagent-promoted ET and PET conditions.
Scheme 1: Comparison of fragmentation reaction pathways of organic radical ions generated under the redox-rea...
![[1860-5397-9-156-i2]](/bjoc/content/inline/1860-5397-9-156-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Using rearrangements of radicals and ions to distinguish mechanistic pathways for ET-reactions.
Scheme 2: Using rearrangements of radicals and ions to distinguish mechanistic pathways for ET-reactions.
In past studies, we developed unique families of substances (exemplified by probes I and II in Figure 1) that act as radical ion probes [20] and found that radical intermediates in their reaction pathways undergo efficient 5-exo hexenyl radical cyclization reactions [21], (Figure 1) [22-30]. For example, PET reactions of probe I with amines were observed to produce a spirocyclic ketone product while its reduction reaction induced by samarium diiodide (SmI2) gives rise to a cyclopropanol (left in Scheme 3) [22,24,27]. On the other hand, the same spirocyclic ketone is obtained in the 9,10-dicyanoanthracene (DCA) and biphenyl (BP) sensitized PET reaction of probe II, while reactions of this substrate with certain oxidants afford ring-expanded ketone and enone products (right in Scheme 3) [23,25,26,28-30].
![[1860-5397-9-156-1]](/bjoc/content/figures/1860-5397-9-156-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Radical anion and cation probe substances I and II, possessing 5-hexenyl structures.
Figure 1: Radical anion and cation probe substances I and II, possessing 5-hexenyl structures.
![[1860-5397-9-156-i3]](/bjoc/content/inline/1860-5397-9-156-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reductive ET reactions of the probe I (left) and oxidative ET reactions of probe II (right).
Scheme 3: Reductive ET reactions of the probe I (left) and oxidative ET reactions of probe II (right).
Careful examination of the reaction of probe II with FeCl3 revealed that a small quantity of the spirocyclic ketone was also formed [23,28]. This observation prompted us to explore the possibility that the free radical rearrangement route becomes more predominant when oxidizing reagents weaker than Fe(III) are used to promote the reaction. Based on a consideration of the redox potentials of Fe and Cu ions (Eº in H2O, V versus NHE), +0.77 for Fe(III)/Fe(II), +0.17 for copper(II)/copper(I) [31], we chose to explore the use of copper(II) reagents in this effort. Although various ET reagents have been employed to promote reactions of cyclopropanol derivatives [32-47], the employment of copper(II) reagents to induce reactions has not been extensively studied [36,39]. In the investigation described below, we have explored copper(II)-salt-promoted oxidative ring-opening reactions of selected bicyclic cyclopropanol derivatives.
Results and Discussion
In the initial phase of this effort, we examined the reaction of cyclopropyl silyl ether 1a (0.40 mmol) with copper(II) acetate, Cu(OAc)2 , (1.1 equiv) for 1 h at room temperature (Scheme 4). Under these conditions no reaction takes place, which we attribute to the steric bulk of the silyl substituent causing interference in the reaction of the substrate with Cu(OAc)2. In accordance with this reasoning, we found that inclusion of n-Bu4NF (1.2 equiv) in the reaction mixture led to a reaction that completely consumes 1a and produced the expected spirocyclic ketone 2, albeit in low yield, and spirocyclic ketone 3 possessing an exo-methylene moiety as the major product. Interestingly, ketone 3 was previously observed as a product of the DCA–BP-sensitized PET reaction of 1a in the presence of Cu(OAc)2 [25]. Only a trace amount of ring-expanded enone 4 along with small amounts of desilylated alcohol 1b (ca. 8%) and ketone 5 were detected in the product mixture by using 1H NMR analysis. Treatment of 1a (0.19 mmol) with n-Bu4NF (2.0 equiv) in THF for 1 h followed by hydrolysis gave a mixture of 1b and 5 (12:88). Therefore, 5 may not result from the copper(II)-oxidation reaction.
![[1860-5397-9-156-i4]](/bjoc/content/inline/1860-5397-9-156-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of silyl ether 1a with Cu(OAc)2 in the absence or presence of n-Bu4NF.
Scheme 4: Reaction of silyl ether 1a with Cu(OAc)2 in the absence or presence of n-Bu4NF.
Based on the above observations, we anticipated that sterically less hindered cyclopropanols would more efficiently undergo copper(II)-induced oxidation reactions than the corresponding silyl ethers. To probe this prediction, cyclopropanols 1, prepared by SmI2-promoted intramolecular Barbier reaction of the corresponding α-bromomethyl cycloalkanones 6 [28], were subjected to reactions promoted by various copper(II) salts, CuX2 (Scheme 5).
![[1860-5397-9-156-i5]](/bjoc/content/inline/1860-5397-9-156-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: SmI2-promoted preparation of 1 and subsequent reaction with CuX2.
Scheme 5: SmI2-promoted preparation of 1 and subsequent reaction with CuX2.
The results of the reaction of 1b with Cu(OAc)2 (Scheme 6) are summarized in Table 1. As expected, this process produces ketone 3 as the major product along with both 2 and ring-expanded enone 4 as minor products. Moreover, the order of addition of 1b and Cu(OAc)2 does not significantly affect the product distribution (compare Table 1, entry 1 to entry 2). An exploration of solvent effects revealed that MeCN is more suitable than DMF while the solubility of Cu(OAc)2 is higher in the latter solvent (compare Table 1, entry 1 to entry 5). In entry 5 (Table 1), ring-opened ketone 5 was obtained. In other experiments (see below), the formations of 5 (see Table 2), and other ring-opened ketones 22 (see Table 3) and 25 (see Scheme 11) are also observed. These products might be formed by deprotonation of the corresponding cyclopropanols 1. It should be noted that THF is not an appropriate solvent for this reaction (Table 1, entry 8), a finding that is in contrast to the previous observation that ether is a better solvent than MeCN and DMF in Cu(BF4)2-promoted ring-opening reactions of cyclopropylsilyl ethers [39]. When CH2Cl2 is employed as solvent, formation of 2 becomes more efficient while the yield of 3 remains moderate (Table 1, entry 7). Although the effect of the quantity of Cu(OAc)2 on the reaction is not great, a decrease in the amount of Cu(OAc)2 causes a small increase in the yield of 2 and a decrease in the yield of 3 (compare Table 1, entry 3 to entry 1). By using more Cu(OAc)2, the yield of 3 is increased in DMF (compare Table 1, entry 6 to entry 5) while it is decreased in MeCN (compare Table 1, entry 4 to entry 2).
![[1860-5397-9-156-i6]](/bjoc/content/inline/1860-5397-9-156-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reaction of cyclopropanol 1b with Cu(OAc)2.
Scheme 6: Reaction of cyclopropanol 1b with Cu(OAc)2.
Table 1: Reaction of cyclopropanol 1b with Cu(OAc)2.a
| entry | Cu(OAc)2 (equiv) | solvent | conv of 1bb (%) | yieldsc (%) | ||
|---|---|---|---|---|---|---|
| 2 | 3 | 4 | ||||
| 1 | 1.1 | MeCN | 91 | 0 | 70 | ~5d |
| 2e | 1.1 | MeCN | 100 | 0 | 70 | ~8d |
| 3 | 0.5 | MeCN | 82 | 5 | 47 | ~2d |
| 4e | 2.2 | MeCN | 100 | 0 | 62 | 4 |
| 5f | 1.1 | DMF | 60 | 0 | 35 | trace |
| 6 | 2.2 | DMF | 69 | 1 | 40 | ~1d |
| 7e | 1.1 | CH2Cl2 | 85 | 10 | 38 | trace |
| 8 | 1.1 | THF | 28 | trace | 6 | trace |
a1b derived from 6b (0.40 mmol) was added to Cu(OAc)2 in a solvent (4 mL). bDetermined by 1H NMR based on the yield of the isolated products (see Experimental). cIsolated or determined by 1H NMR. dCrude yields. eCu(OAc)2 was added to 1b in a solvent. fKetone 5 (~5%) was obtained.
The observations described above suggest that the mechanism for this reaction shown in Scheme 7 is plausible. Because copper(II) is a relatively weak outer-sphere SET oxidant [1], addition of the hydroxy group of 1b to Cu(OAc)2 takes place initially to produce Lewis base–acid complex 7, followed by inner-sphere ET involving elimination of CuOAc and AcOH, which gives cyclopropoxy radical 8. Either external or internal bond cleavage of 8 generates the respective primary alkyl radical 9 or tertiary alkyl radical 10. An equilibrium interconverting 9 and 10 through 8 [22-30] might occur (see below). A mechanism on the fragmentation of initially formed metal–organic complexes, giving β-ketoalkyl radicals [40], cyclopropoxy radicals [25,28,48-50], or β-metalated carbonyls [39], is still controversial [35-47]. Thus, we believe the reaction follows the pathways shown in Scheme 7 although the possibility of direct formations of 9 and 10, a concerted ET and cyclopropane ring opening, cannot be ruled out. Rapid 5-exo cyclization of hexenyl radical moiety in 9 produces spirocyclic primary alkyl radical 11. Hydrogen-atom abstraction by 11 then leads to formation of spirocyclic ketone product 2, while trapping of 11 by CuOAc followed by β-H elimination (either hydride elimination or deprotonation) [39] of the resulting organocopper intermediate 12 generates the exocyclic methylene analogue 3 as the major product [25]. Protonation of 12 might be an alternative route for the formation of 2 (not shown in Scheme 7). Reactions of alkyl radicals with copper(II) are well documented [51,52], and it has been also suggested that copper(I) efficiently reacts with alkyl radicals [39]. As described, 1.1 equiv of Cu(OAc)2 leads to nearly complete reaction of 1b (see entry 1 and entry 2 in Table 1). Thus, CuOAc which is generated after initial ET between Cu(OAc)2 and 1b may capture the primary alkyl radical 11. In addition, although not predominant, oxidation of 10 by Cu(OAc)2 gives rise to tertiary carbocation 13 [51,52], which is then deprotonated to form enone 4.
![[1860-5397-9-156-i7]](/bjoc/content/inline/1860-5397-9-156-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Plausible reaction pathways for the reaction of 1b with Cu(OAc)2.
Scheme 7: Plausible reaction pathways for the reaction of 1b with Cu(OAc)2.
Studies of the effect of the counter ion on copper(II)-promoted reactions of 1b (Scheme 8) gave the results summarized in Table 2. While no reaction occurred when copper(II) acetylacetonate, Cu(acac)2, is used, (Table 2, entry 1), copper(II) 2-ethylhexanoate, Cu(ehex)2, serves as an effective oxidant in transforming 1b to 3 in a yield that is comparable to the process promoted by Cu(OAc)2 (compare Table 2, entry 2 to entry 3). Noticeable amounts of 2 are also generated in this reaction. When CuCl2 is employed to oxidize 1b, only ring-expanded ketones 4 and 14 are produced along with a lesser amount of chloro ketone 15, and competitive formation of 2 and 3 does not occur (Table 2, entry 4). An increase in the amount of CuCl2 causes a slight increase in the conversion of 1b and the total yield of ring-expanded products 4 and 14 (compare Table 2, entry 5 to entry 4). Interestingly, CuCl2 (1.1 equiv) could also promote the reaction of silyl ether 1a to produce 4 (23%), 14 (4%) and 15 (3%) at 89% conversion of 1a. Although the origin of 15 is uncertain, one possibility is that it is formed by halogen substitution of unconverted bromide 6b to 1b by SmI2. The formation of chloro ketone 23 (see Table 3) may be similarly explained. Finally, reaction of 1b with Cu(OTf)2 leads to formation of ring-expanded products 4 and 16 and a negligible amount of 2 (Table 2, entry 6). Acetamide 16 is probably produced in this process through a Ritter reaction between cation 13 and the solvent acetonitrile (Scheme 9).
![[1860-5397-9-156-i8]](/bjoc/content/inline/1860-5397-9-156-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Reaction of cyclopropanol 1b with various copper(II) salts (CuX2).
Scheme 8: Reaction of cyclopropanol 1b with various copper(II) salts (CuX2).
Table 2: Reaction of cyclopropanol 1b with various copper(II) salts (CuX2).a
| entry | X | Conv of 1bb (%) | yieldsc (%) | ||
|---|---|---|---|---|---|
| 2 | 3 | 4 | |||
| 1 | acetyl acetonate (acac) | 0 | No reaction | ||
| 2d | 2-ethyl hexanoate (ehex) | 94 | 5 | 63 | ~4e |
| 3f | OAc | 91 | 0 | 70 | ~5e |
| 4g | Cl | 63 | 0 | 0 | ~25e(9)h |
| 5i | Cl | 71 | 0 | 0 | 34(6)h |
| 6j | OTf | 77 | trace | 0 | ~11e(34)k |
a1b derived from 6b (0.40 mmol) was added to CuX2 (1.1 equiv for entries 1–4,6; 2.2 equiv for entry 5) in MeCN (4 mL). bDetermined by 1H NMR based on the yield of the isolated products (see Experimental). cIsolated or determined by 1H NMR. dKetone 5 (13%) was obtained. eCrude yield. fSame as entry 1 in Table 1. gKetone 5 (9%) and chloro ketone 15 (4%) were obtained. hNumber in the parenthesis is the yield of chloro adduct 14. iKetone 5 (13%) and chloro ketone 15 (11%) were obtained. jKetone 5 (~2%) was obtained. kNumber in parentheses is the yield of acetoamide 16.
![[1860-5397-9-156-i9]](/bjoc/content/inline/1860-5397-9-156-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Formation of acetoamide 16 from the cation 13.
Scheme 9: Formation of acetoamide 16 from the cation 13.
Hypothetically, both the Lewis acidity and oxidizing ability of CuX2 should depend on the basicity of the counter ion (X−: conjugate base of HX). Based on the acidity order HX, TfOH > HCl > AcOH ~ 2-ethyl hexanoic acid > acetylacetone [53,54], it is possible to assign Cu(acac)2, which is ineffective in promoting the reaction, as the weakest oxidant. On the other hand, CuCl2 and Cu(OTf)2 induce reactions that follow a different pathway from those promoted by copper(II) carboxylates. These observations suggest that a rapid equilibrium does indeed exist between isomeric radical intermediates 9 and 10 (Scheme 7) and that the thermodynamically less stable isomer 9 undergoes fast hexenyl-radical cyclization leading to the formation of 11 in reactions promoted by copper(II) carboxylates. On the other hand, a fast oxidation of the more stable isomer 10 by stronger oxidants such as CuCl2 or Cu(OTf)2 occurs to give the stable tertiary carbocation 13, which is then captured by Cl− or MeCN.
In order to explore the generality of the proposed counter-anion-dependent reactivity switch in the nature of copper(II)-promoted reactions of 1, the pentenyl-substituted cyclopropanol 1c was employed as the substrate (Scheme 10 and Table 3). A major product of the reaction of 1c promoted by Cu(OAc)2 was observed to be the exo-methylene containing spirocyclic ketone 19 (Table 3, entry 1), which is produced in the DCA–BP sensitized PET reaction of silyl ether of 1c in the presence of Cu(OAc)2 [25]. Contrary to the expectation that a base could assist the deprotonation of the complex between copper and 1c (similar to 7 in Scheme 7), the addition of pyridine was found to decelerate the reaction (Table 3, entry 2). This observation suggests that coordination of pyridine to copper reduces the oxidizing ability of Cu(OAc)2. Cu(ehex)2 was also effective to give 19 although the yield was relatively low (Table 3, entry 3). Reaction of 1c with CuCl2 was observed to form ring-expanded ketones 20 and 21, along with small amounts of 22 and 23. However, competitive generation of 19 does not take place (Table 3, entry 4). Finally, reaction of 1c with Cu(OTf)2 leads to the formation of ring-expanded enone 20 and acetoamide 24 (Table 3, entry 5).
![[1860-5397-9-156-i10]](/bjoc/content/inline/1860-5397-9-156-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Reaction of cyclopropanol 1c with various copper(II) salts (CuX2).
Scheme 10: Reaction of cyclopropanol 1c with various copper(II) salts (CuX2).
Table 3: Reaction of cyclopropanol 1c with various copper(II) salts (CuX2).a
| entry | X | additive | conv of 1cb (%) | yieldsc (%) | |
|---|---|---|---|---|---|
| 19 | 20 | ||||
| 1 | OAc | – | 95 | 55 | 0 |
| 2 | OAc | pyridine (1.2 equiv) | ~65d | 33 | 0 |
| 3 | ehex | – | 100 | 33 | 0 |
| 4e | Cl | – | 63 | 0 | 28(8)f |
| 5 | OTf | – | ~93d | 0 | 13(33)g |
aCuX2 (1.1 equiv) was added to 1c derived from 6c (0.4 mmol) in MeCN (4 mL). bDetermined by 1H NMR based on the yield of the isolated products (see Experimental). cIsolated or determined by 1H NMR. dBased on the crude yield of 1c. eKetone 22 (14%) and chloro ketone 23 (5%) were obtained. fNumber in parentheses is the yield of chloro adduct 21. gNumber in parentheses is the yield of acetoamide 24.
As described above, observation of the occurrence of hexenyl-radical cyclization processes serves as good evidence for the involvement of radical intermediates in mechanistic pathways for reactions of 1b and 1c. In order to gain more information about these processes, we explored an oxidation reaction of substrate 1d, which does not contain an alkene tether and whose reaction pathway, thus, cannot involve radical intermediates that undergo hexenyl-radical cyclization. We observed that reaction of the methyl-substituted cyclopropanol 1d with Cu(OAc)2 leads to formation of the ring-expanded enone 25 as a major product along with a trace amount of ketone 26 (Scheme 11).
![[1860-5397-9-156-i11]](/bjoc/content/inline/1860-5397-9-156-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Reaction of cyclopropanol 1d with various Cu(OAc)2.
Scheme 11: Reaction of cyclopropanol 1d with various Cu(OAc)2.
The Cu(OAc)2-promoted reactions of 1c and 1d are compared in Scheme 12. The ring-expanded tertiary alkyl radical 27, formed as an intermediate in the reaction of 8 (R = (CH2)3CH=CH2), undergoes rapid 5-exo hexenyl cyclization along the route for the production of spirocyclic ketone 19. Thus, oxidation of 27 followed by deprotonation to give enone 20 is a minor contributor. If an external bond cleavage of 8 occurs, cyclization of heptenyl-radical moiety in the resulting primary alkyl radical (not shown in Scheme 12) is expected. However, the exo-cyclization of heptenyl radical is two orders of magnitude slower than that of the hexenyl radical [55]. In contrast, because no competitive radical-rearrangement process exists, the corresponding radical intermediate 28 formed from 8 (R = Me) undergoes sequential oxidation and deprotonation to give enone 25 as a major product.
![[1860-5397-9-156-i12]](/bjoc/content/inline/1860-5397-9-156-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Comparison of reaction pathways of ring-expanded radical 27 and 28.
Scheme 12: Comparison of reaction pathways of ring-expanded radical 27 and 28.
Conclusion
Various copper(II) salts promote ring-opening reactions of bicyclic cyclopropanol derivatives. Using substrates that possess hexenyl moieties, we observed that the nature of the counter anion of copper(II) salts has a significant impact on the product distributions. The results suggest that reaction pathways followed by radical intermediates derived from these substrates are strongly influenced by post ring-opening steps. Thus, cyclopropane bond cleavage, which is reversible, does not serve as a product-determining step if a rapid follow-up reaction like hexenyl-radical cyclization does not exist. The results show that by using a proper choice of copper(II) salts it is possible to control the reaction pathways followed by radical and ionic intermediates derived from the initially formed Lewis base–acid complexes if the radicals and ions are capable of undergoing different rearrangement reactions.
Experimental
General: NMR spectra were recorded in CDCl3 with Me4Si as an internal standard at 400 MHz for 1H NMR and 100 MHz for 13C NMR. Column chromatography was performed with silica gel (Wakogel C-200). Preparative TLC was performed on 20 cm × 20 cm plates coated with silica gel (Wakogel B-5F). MeCN was distilled over P2O5 and subsequently distilled with K2CO3. CH2Cl2 was treated with H2SO4, water, 5% NaOH, water, and CaCl2, and then distilled with CaH2. THF was distilled over sodium benzophenone under N2. Anhydrous DMF was purchased and used without distillation. Other reagents and solvents were purchased and used without further purification. Substrates 1a [25], 1b [29], 1d [29], 6b [24], and 6d [28] and products 2 [24], 3 [25], 4 [25], 5 [29], 19 [25], 20 [26], 25 [25], and 26 [25] are known compounds. Spectral data of 1c, 6c, 14, 15, 16, 21, 22, and 23 are presented below.
Preparation of cyclopropanols 1: Cyclopropanol derivatives 1 were prepared from the corresponding bromo ketones 6 by using SmI2 following previously reported procedures [25,28]. Silyl ether 1a was prepared by the treatment of alcohol 1b with TMSCl and Et3N. The synthesized alcohols 1b, 1c and 1d were directly used for the reactions owing to their instabilities during silica-gel chromatography.
1-Hydroxy-3-(4-pentenyl)-6,7-benzobicyclo[4.1.0]heptane (1c): White solid; mp 71.5–72.9 °C; 1H NMR (400 MHz, CDCl3) δ 7.67–7.64 (m, 1H), 7.23–7.17 (m, 1H), 7.11–7.05 (m, 1H), 7.03–6.99 (m, 1H), 5.88–5.76 (m, 1H), 5.05–4.93 (m, 2H), 2.62 (ddd, J = 15.2, 5.2, 1.6 Hz, 1H), 2.52 (bs, 1H), 2.38 (td, J = 15.2, 5.2 Hz, 1H), 2.14–2.04 (m, 2H), 1.96 (ddd, J = 12.8, 5.6, 2.0 Hz, 1H), 1.66–1.46 (m, 5H), 1.20 (d, J = 6.0 Hz, 1H), 0.81 (d, J = 5.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 140.8, 138.9, 133.0, 127.9, 126.2, 125.3, 123.8, 114.4, 58.6, 33.9, 32.1, 30.5, 26.9, 26.4, 23.3, 21.3; IR (neat) νmax (cm−1): 3278, 3188, 3072, 2921, 1640, 1444, 1278, 1228, 1194, 990, 908, 740; LRMS–EI m/z (% relative intensity): 228 (M+, 6), 160 (100); HRMS–EI (m/z): [M]+ calcd for C16H20O, 228.1514; found, 228.1511.
2-Bromomethyl-2-(4-pentenyl)-1-tetralone (6c): Pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 8.05–8.02 (m, 1H), 7.51–7.46 (m, 1H), 7.34–7.23 (m, 2H), 5.77–5.70 (m, 1H), 5.00–4.91 (m, 2H), 3.77 (d, J = 10.4 Hz, 1H), 3.64 (d, J = 10.4 Hz, 1H), 3.13–2.90 (m, 2H), 2.34–2.16 (m, 2H), 2.04–1.98 (m, 2H), 1.78–1.24 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 198.7, 143.0, 138.0, 133.5, 131.3, 128.8, 128.1, 126.8, 115.0, 48.6, 39.3, 33.9, 32.7, 30.9, 24.8, 22.7; IR (neat) νmax (cm−1): 2938, 1680, 1600, 1454, 1304, 1224, 991, 910, 743; HRMS–ESI (m/z): [M + H]+ calcd for C16H19O79Br, 307.0692; found, 307.0687; [M + H]+ calcd for C16H19O81Br, 309.0672; found, 306.0665.
Reaction of cyclopropanols 1 with copper(II) salts: A typical experiment using 1b is described (Table 1, entry 1). To Cu(OAc)2 (79.9 mg, 0.44 mmol) in MeCN (4 mL) was added 1b (85.7 mg, 0.40 mmol). In some experiments, the order of addition was reversed (see entry 2 in Table 1 and Table 3). The resulting mixture was stirred under N2 at room temperature for 1 h, diluted with water and extracted with Et2O. The extract was washed with water, saturated aqueous Na2S2O3, saturated aqueous NaHCO3, and brine, dried over anhydrous MgSO4, and concentrated in vacuo giving a residue that was subjected to TLC (AcOEt:n-hexane 20/1), and 3 (59.3 mg, 0.28 mmol, 70%) and 4 (~5 mg, ~0.02 mmol, ~5%) were obtained. Other reactions were performed in a similar manner. Because cyclopropanols 1 have a tendency to partially decompose during silica-gel chromatography, their conversion in reactions was determined by using 1H NMR analysis of the crude reaction mixtures. When product isolations were not performed, yields were also determined by 1H NMR, and crude yields are reported in some cases.
3-(3-Butenyl)-3-chloro-1-benzosuberone (14): Brown liquid; 1H NMR (400 MHz, CDCl3) δ 7.80 (d, J = 7.6 Hz, 1H), 7.41 (t, J = 6.6 Hz, 1H), 7.32–7.29 (m, 2H), 5.88–5.78 (m, 1H), 5.10–4.98 (m, 2H), 3.51–3.40 (m, 2H), 3.16 (dd, J = 12.0, 1.6 Hz, 1H), 3.03 (dd, J = 17.0, 8.2 Hz, 1H), 2.55 (dd, J = 15.4, 8.2 Hz, 1H), 2.47–2.38 (m, 1H), 2.34–2.25 (m, 1H), 2.10–1.96 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 197.8, 143.9, 137.5, 137.2, 132.0, 130.3, 128.9, 126.5, 115.2, 72.8, 55.8, 43.1, 42.8, 31.0, 28.7; IR (neat) νmax (cm−1): 2939, 1681, 1602, 1453, 1299, 1226, 915, 749; HRMS–ESI (m/z): [M + H]+ calcd for C15H17O35Cl, 249.1041; found, 249.1038; [M + H]+ calcd for C15H17O37Cl, 250.1074; found, 250.1071.
2-(3-Butenyl)-2-chloromethyl-1-tetralone (15): Colorless oil; 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 8.4 Hz, 1H), 7.48 (t, J = 6.8 Hz, 1H), 7.31 (t, J = 7.6 Hz, 1H), 7.23 (d, J = 7.6 Hz, 1H), 5.78–5.68 (m, 1H), 5.01–4.91 (m, 2H), 3.87 (d, J = 11.6 Hz, 1H), 3.79 (d, J = 11.2 Hz, 1H), 3.13–3.05 (m, 1H), 2.96 (dt, J = 17.4, 5.0 Hz, 1H), 2.35–2.28 (m, 1H), 2.22–2.06 (m, 2H), 2.01–1.92 (m, 1H), 1.88–1.72 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 198.8, 142.8, 137.6, 133.5, 131.3, 128.7, 128.0, 126.8, 115.0, 49.1, 49.0, 31.6, 29.9, 27.7, 24.7; IR (neat) νmax (cm−1) 2940, 1680, 1601, 1453, 1225, 914, 748; HRMS–ESI (m/z): [M + H]+ calcd for C15H17O35Cl, 249.1041; found, 249.1041; [M + H]+ calcd for C15H17O37Cl, 251.1011; found, 251.1006.
3-(Acetylamino)-3-(3-butenyl)-1-benzosuberone (16): Yellow solid; mp 105.0–107.0 °C; 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 6.4 Hz, 1H), 7.41 (t, J = 6.8 Hz, 1H), 7.30 (t, J = 6.4 Hz, 1H), 7.25 (d, J = 8.0 Hz, 1H), 5.87–5.77 (m, 1H), 5.56 (bs, 1H), 5.06–4.95 (m, 2H), 3.12–2.97 (m, 4H), 2.46–2.40 (m, 1H), 2.27–2.20 (m, 1H), 2.10–2.03 (m, 3H), 1.99–1.95 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 201.3, 169.9, 144.2, 138.2, 138.1, 132.1, 130.3, 128.6, 126.6, 115.0, 57.5, 50.9, 39.3, 36.1, 31.2, 28.3, 24.2; IR (neat) νmax (cm−1) 3308, 3209, 2246, 1665, 1599, 1548, 1450, 1298, 1232, 912, 732; HRMS–ESI (m/z): [M + Na]+ calcd for C17H21NO2, 271.1567; found, 294.1463.
3-Chloro-3-(4-pentenyl)-1-benzosuberone (21): Brown oil; 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 7.6 Hz, 1H), 7.42 (t, J = 7.6 Hz, 1H), 7.32–7.26 (m, 2H) , 5.87–5.76 (m, 1H), 5.07–4.96 (m, 2H), 3.51–3.41 (m, 2H), 3.16 (dd, J = 12.0, 1.6 Hz, 1H), 3.03 (dd, J = 16.0, 8.0 Hz, 1H), 2.55 (dd, J = 15.4, 8.4 Hz, 1H), 2.13–2.08 (m, 2H), 1.98–1.62 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 198.0, 144.0, 138.0, 137.6, 132.0, 130.3, 128.9, 126.5, 115.0, 73.3, 55.8, 43.2, 43.1, 33.4, 31.1, 23.6; IR (neat) νmax (cm−1) 2943, 1680, 1600, 1449, 1297, 913, 751; HRMS–ESI (m/z): [M + H]+ calcd for C16H19O35Cl, 263.1197; found, 263.1191; [M + H]+ calcd for C16H19O37Cl, 265.1168; found, 265.1168.
2-Methyl-2-(4-pentenyl)-1-tetralone (22): Pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 8.0 Hz, 1H), 7.45 (t, J = 7.6 Hz, 1H), 7.30 (t, J = 8.0 Hz, 1H), 7.21 (d, J = 8.0 Hz, 1H), 5.82–5.72 (m, 1H), 3.00–2.94 (m, 2H), 2.11–2.00 (m, 1H), 1.96–1.89 (m, 3H), 1.71–1.62 (m, 1H), 1.56–1.48 (m, 1H), 1.43–1.34 (m, 2H), 1.18 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 202.6, 143.3, 138.6, 132.9, 131.7, 128.6, 128.0, 126.6, 114.6, 44.6, 35.9, 34.2, 33.6, 25.4, 23.3, 22.2; IR (neat) νmax (cm−1): 2933, 2859, 1682, 1601, 1454, 1222, 909, 741; HRMS–ESI (m/z): [M + H]+ calcd for C16H20O, 229.1587; found, 229.1593.
2-Chloromethyl-2-(4-pentenyl)-1-tetralone (23): Pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 8.0 Hz, 1H), 7.48 (t, J = 7.6 Hz, 1H), 7.31 (t, J = 8.0 Hz, 1H), 7.23 (d, J = 7.6 Hz, 1H), 5.78–5.68 (m, 1H), 5.00–4.90 (m, 2H), 3.82 (d, J = 11.2 Hz, 1H), 3.77 (d, J = 11.2 Hz, 1H), 3.12–3.04 (m, 1H), 2.95 (dt, J = 18.0, 4.8 Hz, 1H), 2.34–2.28 (m, 1H), 2.22–2.16 (m, 2H), 2.03–1.97 (m, 2H), 1.78–1.72 (m, 2H), 1.45–1.30 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 199.1, 142.9, 138.0, 133.5, 131.4, 128.7, 128.0, 126.8, 115.0, 49.2, 49.1, 33.9, 31.9, 29.9, 24.7, 22.7; IR (neat) νmax (cm−1): 2939, 1680, 1600, 1454, 1222, 911, 746; HRMS–ESI (m/z): [M + H]+ calcd for C16H19O35Cl, 263.1195; found, 263.1197; [M + H]+ calcd for C16H19O37Cl, 265.1168; found, 265.1168.
3-(N-Acetylamino)-3-(4-pentenyl)-1-benzosuberone (24): Viscous yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 7.6 Hz, 1H), 7.40 (t, J = 7.6 Hz, 1H), 7.28 (t, J = 7.6 Hz, 1H), 7.24 (d, J = 7.2 Hz, 1H), 5.84–5.74 (m, 1H), 5.55 (bs, 1H), 5.04–4.94 (m, 2H), 3.11–2.96 (m, 4H), 2.42–2.36 (m, 1H), 2.11–2.04 (m, 4H), 1.93 (s, 3H), 1.91–1.83 (m, 1H) , 1.43–1.35 (m, 2H) ppm; 13C NMR (100 MHz, CDCl3) δ 201.3, 169.8, 144.2, 138.4, 138.2, 132.0, 130.3, 128.5, 126.5, 114.8, 57.6, 50.9, 39.2, 36.6, 33.7, 31.2, 24.2, 23.1 ppm; IR (neat) νmax (cm−1): 3301, 3204, 2246, 1660, 1599, 1547, 1449, 1298, 1229, 912, 731; HRMS–ESI (m/z): [M + Na]+ calcd for C18H23NO2, 308.1621; found, 308.1622.
Acknowledgements
This study was partly supported by a grant from the Sasaki Environment Technology Foundation. We thank Professor Masaki Kamata (Niigata University) for his useful comments, and Professor James M. Tanko (Virginia Polytechnic Institute and State University, USA) for his suggestion on radical ion probes. We also thank Mr. Junichi Sakai (Niigata University) for his assistance in making HRMS (EI) measurements.
References
-
Eberson, L. Electron Transfer Reactions in Organic Chemistry; Reactivity and Structure Concepts in Organic Chemistry, Vol. 25; Springer Verlag: Berlin, Germany, 1987. doi:10.1007/978-3-642-72544-9
Return to citation in text: [1] [2] [3] -
Mariano, P. S., Ed. Advances in Electron Transfer Chemistry; JAI Press: Greenwich, 1991–1999; Vol. 1–6.
Return to citation in text: [1] [2] -
Balzani, V., Ed. Electron Transfer in Chemistry; Wiley-VCH: Weinheim, Germany, 2001; Vol. 1–5.
Return to citation in text: [1] [2] -
Lund, H.; Hammerich, O. Organic Electrochemistry, 4th ed.; Marcel Dekker: New York, NY, 2001.
Return to citation in text: [1] [2] -
Procter, D. J.; Flowers, R. A., Eds. Electron Transfer Reagents in Organic Synthesis. Tetrahedron 2009, 65, 10725–10950.
Return to citation in text: [1] [2] -
Schmittel, M.; Burghart, A. Angew. Chem., Int. Ed. Engl. 1997, 36, 2550–2589. doi:10.1002/anie.199725501
Return to citation in text: [1] [2] -
Berger, D. J.; Tanko, J. M. In The Chemistry of Double-Bonded Functional Groups; Patai, S., Ed.; John Wiley & Sons: New York, NY, 1997; pp 1281–1354. doi:10.1002/0470857234.ch22
Return to citation in text: [1] [2] -
Roth, H. D. In Reactive Intermediate Chemistry; Moss, R. A.; Platz, M. S.; Jones, M., Jr., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2004; pp 205–272.
Return to citation in text: [1] [2] -
Floreancig, P. E., Ed. The Chemistry of Radical Ions. Tetrahedron 2006, 62, 6447–6594.
Return to citation in text: [1] [2] -
Todres, Z. V. Ion-Radical Organic Chemistry Principles and Applications, 2nd ed.; CRC Press: Boca Raton, FL, 2009.
Return to citation in text: [1] [2] -
Fox, M. A.; Channon, M., Eds. Photoinduced Electron Transfer; Elsevier: Amsterdam, The Netherlands, 1988.
Parts A–D.
Return to citation in text: [1] -
Kavarnos, G. J. Fundamental of Photoinduced Electron Transfer; VCH Press: New York, NY, USA, 1993.
Return to citation in text: [1] -
Horspool, W. M.; Lenci, F., Eds. CRC Handbook of Organic Photochemistry and Photobiology, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2004.
Return to citation in text: [1] -
Hasegawa, E. J. Photosci. 2003, 10, 61–69.
Return to citation in text: [1] -
Cossy, J.; Beloti, D. Tetrahedron 2006, 62, 6459–6470. doi:10.1016/j.tet.2006.03.062
Return to citation in text: [1] -
Griesbeck, A. G.; Hoffmann, N.; Warzecha, K.-D. Acc. Chem. Res. 2007, 40, 128–140. doi:10.1021/ar068148w
Return to citation in text: [1] -
Floreancig, P. E. Synlett 2007, 191–203. doi:10.1055/s-2007-968021
Return to citation in text: [1] -
Waske, P. A.; Tzvetkov, N. T.; Mattay, J. Synlett 2007, 669–685. doi:10.1055/s-2007-970777
Return to citation in text: [1] -
Cho, D. W.; Yoon, U. C.; Mariano, P. S. Acc. Chem. Res. 2011, 44, 204–215. doi:10.1021/ar100125j
Return to citation in text: [1] -
Tanko, J. M.; Drumright, R. E. J. Am. Chem. Soc. 1990, 112, 5362–5363. doi:10.1021/ja00169a060
See for the originally proposed term radical ion probe.
Return to citation in text: [1] -
Griller, D.; Ingold, K. U. Acc. Chem. Res. 1980, 13, 317–323. doi:10.1021/ar50153a004
Return to citation in text: [1] -
Hasegawa, E.; Takizawa, S.; Iwaya, K.; Kurokawa, M.; Chiba, N.; Yamamichi, K. Chem. Commun. 2002, 1966–1967. doi:10.1039/B205781F
Return to citation in text: [1] [2] [3] -
Hasegawa, E.; Tsuchida, H.; Tamura, M. Chem. Lett. 2005, 34, 1688–1689. doi:10.1246/cl.2005.1688
Return to citation in text: [1] [2] [3] [4] -
Hasegawa, E.; Takizawa, S.; Seida, T.; Yamaguchi, A.; Yamaguchi, N.; Chiba, N.; Takahashi, T.; Ikeda, H.; Akiyama, K. Tetrahedron 2006, 62, 6581–6588. doi:10.1016/j.tet.2006.03.061
Return to citation in text: [1] [2] [3] [4] [5] -
Hasegawa, E.; Yamaguchi, N.; Muraoka, H.; Tsuchida, H. Org. Lett. 2007, 9, 2811–2814. doi:10.1021/ol0709937
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] -
Hasegawa, E.; Ogawa, Y.; Kakinuma, K.; Tsuchida, H.; Tosaka, E.; Takizawa, S.; Muraoka, H.; Saikawa, T. Tetrahedron 2008, 64, 7724–7728. doi:10.1016/j.tet.2008.06.012
Return to citation in text: [1] [2] [3] [4] -
Hasegawa, E.; Hirose, H.; Sasaki, K.; Takizawa, S.; Seida, T.; Chiba, N. Heterocycles 2009, 77, 1147–1161. doi:10.3987/COM-08-S(F)94
Return to citation in text: [1] [2] [3] -
Tsuchida, H.; Tamura, M.; Hasegawa, E. J. Org. Chem. 2009, 74, 2467–2475. doi:10.1021/jo802749g
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Hasegawa, E.; Kakinuma, K.; Yanaki, T.; Komata, S. Tetrahedron 2009, 65, 10876–10881. doi:10.1016/j.tet.2009.09.108
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Tsuchida, H.; Hasegawa, E. Tetrahedron 2010, 66, 3447–3451. doi:10.1016/j.tet.2010.03.029
Return to citation in text: [1] [2] [3] -
Lange, N. A. In Lange's Handbook of Chemistry, 13th ed.; Dean, J. A., Ed.; McGraw-Hill: New York, NY, USA, 1985.
Section 6.
Return to citation in text: [1] -
Gibson, D. H.; DePuy, C. H. Chem. Rev. 1974, 74, 605–623. doi:10.1021/cr60292a001
Return to citation in text: [1] -
Kuwajima, I.; Nakamura, E. Metal homoenolates from siloxycyclopropanes. In Small Ring Compounds in Organic Synthesis IV; de Meijere, A., Ed.; Topics in Current Chemistry, Vol. 155; Springer Verlag: Berlin, Germany, 1990; pp 1–39. doi:10.1007/3-540-52422-3_1
Return to citation in text: [1] -
Kulinkovich, O. G. Chem. Rev. 2003, 103, 2597–2632. doi:10.1021/cr010012i
Return to citation in text: [1] -
Ito, Y.; Fujii, S.; Saegusa, T. J. Org. Chem. 1976, 41, 2073–2074. doi:10.1021/jo00873a055
Return to citation in text: [1] [2] -
Ryu, I.; Ando, M.; Ogawa, A.; Murai, S.; Sonoda, N. J. Am. Chem. Soc. 1983, 105, 7192–7194. doi:10.1021/ja00362a041
Return to citation in text: [1] [2] [3] -
Paolobelli, A. B.; Gioacchini, F.; Ruzziconi, R. Tetrahedron Lett. 1993, 34, 6333–6336. doi:10.1016/S0040-4039(00)73745-2
Return to citation in text: [1] [2] -
Iwasawa, N.; Hayakawa, S.; Funahashi, M.; Isobe, K.; Narasaka, K. Bull. Chem. Soc. Jpn. 1993, 66, 819–827. doi:10.1246/bcsj.66.819
Return to citation in text: [1] [2] -
Ryu, I.; Matsumoto, K.; Kameyama, Y.; Ando, M.; Kusumoto, N.; Ogawa, A.; Kambe, N.; Murai, S.; Sonoda, N. J. Am. Chem. Soc. 1993, 115, 12330–12339. doi:10.1021/ja00079a013
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Booker-Milburn, K. I.; Thompson, D. F. J. Chem. Soc., Perkin Trans. 1 1995, 2315–2321. doi:10.1039/P19950002315
Return to citation in text: [1] [2] [3] -
Iwasawa, N.; Funahashi, M.; Hayakawa, S.; Ikeno, T.; Narasaka, K. Bull. Chem. Soc. Jpn. 1999, 72, 85–97. doi:10.1246/bcsj.72.85
Return to citation in text: [1] [2] -
Highton, A.; Volpicelli, R.; Simpkins, N. S. Tetrahedron Lett. 2004, 45, 6679–6683. doi:10.1016/j.tetlet.2004.06.115
Return to citation in text: [1] [2] -
Kirihara, M.; Kakuda, H.; Ichinose, M.; Ochiai, Y.; Takizawa, S.; Mokuya, A.; Okubo, K.; Hatano, A.; Shiro, M. Tetrahedron 2005, 61, 4831–4839. doi:10.1016/j.tet.2005.03.033
Return to citation in text: [1] [2] -
Chiba, S.; Cao, Z.; El Bialy, S. A. A.; Narasaka, K. Chem. Lett. 2006, 35, 18–19. doi:10.1246/cl.2006.18
Return to citation in text: [1] [2] -
Jiao, J.; Nguyen, L. X.; Patterson, D. R.; Flowers, R. A., II. Org. Lett. 2007, 9, 1323–1326. doi:10.1021/ol070159h
Return to citation in text: [1] [2] -
Jida, M.; Guillot, R.; Ollivier, J. Tetrahedron Lett. 2007, 48, 8765–8767. doi:10.1016/j.tetlet.2007.10.003
Return to citation in text: [1] [2] -
Wang, Y. F.; Toh, K. K.; Ng, E. P. J.; Chiba, S. J. Am. Chem. Soc. 2011, 133, 6411–6421. doi:10.1021/ja200879w
Return to citation in text: [1] [2] -
Ryu, I.; Fukushima, H.; Okuda, T.; Matsu, K.; Kambe, N.; Sonoda, N.; Komatsu, M. Synlett 1997, 1265–1268. doi:10.1055/s-1997-1554
Return to citation in text: [1] -
Chatgilialoglu, C.; Ferreri, C.; Lucarini, M.; Venturini, A.; Zavitsas, A. Chem.–Eur. J. 1997, 3, 376–387. doi:10.1002/chem.19970030309
Return to citation in text: [1] -
Cooksy, A. L.; King, H. F.; Richardson, W. H. J. Org. Chem. 2003, 68, 9441–9452. doi:10.1021/jo035085b
Return to citation in text: [1] -
Kochi, J. K. Acc. Chem. Res. 1974, 7, 351–360. doi:10.1021/ar50082a006
Return to citation in text: [1] [2] -
Snider, B. B. Chem. Rev. 1996, 96, 339–364. doi:10.1021/cr950026m
Return to citation in text: [1] [2] -
Smith, M. B.; March, J. March’s Advanced Organic Chemistry, 6th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2007.
Return to citation in text: [1] -
Howells, R. D.; McCown, J. D. Chem. Rev. 1977, 77, 69–92. doi:10.1021/cr60305a005
Return to citation in text: [1] -
Beckwith, A. L. J.; Schiesser, C. H. Tetrahedron 1985, 41, 3925–3941. doi:10.1016/S0040-4020(01)97174-1
Return to citation in text: [1]
| 53. | Smith, M. B.; March, J. March’s Advanced Organic Chemistry, 6th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2007. |
| 54. | Howells, R. D.; McCown, J. D. Chem. Rev. 1977, 77, 69–92. doi:10.1021/cr60305a005 |
| 25. | Hasegawa, E.; Yamaguchi, N.; Muraoka, H.; Tsuchida, H. Org. Lett. 2007, 9, 2811–2814. doi:10.1021/ol0709937 |
| 55. | Beckwith, A. L. J.; Schiesser, C. H. Tetrahedron 1985, 41, 3925–3941. doi:10.1016/S0040-4020(01)97174-1 |
| 1. | Eberson, L. Electron Transfer Reactions in Organic Chemistry; Reactivity and Structure Concepts in Organic Chemistry, Vol. 25; Springer Verlag: Berlin, Germany, 1987. doi:10.1007/978-3-642-72544-9 |
| 2. | Mariano, P. S., Ed. Advances in Electron Transfer Chemistry; JAI Press: Greenwich, 1991–1999; Vol. 1–6. |
| 3. | Balzani, V., Ed. Electron Transfer in Chemistry; Wiley-VCH: Weinheim, Germany, 2001; Vol. 1–5. |
| 4. | Lund, H.; Hammerich, O. Organic Electrochemistry, 4th ed.; Marcel Dekker: New York, NY, 2001. |
| 5. | Procter, D. J.; Flowers, R. A., Eds. Electron Transfer Reagents in Organic Synthesis. Tetrahedron 2009, 65, 10725–10950. |
| 20. |
Tanko, J. M.; Drumright, R. E. J. Am. Chem. Soc. 1990, 112, 5362–5363. doi:10.1021/ja00169a060
See for the originally proposed term radical ion probe. |
| 28. | Tsuchida, H.; Tamura, M.; Hasegawa, E. J. Org. Chem. 2009, 74, 2467–2475. doi:10.1021/jo802749g |
| 25. | Hasegawa, E.; Yamaguchi, N.; Muraoka, H.; Tsuchida, H. Org. Lett. 2007, 9, 2811–2814. doi:10.1021/ol0709937 |
| 11. |
Fox, M. A.; Channon, M., Eds. Photoinduced Electron Transfer; Elsevier: Amsterdam, The Netherlands, 1988.
Parts A–D. |
| 12. | Kavarnos, G. J. Fundamental of Photoinduced Electron Transfer; VCH Press: New York, NY, USA, 1993. |
| 13. | Horspool, W. M.; Lenci, F., Eds. CRC Handbook of Organic Photochemistry and Photobiology, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2004. |
| 14. | Hasegawa, E. J. Photosci. 2003, 10, 61–69. |
| 15. | Cossy, J.; Beloti, D. Tetrahedron 2006, 62, 6459–6470. doi:10.1016/j.tet.2006.03.062 |
| 16. | Griesbeck, A. G.; Hoffmann, N.; Warzecha, K.-D. Acc. Chem. Res. 2007, 40, 128–140. doi:10.1021/ar068148w |
| 17. | Floreancig, P. E. Synlett 2007, 191–203. doi:10.1055/s-2007-968021 |
| 18. | Waske, P. A.; Tzvetkov, N. T.; Mattay, J. Synlett 2007, 669–685. doi:10.1055/s-2007-970777 |
| 19. | Cho, D. W.; Yoon, U. C.; Mariano, P. S. Acc. Chem. Res. 2011, 44, 204–215. doi:10.1021/ar100125j |
| 39. | Ryu, I.; Matsumoto, K.; Kameyama, Y.; Ando, M.; Kusumoto, N.; Ogawa, A.; Kambe, N.; Murai, S.; Sonoda, N. J. Am. Chem. Soc. 1993, 115, 12330–12339. doi:10.1021/ja00079a013 |
| 25. | Hasegawa, E.; Yamaguchi, N.; Muraoka, H.; Tsuchida, H. Org. Lett. 2007, 9, 2811–2814. doi:10.1021/ol0709937 |
| 1. | Eberson, L. Electron Transfer Reactions in Organic Chemistry; Reactivity and Structure Concepts in Organic Chemistry, Vol. 25; Springer Verlag: Berlin, Germany, 1987. doi:10.1007/978-3-642-72544-9 |
| 2. | Mariano, P. S., Ed. Advances in Electron Transfer Chemistry; JAI Press: Greenwich, 1991–1999; Vol. 1–6. |
| 3. | Balzani, V., Ed. Electron Transfer in Chemistry; Wiley-VCH: Weinheim, Germany, 2001; Vol. 1–5. |
| 4. | Lund, H.; Hammerich, O. Organic Electrochemistry, 4th ed.; Marcel Dekker: New York, NY, 2001. |
| 5. | Procter, D. J.; Flowers, R. A., Eds. Electron Transfer Reagents in Organic Synthesis. Tetrahedron 2009, 65, 10725–10950. |
| 6. | Schmittel, M.; Burghart, A. Angew. Chem., Int. Ed. Engl. 1997, 36, 2550–2589. doi:10.1002/anie.199725501 |
| 7. | Berger, D. J.; Tanko, J. M. In The Chemistry of Double-Bonded Functional Groups; Patai, S., Ed.; John Wiley & Sons: New York, NY, 1997; pp 1281–1354. doi:10.1002/0470857234.ch22 |
| 8. | Roth, H. D. In Reactive Intermediate Chemistry; Moss, R. A.; Platz, M. S.; Jones, M., Jr., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2004; pp 205–272. |
| 9. | Floreancig, P. E., Ed. The Chemistry of Radical Ions. Tetrahedron 2006, 62, 6447–6594. |
| 10. | Todres, Z. V. Ion-Radical Organic Chemistry Principles and Applications, 2nd ed.; CRC Press: Boca Raton, FL, 2009. |
| 36. | Ryu, I.; Ando, M.; Ogawa, A.; Murai, S.; Sonoda, N. J. Am. Chem. Soc. 1983, 105, 7192–7194. doi:10.1021/ja00362a041 |
| 39. | Ryu, I.; Matsumoto, K.; Kameyama, Y.; Ando, M.; Kusumoto, N.; Ogawa, A.; Kambe, N.; Murai, S.; Sonoda, N. J. Am. Chem. Soc. 1993, 115, 12330–12339. doi:10.1021/ja00079a013 |
| 28. | Tsuchida, H.; Tamura, M.; Hasegawa, E. J. Org. Chem. 2009, 74, 2467–2475. doi:10.1021/jo802749g |
| 6. | Schmittel, M.; Burghart, A. Angew. Chem., Int. Ed. Engl. 1997, 36, 2550–2589. doi:10.1002/anie.199725501 |
| 7. | Berger, D. J.; Tanko, J. M. In The Chemistry of Double-Bonded Functional Groups; Patai, S., Ed.; John Wiley & Sons: New York, NY, 1997; pp 1281–1354. doi:10.1002/0470857234.ch22 |
| 8. | Roth, H. D. In Reactive Intermediate Chemistry; Moss, R. A.; Platz, M. S.; Jones, M., Jr., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2004; pp 205–272. |
| 9. | Floreancig, P. E., Ed. The Chemistry of Radical Ions. Tetrahedron 2006, 62, 6447–6594. |
| 10. | Todres, Z. V. Ion-Radical Organic Chemistry Principles and Applications, 2nd ed.; CRC Press: Boca Raton, FL, 2009. |
| 25. | Hasegawa, E.; Yamaguchi, N.; Muraoka, H.; Tsuchida, H. Org. Lett. 2007, 9, 2811–2814. doi:10.1021/ol0709937 |
| 24. | Hasegawa, E.; Takizawa, S.; Seida, T.; Yamaguchi, A.; Yamaguchi, N.; Chiba, N.; Takahashi, T.; Ikeda, H.; Akiyama, K. Tetrahedron 2006, 62, 6581–6588. doi:10.1016/j.tet.2006.03.061 |
| 23. | Hasegawa, E.; Tsuchida, H.; Tamura, M. Chem. Lett. 2005, 34, 1688–1689. doi:10.1246/cl.2005.1688 |
| 25. | Hasegawa, E.; Yamaguchi, N.; Muraoka, H.; Tsuchida, H. Org. Lett. 2007, 9, 2811–2814. doi:10.1021/ol0709937 |
| 26. | Hasegawa, E.; Ogawa, Y.; Kakinuma, K.; Tsuchida, H.; Tosaka, E.; Takizawa, S.; Muraoka, H.; Saikawa, T. Tetrahedron 2008, 64, 7724–7728. doi:10.1016/j.tet.2008.06.012 |
| 28. | Tsuchida, H.; Tamura, M.; Hasegawa, E. J. Org. Chem. 2009, 74, 2467–2475. doi:10.1021/jo802749g |
| 29. | Hasegawa, E.; Kakinuma, K.; Yanaki, T.; Komata, S. Tetrahedron 2009, 65, 10876–10881. doi:10.1016/j.tet.2009.09.108 |
| 30. | Tsuchida, H.; Hasegawa, E. Tetrahedron 2010, 66, 3447–3451. doi:10.1016/j.tet.2010.03.029 |
| 31. |
Lange, N. A. In Lange's Handbook of Chemistry, 13th ed.; Dean, J. A., Ed.; McGraw-Hill: New York, NY, USA, 1985.
Section 6. |
| 29. | Hasegawa, E.; Kakinuma, K.; Yanaki, T.; Komata, S. Tetrahedron 2009, 65, 10876–10881. doi:10.1016/j.tet.2009.09.108 |
| 22. | Hasegawa, E.; Takizawa, S.; Iwaya, K.; Kurokawa, M.; Chiba, N.; Yamamichi, K. Chem. Commun. 2002, 1966–1967. doi:10.1039/B205781F |
| 24. | Hasegawa, E.; Takizawa, S.; Seida, T.; Yamaguchi, A.; Yamaguchi, N.; Chiba, N.; Takahashi, T.; Ikeda, H.; Akiyama, K. Tetrahedron 2006, 62, 6581–6588. doi:10.1016/j.tet.2006.03.061 |
| 27. | Hasegawa, E.; Hirose, H.; Sasaki, K.; Takizawa, S.; Seida, T.; Chiba, N. Heterocycles 2009, 77, 1147–1161. doi:10.3987/COM-08-S(F)94 |
| 32. | Gibson, D. H.; DePuy, C. H. Chem. Rev. 1974, 74, 605–623. doi:10.1021/cr60292a001 |
| 33. | Kuwajima, I.; Nakamura, E. Metal homoenolates from siloxycyclopropanes. In Small Ring Compounds in Organic Synthesis IV; de Meijere, A., Ed.; Topics in Current Chemistry, Vol. 155; Springer Verlag: Berlin, Germany, 1990; pp 1–39. doi:10.1007/3-540-52422-3_1 |
| 34. | Kulinkovich, O. G. Chem. Rev. 2003, 103, 2597–2632. doi:10.1021/cr010012i |
| 35. | Ito, Y.; Fujii, S.; Saegusa, T. J. Org. Chem. 1976, 41, 2073–2074. doi:10.1021/jo00873a055 |
| 36. | Ryu, I.; Ando, M.; Ogawa, A.; Murai, S.; Sonoda, N. J. Am. Chem. Soc. 1983, 105, 7192–7194. doi:10.1021/ja00362a041 |
| 37. | Paolobelli, A. B.; Gioacchini, F.; Ruzziconi, R. Tetrahedron Lett. 1993, 34, 6333–6336. doi:10.1016/S0040-4039(00)73745-2 |
| 38. | Iwasawa, N.; Hayakawa, S.; Funahashi, M.; Isobe, K.; Narasaka, K. Bull. Chem. Soc. Jpn. 1993, 66, 819–827. doi:10.1246/bcsj.66.819 |
| 39. | Ryu, I.; Matsumoto, K.; Kameyama, Y.; Ando, M.; Kusumoto, N.; Ogawa, A.; Kambe, N.; Murai, S.; Sonoda, N. J. Am. Chem. Soc. 1993, 115, 12330–12339. doi:10.1021/ja00079a013 |
| 40. | Booker-Milburn, K. I.; Thompson, D. F. J. Chem. Soc., Perkin Trans. 1 1995, 2315–2321. doi:10.1039/P19950002315 |
| 41. | Iwasawa, N.; Funahashi, M.; Hayakawa, S.; Ikeno, T.; Narasaka, K. Bull. Chem. Soc. Jpn. 1999, 72, 85–97. doi:10.1246/bcsj.72.85 |
| 42. | Highton, A.; Volpicelli, R.; Simpkins, N. S. Tetrahedron Lett. 2004, 45, 6679–6683. doi:10.1016/j.tetlet.2004.06.115 |
| 43. | Kirihara, M.; Kakuda, H.; Ichinose, M.; Ochiai, Y.; Takizawa, S.; Mokuya, A.; Okubo, K.; Hatano, A.; Shiro, M. Tetrahedron 2005, 61, 4831–4839. doi:10.1016/j.tet.2005.03.033 |
| 44. | Chiba, S.; Cao, Z.; El Bialy, S. A. A.; Narasaka, K. Chem. Lett. 2006, 35, 18–19. doi:10.1246/cl.2006.18 |
| 45. | Jiao, J.; Nguyen, L. X.; Patterson, D. R.; Flowers, R. A., II. Org. Lett. 2007, 9, 1323–1326. doi:10.1021/ol070159h |
| 46. | Jida, M.; Guillot, R.; Ollivier, J. Tetrahedron Lett. 2007, 48, 8765–8767. doi:10.1016/j.tetlet.2007.10.003 |
| 47. | Wang, Y. F.; Toh, K. K.; Ng, E. P. J.; Chiba, S. J. Am. Chem. Soc. 2011, 133, 6411–6421. doi:10.1021/ja200879w |
| 24. | Hasegawa, E.; Takizawa, S.; Seida, T.; Yamaguchi, A.; Yamaguchi, N.; Chiba, N.; Takahashi, T.; Ikeda, H.; Akiyama, K. Tetrahedron 2006, 62, 6581–6588. doi:10.1016/j.tet.2006.03.061 |
| 22. | Hasegawa, E.; Takizawa, S.; Iwaya, K.; Kurokawa, M.; Chiba, N.; Yamamichi, K. Chem. Commun. 2002, 1966–1967. doi:10.1039/B205781F |
| 23. | Hasegawa, E.; Tsuchida, H.; Tamura, M. Chem. Lett. 2005, 34, 1688–1689. doi:10.1246/cl.2005.1688 |
| 24. | Hasegawa, E.; Takizawa, S.; Seida, T.; Yamaguchi, A.; Yamaguchi, N.; Chiba, N.; Takahashi, T.; Ikeda, H.; Akiyama, K. Tetrahedron 2006, 62, 6581–6588. doi:10.1016/j.tet.2006.03.061 |
| 25. | Hasegawa, E.; Yamaguchi, N.; Muraoka, H.; Tsuchida, H. Org. Lett. 2007, 9, 2811–2814. doi:10.1021/ol0709937 |
| 26. | Hasegawa, E.; Ogawa, Y.; Kakinuma, K.; Tsuchida, H.; Tosaka, E.; Takizawa, S.; Muraoka, H.; Saikawa, T. Tetrahedron 2008, 64, 7724–7728. doi:10.1016/j.tet.2008.06.012 |
| 27. | Hasegawa, E.; Hirose, H.; Sasaki, K.; Takizawa, S.; Seida, T.; Chiba, N. Heterocycles 2009, 77, 1147–1161. doi:10.3987/COM-08-S(F)94 |
| 28. | Tsuchida, H.; Tamura, M.; Hasegawa, E. J. Org. Chem. 2009, 74, 2467–2475. doi:10.1021/jo802749g |
| 29. | Hasegawa, E.; Kakinuma, K.; Yanaki, T.; Komata, S. Tetrahedron 2009, 65, 10876–10881. doi:10.1016/j.tet.2009.09.108 |
| 30. | Tsuchida, H.; Hasegawa, E. Tetrahedron 2010, 66, 3447–3451. doi:10.1016/j.tet.2010.03.029 |
| 25. | Hasegawa, E.; Yamaguchi, N.; Muraoka, H.; Tsuchida, H. Org. Lett. 2007, 9, 2811–2814. doi:10.1021/ol0709937 |
| 21. | Griller, D.; Ingold, K. U. Acc. Chem. Res. 1980, 13, 317–323. doi:10.1021/ar50153a004 |
| 23. | Hasegawa, E.; Tsuchida, H.; Tamura, M. Chem. Lett. 2005, 34, 1688–1689. doi:10.1246/cl.2005.1688 |
| 28. | Tsuchida, H.; Tamura, M.; Hasegawa, E. J. Org. Chem. 2009, 74, 2467–2475. doi:10.1021/jo802749g |
| 29. | Hasegawa, E.; Kakinuma, K.; Yanaki, T.; Komata, S. Tetrahedron 2009, 65, 10876–10881. doi:10.1016/j.tet.2009.09.108 |
| 40. | Booker-Milburn, K. I.; Thompson, D. F. J. Chem. Soc., Perkin Trans. 1 1995, 2315–2321. doi:10.1039/P19950002315 |
| 1. | Eberson, L. Electron Transfer Reactions in Organic Chemistry; Reactivity and Structure Concepts in Organic Chemistry, Vol. 25; Springer Verlag: Berlin, Germany, 1987. doi:10.1007/978-3-642-72544-9 |
| 29. | Hasegawa, E.; Kakinuma, K.; Yanaki, T.; Komata, S. Tetrahedron 2009, 65, 10876–10881. doi:10.1016/j.tet.2009.09.108 |
| 22. | Hasegawa, E.; Takizawa, S.; Iwaya, K.; Kurokawa, M.; Chiba, N.; Yamamichi, K. Chem. Commun. 2002, 1966–1967. doi:10.1039/B205781F |
| 23. | Hasegawa, E.; Tsuchida, H.; Tamura, M. Chem. Lett. 2005, 34, 1688–1689. doi:10.1246/cl.2005.1688 |
| 24. | Hasegawa, E.; Takizawa, S.; Seida, T.; Yamaguchi, A.; Yamaguchi, N.; Chiba, N.; Takahashi, T.; Ikeda, H.; Akiyama, K. Tetrahedron 2006, 62, 6581–6588. doi:10.1016/j.tet.2006.03.061 |
| 25. | Hasegawa, E.; Yamaguchi, N.; Muraoka, H.; Tsuchida, H. Org. Lett. 2007, 9, 2811–2814. doi:10.1021/ol0709937 |
| 26. | Hasegawa, E.; Ogawa, Y.; Kakinuma, K.; Tsuchida, H.; Tosaka, E.; Takizawa, S.; Muraoka, H.; Saikawa, T. Tetrahedron 2008, 64, 7724–7728. doi:10.1016/j.tet.2008.06.012 |
| 27. | Hasegawa, E.; Hirose, H.; Sasaki, K.; Takizawa, S.; Seida, T.; Chiba, N. Heterocycles 2009, 77, 1147–1161. doi:10.3987/COM-08-S(F)94 |
| 28. | Tsuchida, H.; Tamura, M.; Hasegawa, E. J. Org. Chem. 2009, 74, 2467–2475. doi:10.1021/jo802749g |
| 29. | Hasegawa, E.; Kakinuma, K.; Yanaki, T.; Komata, S. Tetrahedron 2009, 65, 10876–10881. doi:10.1016/j.tet.2009.09.108 |
| 30. | Tsuchida, H.; Hasegawa, E. Tetrahedron 2010, 66, 3447–3451. doi:10.1016/j.tet.2010.03.029 |
| 25. | Hasegawa, E.; Yamaguchi, N.; Muraoka, H.; Tsuchida, H. Org. Lett. 2007, 9, 2811–2814. doi:10.1021/ol0709937 |
| 26. | Hasegawa, E.; Ogawa, Y.; Kakinuma, K.; Tsuchida, H.; Tosaka, E.; Takizawa, S.; Muraoka, H.; Saikawa, T. Tetrahedron 2008, 64, 7724–7728. doi:10.1016/j.tet.2008.06.012 |
| 39. | Ryu, I.; Matsumoto, K.; Kameyama, Y.; Ando, M.; Kusumoto, N.; Ogawa, A.; Kambe, N.; Murai, S.; Sonoda, N. J. Am. Chem. Soc. 1993, 115, 12330–12339. doi:10.1021/ja00079a013 |
| 51. | Kochi, J. K. Acc. Chem. Res. 1974, 7, 351–360. doi:10.1021/ar50082a006 |
| 52. | Snider, B. B. Chem. Rev. 1996, 96, 339–364. doi:10.1021/cr950026m |
| 25. | Hasegawa, E.; Yamaguchi, N.; Muraoka, H.; Tsuchida, H. Org. Lett. 2007, 9, 2811–2814. doi:10.1021/ol0709937 |
| 51. | Kochi, J. K. Acc. Chem. Res. 1974, 7, 351–360. doi:10.1021/ar50082a006 |
| 52. | Snider, B. B. Chem. Rev. 1996, 96, 339–364. doi:10.1021/cr950026m |
| 35. | Ito, Y.; Fujii, S.; Saegusa, T. J. Org. Chem. 1976, 41, 2073–2074. doi:10.1021/jo00873a055 |
| 36. | Ryu, I.; Ando, M.; Ogawa, A.; Murai, S.; Sonoda, N. J. Am. Chem. Soc. 1983, 105, 7192–7194. doi:10.1021/ja00362a041 |
| 37. | Paolobelli, A. B.; Gioacchini, F.; Ruzziconi, R. Tetrahedron Lett. 1993, 34, 6333–6336. doi:10.1016/S0040-4039(00)73745-2 |
| 38. | Iwasawa, N.; Hayakawa, S.; Funahashi, M.; Isobe, K.; Narasaka, K. Bull. Chem. Soc. Jpn. 1993, 66, 819–827. doi:10.1246/bcsj.66.819 |
| 39. | Ryu, I.; Matsumoto, K.; Kameyama, Y.; Ando, M.; Kusumoto, N.; Ogawa, A.; Kambe, N.; Murai, S.; Sonoda, N. J. Am. Chem. Soc. 1993, 115, 12330–12339. doi:10.1021/ja00079a013 |
| 40. | Booker-Milburn, K. I.; Thompson, D. F. J. Chem. Soc., Perkin Trans. 1 1995, 2315–2321. doi:10.1039/P19950002315 |
| 41. | Iwasawa, N.; Funahashi, M.; Hayakawa, S.; Ikeno, T.; Narasaka, K. Bull. Chem. Soc. Jpn. 1999, 72, 85–97. doi:10.1246/bcsj.72.85 |
| 42. | Highton, A.; Volpicelli, R.; Simpkins, N. S. Tetrahedron Lett. 2004, 45, 6679–6683. doi:10.1016/j.tetlet.2004.06.115 |
| 43. | Kirihara, M.; Kakuda, H.; Ichinose, M.; Ochiai, Y.; Takizawa, S.; Mokuya, A.; Okubo, K.; Hatano, A.; Shiro, M. Tetrahedron 2005, 61, 4831–4839. doi:10.1016/j.tet.2005.03.033 |
| 44. | Chiba, S.; Cao, Z.; El Bialy, S. A. A.; Narasaka, K. Chem. Lett. 2006, 35, 18–19. doi:10.1246/cl.2006.18 |
| 45. | Jiao, J.; Nguyen, L. X.; Patterson, D. R.; Flowers, R. A., II. Org. Lett. 2007, 9, 1323–1326. doi:10.1021/ol070159h |
| 46. | Jida, M.; Guillot, R.; Ollivier, J. Tetrahedron Lett. 2007, 48, 8765–8767. doi:10.1016/j.tetlet.2007.10.003 |
| 47. | Wang, Y. F.; Toh, K. K.; Ng, E. P. J.; Chiba, S. J. Am. Chem. Soc. 2011, 133, 6411–6421. doi:10.1021/ja200879w |
| 25. | Hasegawa, E.; Yamaguchi, N.; Muraoka, H.; Tsuchida, H. Org. Lett. 2007, 9, 2811–2814. doi:10.1021/ol0709937 |
| 28. | Tsuchida, H.; Tamura, M.; Hasegawa, E. J. Org. Chem. 2009, 74, 2467–2475. doi:10.1021/jo802749g |
| 39. | Ryu, I.; Matsumoto, K.; Kameyama, Y.; Ando, M.; Kusumoto, N.; Ogawa, A.; Kambe, N.; Murai, S.; Sonoda, N. J. Am. Chem. Soc. 1993, 115, 12330–12339. doi:10.1021/ja00079a013 |
| 25. | Hasegawa, E.; Yamaguchi, N.; Muraoka, H.; Tsuchida, H. Org. Lett. 2007, 9, 2811–2814. doi:10.1021/ol0709937 |
| 28. | Tsuchida, H.; Tamura, M.; Hasegawa, E. J. Org. Chem. 2009, 74, 2467–2475. doi:10.1021/jo802749g |
| 48. | Ryu, I.; Fukushima, H.; Okuda, T.; Matsu, K.; Kambe, N.; Sonoda, N.; Komatsu, M. Synlett 1997, 1265–1268. doi:10.1055/s-1997-1554 |
| 49. | Chatgilialoglu, C.; Ferreri, C.; Lucarini, M.; Venturini, A.; Zavitsas, A. Chem.–Eur. J. 1997, 3, 376–387. doi:10.1002/chem.19970030309 |
| 50. | Cooksy, A. L.; King, H. F.; Richardson, W. H. J. Org. Chem. 2003, 68, 9441–9452. doi:10.1021/jo035085b |
| 25. | Hasegawa, E.; Yamaguchi, N.; Muraoka, H.; Tsuchida, H. Org. Lett. 2007, 9, 2811–2814. doi:10.1021/ol0709937 |
| 39. | Ryu, I.; Matsumoto, K.; Kameyama, Y.; Ando, M.; Kusumoto, N.; Ogawa, A.; Kambe, N.; Murai, S.; Sonoda, N. J. Am. Chem. Soc. 1993, 115, 12330–12339. doi:10.1021/ja00079a013 |
| 25. | Hasegawa, E.; Yamaguchi, N.; Muraoka, H.; Tsuchida, H. Org. Lett. 2007, 9, 2811–2814. doi:10.1021/ol0709937 |
© 2013 Hasegawa et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)