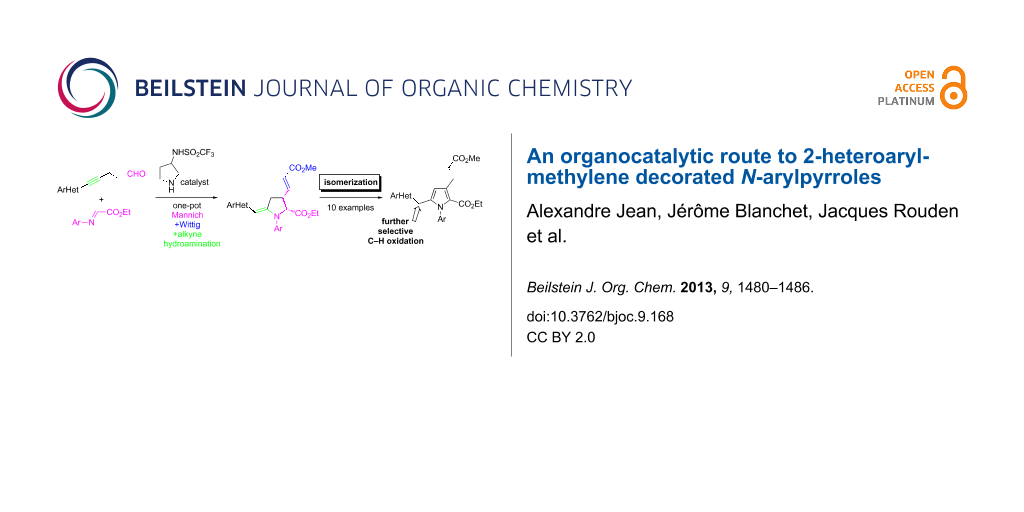
Abstract
A concise and regioselective preparation of 2-heteroarylmethylene decorated N-arylpyrroles is described through a metal-free Mannich/Wittig/hydroamination sequence followed by isomerization of the N-arylpyrrolidine adducts. Furthermore, the C–H regioselective oxidation of these substrates is demonstrated, extending the molecular diversity and versatility of these scaffolds.

Graphical Abstract
Introduction
Due to their presence in some natural products [1] and pharmaceuticals [2-4], the preparation of N-arylpyrroles is an active field of investigation [5]. Depending on their substituents, N-arylpyrroles could also be electron donor/acceptor molecules with a dual fluorescence ability suggesting attractive optoelectronic applications [6,7]. If the N-arylation of pyrroles is possible by Ullmann-type condensation [8-10], the regioselective functionalization of pyrroles is less trivial when asymmetric substrates are targeted. An indirect solution, based on the construction of substituted pyrrolidines that oxidize into elaborated pyrroles, can be employed fruitfully [11,12]. We recently described a one-pot organo-catalyzed synthesis of N-heteroarylmethylene pyrrolidines 4 [13] from readily available aldehydes 1 and imine 2 by a sequence of Mannich coupling [14-24], Wittig olefination with phosphonium 3, and proton-mediated hydroamination (Scheme 1). In the course of our investigations, we observed that pyrrolidine 4 could be converted into the corresponding pyrrole 5 by a simple isomerization, avoiding the use of oxidants. We describe herein the details of these observations and the scope of this methodology for the concise preparation of substituted 2-heteroaromatic decorated N-arylpyrroles.
![[1860-5397-9-168-i1]](/bjoc/content/inline/1860-5397-9-168-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic approach toward N-arylpyrroles.
Scheme 1: Synthetic approach toward N-arylpyrroles.
Results and Discussion
We first observed the unexpected formation of pyrrole 5a in 50% yield after treatment of pyrrolidine 4a with KCN in DMF (Scheme 2, conditions a). Although obtained in modest yield, we found the original and unique structure of the substituted pyrrole 5a interesting, especially with the 2-pyridylmethylene decoration. In an attempt to rationalize the formation of 5a, we hypothesized that KCN acted as a nucleophilic and weak base since the level of oxidation of 4a and 5a was the same. To improve the efficiency of the transformation, a stronger nucleophilic base such as DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) was tested [25]. Pleasingly, when pyrrolidine 4a was exposed to DBU in CH2Cl2, 5a was obtained in excellent yield (98%, 1 h, conditions b; Scheme 2). The reaction can also be promoted by a catalytic amount of DBU (0.2 equiv) delivering 5a (96%) after prolonged reaction time (22 h, conditions c, Scheme 2). Interestingly and despite its strong nucleophilic character, DABCO (1,4-diazabicyclo[2.2.2]octane) was unable to promote the isomerization (conditions d, Scheme 2) and the starting material was recovered.
![[1860-5397-9-168-i2]](/bjoc/content/inline/1860-5397-9-168-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Isomerization of pyrrolidine 4a (PMP: 4-methoxyphenyl).
Scheme 2: Isomerization of pyrrolidine 4a (PMP: 4-methoxyphenyl).
As presented in Scheme 3, the methodology was next attempted in a one-pot process. Hence, the transformation of aldehyde 1a and imine 2a into pyrrolidine 4a was followed by the introduction of DBU leading to pyrrole 5a in 26% yield. However, proceeding stepwise and isolating the pyrrolidine 4a by a simple filtration on silica gel before isomerization is more rewarding: following this route, the global yield for the whole process reaches 59% yield. Applying this procedure, various 2-heteroarylmethylenepyrrolidines 4b–h prepared from aldehydes 1b–h and imine 2a were exposed to DBU (1.1 equiv). Pleasingly, pyrrolidines 4b–h were transformed into the corresponding pyrroles 5b–i with homogeneous efficiency. Hence, the chemistry proved to be compatible with substrates containing meta-, para-pyridyl and quinolinyl substituents, allowing the preparation of 5b (81%), 5c (78%) and 5d (98%). Pyrrolidine 4e containing an electronically deficient pyridyl residue was also converted into 5e (80%) while pyrrolidine 4f bearing a pyrazine core underwent aromatization with high efficiency to give 5f (97%). The C2-symmetric scaffold 4g was efficiently converted into 5g (86%) and similar treatment of pyrimidine 4h provided pyrrole 5h in high yield (91%).
![[1860-5397-9-168-i3]](/bjoc/content/inline/1860-5397-9-168-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of N-arylpyrroles 5a–h (unless otherwise specified, yields in brackets refer to the isomerization step while yields in square brackets refer to the two-step procedure from the corresponding aldehyde).
Scheme 3: Preparation of N-arylpyrroles 5a–h (unless otherwise specified, yields in brackets refer to the iso...
The Mannich coupling was next attempted with different imines 2b–e in order to modulate the nature of the aryl moiety (Scheme 4). The electronic nature of the aniline being crucial for the stability of the imine and the hydroamination step, electronically rich anilines were selected to form imines 2b,c. Hence, when imines 2b,c were exposed to aldehyde 1a in the presence of catalyst 6 (available in racemic form), the Mannich adducts 7i,j were obtained and directly reacted with phosphonium salt 3. In line with our procedure, the resulting acyclic anilines 8i,j were then exposed to TFA to promote the cyclization into pyrrolidines 4i,j which upon treatment with DBU were converted into pyrroles 5i,j in 41% and 52% overall yields. While p-alkoxy substituted (R = OAllyl, OBn) anilines are compatible, the methodology proved troublesome with o-alkoxy substituted anilines, the main limitation being the formation of the corresponding imines. Similarly, imine 2d prepared from para-bromoaniline was found to be unstable and only degradation was observed during the Mannich reaction. When imine 2e, derived from the para-iodoaniline, was engaged in the process, the hydroamination step turned out to be problematic, which prevented the isolation of the corresponding pyrrolidine.
![[1860-5397-9-168-i4]](/bjoc/content/inline/1860-5397-9-168-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Even if not completely elucidated, a mechanism of the isomerization can be suggested in which the acrylate moiety is crucial. Indeed, without this unsaturation, it was not possible to observe the isomerization of the exo-enamine into the endo compound under basic treatment [26]. These observations suggest that DBU or KCN behave as base to promote the deconjugation of the acrylate moiety of 4a [27]. The resulting product 4a’ would lead under basic treatment to pyrroline 4a” from which aromatization to 5a would be expected to follow (Scheme 5).
![[1860-5397-9-168-i5]](/bjoc/content/inline/1860-5397-9-168-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Having established a practical methodology for the preparation of substituted N-arylpyrroles, we next undertook synthetic transformations to extend the molecular diversity of the substrates. While attempts to perform an oxidation of the bis(heteroaryl)methylene position with elemental sulfur [28] or SeO2 failed, the oxidation of this methylene position was regioselectively carried out by treatment of 5a–e with (NH4)2Ce(NO3)6 (CAN), delivering the alcohols 9a–e (Scheme 6). The methylene oxidation was especially efficient with substrates containing mononitrogenated heteroaryl substituents, with yields ranging from 64–87%. Oxidation under the same conditions was found to be more troublesome with pyrazine 5f since alcohol 9f was isolated in only 20% yield. Similar treatment of pyrazine 5g and pyrimidine 5h gave a complex mixture of products. While the oxidation of the bis(aryl)methylene position with CAN has been reported [29], this is the first example of bis(heteroaryl)methylene oxidation employing this reagent [30]. In order to increase the local electron deficiency of the scaffold, 9a was oxidized with 2-iodoxybenzoic acid (IBX) into ketone 10a (98%), which presents an ideal push–pull configuration tunable with the pH by protonation of the pyridine ring. This is likely to lead to applications of 10a such as for new water-soluble molecular probes.
![[1860-5397-9-168-i6]](/bjoc/content/inline/1860-5397-9-168-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Bis(heteroaryl)methylene oxidation of 5a–f.
Scheme 6: Bis(heteroaryl)methylene oxidation of 5a–f.
Conclusion
A new catalytic and regioselective preparation of substituted N-arylpyrroles decorated with various 2-heteroaromatic scaffolds is reported. Based on the isomerization of pyrrolidines prepared by a simple and efficient sequence of Mannich/Wittig olefination/hydroamination reactions, no oxidant or metallic salts were employed [31]. This study also led us to investigate the feasibility of this process with different anilines and enlarge the molecular diversity of the scaffold. So far the methodology is limited to electron-rich anilines due to the formation and reactivity of the corresponding imines and the stability of the Mannich adduct for the hydroamination step. However, this electronic configuration is ideal for the preparation of electron donor/acceptor N-arylpyrroles as demonstrated in this study. In addition, we documented an efficient C–H oxidation of the bis(heteroaryl)methylene position promoted by CAN.
Experimental
General: 1H and 13C NMR spectra were recorded in deuterated chloroform on Bruker Avance DPX 400 or 300 spectrometers and were referenced to residual chloroform (7.26 ppm, 1H; 77.00 ppm, 13C). Chemical shifts are expressed in parts per million (ppm). Data for 1H are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintuplet, sept = septuplet, m = multiplet), coupling constant (Hz), integration. Mass spectra and high-resolution mass spectra (HRMS) were obtained on a Waters-Micromass Q-Tof micro instrument. IR data were obtained on a PerkinElmer Spectrum 100 FTIR-spectrometer with only major peaks being reported. Thin-layer chromatography (TLC) was performed on silica gel 60 F254 plates (0.1 mm, Merck). Visualization was accomplished with UV (254 nm) or KMnO4 staining solutions. Chromatographic separations were achieved on silica-gel columns (Kieselgel 60, 40–63 μm, Merck).
Technical grade N,N-dimethylformamide and dichloromethane were used for this work. Following our procedure [13], catalyst 6 was prepared from (±)-1-benzyl-3-aminopyrrolidine [18471-40-4]. N-Arylimino ethyl glyoxylates 2a–c were prepared by a condensation of ethyl glyoxylate and arylamines in toluene (c = 1 M) with MgSO4 at room temperature. IBX (2-iodoxybenzoic acid) was prepared according to standard procedures.
Ethyl 3-(3-methoxy-3-oxopropyl)-1-(4-methoxyphenyl)-5-(pyridin-2-ylmethyl)-1H-pyrrole-2-carboxylate (5a): Representative procedure: In a flask containing a stirred solution of 4a (432 mg, 1.02 mmol, 1.0 equiv) in CH2Cl2 (10.2 mL) at room temperature was introduced DBU (168 μL, 1.12 mmol). The mixture was allowed to react at this temperature for 1 h. Then, the volatiles were removed under reduced pressure and the crude was purified by flash column chromatography (CH2Cl2/MeOH 99:1) on silica gel to yield 5a (432 mg, 99%) as an orange oil. 1H NMR (300 MHz, CDCl3) 1.08 (t, J = 7.1 Hz, 3H, OCH2CH3), 2.65 (br t, J = 7.9 Hz, 2H), 3.11 (br t, J = 7.9 Hz, 2H), 3.67 (s, 3H), 3.81 (s, 3H), 3.83 (s, 2H), 4.06 (q, J = 7.1 Hz, 2H), 5.92 (s, 1H), 6.83 (d, J = 8.8 Hz, 2H), 6.86 (d, J = 7.8 Hz, 1H), 6.98 (d, J = 8.8 Hz, 2H), 7.06 (m, 1H), 7.52 (td, J = 1.5, 7.8 Hz, 1H), 8.45 (d, J = 4.3 Hz, 1H) ppm; 13C NMR (75 MHz, CDCl3) 173.7, 160.8, 158.9, 158.4, 149.1, 137.6, 136.3, 132.4, 132.1, 128.8 (2*CH), 122.8, 121.3, 120.7, 113.5 (2*CH), 110.4, 59.3, 55.2, 51.3, 35.8, 34.9, 23.5, 13.9 ppm; IR: 2920, 1690, 1512, 1438, 1245, 1169, 1080, 910, 727 cm−1; HRMS (ESI+): (M + H)+ calcd for C24H27N2O5, 423.1920; found, 423.1926; Rf 0.15 (CH2Cl2/MeOH 99:1).
Ethyl 5-(hydroxy(pyridin-2-yl)methyl)-3-(3-methoxy-3-oxopropyl)-1-(4-methoxyphenyl)-1H-pyrrole-2-carboxylate (9a): Representative procedure: In a flask containing a well stirred solution of CAN (269 mg, 0.490 mmol, 3.0 equiv) in H2O (3.1 mL) at 0 °C was introduced dropwise a solution of 5a (69 mg, 0.164 mmol in 2.4 mL of CH3CN) over 10 min. Then, the mixture was allowed to react at this temperature for 3 h and was quenched by the addition of an aqueous solution of Na2S2O3 (1 M). The resulting mixture was extracted with AcOEt (3×), and the combined organic layers were washed with brine, dried over MgSO4, and filtered. The volatiles were removed under reduced pressure to give 58 mg of the crude product, which was purified by flash column chromatography (CH2Cl2/MeOH 99:1) on silica gel to yield 9a (49 mg, 71%) as an orange oil. 1H NMR (300 MHz, CDCl3) 1.08 (t, J = 7.1 Hz, 3H, OCH2CH3), 2.60 (br t, J = 7.9 Hz, 2H), 3.06 (m, 2H), 3.64 (s, 3H), 3.82 (s, 3H), 4.06 (q, J = 7.1 Hz, 2H), 5.43 (s, 1H), 5.85 (s, 1H), 6.87 (ddd, J = 2.8, 8.6, 11.4 Hz, 2H), 7.05 (d, J = 7.8 Hz, 1H), 7.08–7.22 (m, 3H), 7.61 (dt, J = 1.6, 7.8 Hz, 1H), 8.45 (d, J = 4.8 Hz, 1H) ppm; 13C NMR (75 MHz, CDCl3) 173.7 (Cq), 160.9 (Cq), 159.1 (Cq), 158.9 (Cq), 147.7 (CH), 141.0 (Cq), 136.6 (CH), 132.0 (Cq), 131.7 (Cq), 129.4 (2*CH), 122.6 (CH), 121.7 (Cq), 121.3 (CH), 113.6 (CH), 113.4 (CH), 110.1 (CH), 67.1 (CH), 59.6 (CH2), 55.4 (CH3), 51.4 (CH3), 35.0 (CH2), 23.5 (CH2), 13.9 (CH3) ppm; IR: 3375, 2927, 1733, 1688, 1512, 1436, 1368, 1295, 1081, 1030, 999, 834, 753 cm−1; HRMS (ESI+): (M + H)+ calcd for C24H27N2O6, 439.1869; found, 439.1886; Rf 0.1 (CH2Cl2/MeOH 9:1).
Ethyl 3-(3-methoxy-3-oxopropyl)-1-(4-methoxyphenyl)-5-picolinoyl-1H-pyrrole-2-carboxylate (10a): A solution of 9a (26 mg, 0.0593 mmol) in AcOEt (0.6 mL) was treated with IBX (50 mg, 0.178 mmol, 3 equiv). The suspension was stirred at 80 °C for 4 h before being brought to rt and filtered. Evaporation of the volatile led to analytically pure 10a which can be further purified by flash chromatography (CH2Cl2/MeOH 99.3:0.7) to yield 24 mg of 10a. 1H NMR (400 MHz, CDCl3) 1.10 (t, J = 7.1 Hz, 3H), 2.69 (br t, J = 7.8 Hz, 2H), 3.14 (br t, J = 7.8 Hz, 2H), 3.67 (s, 3H), 3.82 (s, 3H), 4.11 (q, J = 7.1 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 7.19 (d, J = 8.8 Hz, 2H), 7.21 (s, 1H), 7.41–7.46 (ddd, J = 1.3, 4.7, 6.1 Hz, 1H), 7.80 (dt, J = 1.6, 7.7 Hz, 1H), 7.86 (br d, J = 7.7 Hz, 1H), 8.68 (d, J = 4.7 Hz, 1H) ppm; 13C NMR (100 MHz, CDCl3) 182.9 (Cq), 173.5 (Cq), 160.7 (Cq), 159.1 (Cq), 155.4 (Cq), 148.3 (CH), 136.9 (CH), 133.3 (Cq), 133.0 (Cq), 130.6 (Cq), 128.5 (2*CH), 127.8 (Cq), 126.2 (CH), 124.0 (CH), 123.0 (CH), 113.3 (2*CH), 60.5 (CH2), 55.3 (CH3), 51.5 (CH3), 34.9 (CH2), 23.2 (CH2), 13.8 (CH3) ppm; IR: 2952, 1716, 1647, 1511, 1437, 1335, 1230, 1164, 1085, 831, 746, 689 cm−1; HRMS (ESI+): (M + H)+ calcd for C24H25N2O6; 437.1713; found, 437.1700; Rf 0.25 (CH2Cl2/MeOH 99:1).
Supporting Information
| Supporting Information File 1: Physical and spectroscopic data of 5b–j, 9b–e and 1H and 13C spectra of all new compounds. | ||
| Format: PDF | Size: 2.6 MB | Download |
References
-
Li, Q.; Jiang, J.; Fan, A.; Cui, Y.; Jia, Y. Org. Lett. 2011, 13, 312. doi:10.1021/ol1027877
Return to citation in text: [1] -
Murugesan, D.; Mital, A.; Kaiser, M.; Shackleford, D. M.; Morizzi, J.; Katneni, K.; Campbell, M.; Hudson, A.; Charman, S. A.; Yeates, C.; Gilbert, I. H. J. Med. Chem. 2013, 56, 2975. doi:10.1021/jm400009c
Return to citation in text: [1] -
He, R.; Zeng, L.-F.; He, Y.; Wu, L.; Gunawan, A. M.; Zhang, Z.-Y. Chem. Commun. 2013, 49, 2064. doi:10.1039/c3cc38961h
Return to citation in text: [1] -
Martinez, G. R.; Hirschfeld, D. R.; Maloney, P. J.; Yang, D. S.; Rosenkranz, R. P.; Walker, K. A. M. J. Med. Chem. 1989, 32, 890. doi:10.1021/jm00124a027
Return to citation in text: [1] -
Patil, N. T.; Yamamoto, Y. ARKIVOC 2007, No. (x), 121. doi:10.3998/ark.5550190.0008.a11
Return to citation in text: [1] -
Yoshihara, T.; Druzhinin, S. I.; Zachariasse, K. A. J. Am. Chem. Soc. 2004, 126, 8535. doi:10.1021/ja049809s
See for some recent applications.
Return to citation in text: [1] -
Delcamp, J. H.; Yella, A.; Holcombe, T. W.; Nazeeruddin, M. K.; Grätzel, M. Angew. Chem., Int. Ed. 2013, 52, 376. doi:10.1002/anie.201205007
See for some recent applications.
Return to citation in text: [1] -
Mederski, W. W. K. R.; Lefort, M.; Germann, M.; Kux, D. Tetrahedron 1999, 55, 12757. doi:10.1016/S0040-4020(99)00752-8
Return to citation in text: [1] -
Cristau, H.-J.; Cellier, P. P.; Spindler, J.-F.; Taillefer, M. Chem.–Eur. J. 2004, 10, 5607. doi:10.1002/chem.200400582
Return to citation in text: [1] -
Ley, S. V.; Thomas, A. W. Angew. Chem., Int. Ed. 2003, 42, 5400. doi:10.1002/anie.200300594
See for a review on the copper promoted N-arylation of azoles.
Return to citation in text: [1] -
Zhang, Y.-Q.; Zhu, D.-Y.; Li, B.-S.; Tu, Y.-Q.; Liu, J.-X.; Lu, Y.; Wang, S.-H. J. Org. Chem. 2012, 77, 4167. doi:10.1021/jo300374v
Return to citation in text: [1] -
Kumar, I.; Mir, N. A.; Ramaraju, P.; Wakhloo, B. P. RSC Adv. 2012, 2, 8922. doi:10.1039/c2ra21258g
Return to citation in text: [1] -
Jean, A.; Blanchet, J.; Rouden, J.; Maddaluno, J.; De Paolis, M. Chem. Commun. 2013, 49, 1651. doi:10.1039/c2cc38954a
Return to citation in text: [1] [2] -
Hoekman, S.; Verkade, J. M. M.; Rutjes, F. P. J. T. In Enantioselective Organocatalyzed Reactions II; Mahrwald, R., Ed.; Springer: Dordrecht, The Netherlands, 2011; pp 343 ff. doi:10.1007/978-90-481-3867-8_5
See for a review on the field.
Return to citation in text: [1] -
Kano, T.; Song, S.; Kubota, Y.; Maruoka, K. Angew. Chem., Int. Ed. 2012, 51, 1191. doi:10.1002/anie.201107375
See for selected contributions.
Return to citation in text: [1] -
Moteki, S. A.; Han, J.; Arimitsu, S.; Akakura, M.; Nakayama, K.; Maruoka, K. Angew. Chem., Int. Ed. 2012, 51, 1187. doi:10.1002/anie.201107239
See for selected contributions.
Return to citation in text: [1] -
Kano, T.; Sakamoto, R.; Akakura, M.; Maruoka, K. J. Am. Chem. Soc. 2012, 134, 7516. doi:10.1021/ja301120z
See for selected contributions.
Return to citation in text: [1] -
Kumar, I.; Mir, N. A.; Gupta, V. K.; Rajnikant. Chem. Commun. 2012, 48, 6975. doi:10.1039/C2CC33103A
See for selected contributions.
Return to citation in text: [1] -
Monaco, M. R.; Renzi, P.; Scarpino Schietroma, D. M.; Bella, M. Org. Lett. 2011, 13, 4546. doi:10.1021/ol2017406
See for selected contributions.
Return to citation in text: [1] -
Yang, J. W.; Chandler, C.; Stadler, M.; Kampen, D.; List, B. Nature 2008, 452, 453. doi:10.1038/nature06740
See for selected contributions.
Return to citation in text: [1] -
Córdova, A.; Watanabe, S.-I.; Tanaka, F.; Notz, W.; Barbas, C. F., III. J. Am. Chem. Soc. 2002, 124, 1866. doi:10.1021/ja017833p
See for selected contributions.
Return to citation in text: [1] -
Pouliquen, M.; Blanchet, J.; Lasne, M.-C.; Rouden, J. Org. Lett. 2008, 10, 1029. doi:10.1021/ol8000975
See for the use of 3-aminopyrrolidine based catalysts to promote asymmetric coupling.
Return to citation in text: [1] -
Kano, T.; Hato, Y.; Yamamoto, A.; Maruoka, K. Tetrahedron 2008, 64, 1197. doi:10.1016/j.tet.2007.11.086
See for the use of 3-aminopyrrolidine based catalysts to promote asymmetric coupling.
Return to citation in text: [1] -
Zhang, H.; Mitsumori, S.; Utsumi, N.; Imai, M.; Garcia-Delgado, N.; Mifsud, M.; Albertshofer, K.; Cheong, P. H.-Y.; Houk, K. N.; Tanaka, F.; Barbas, C. F., III. J. Am. Chem. Soc. 2008, 130, 875. doi:10.1021/ja074907+
See for the use of 3-aminopyrrolidine based catalysts to promote asymmetric coupling.
Return to citation in text: [1] -
Maji, B.; Baidya, M.; Ammer, J.; Kobayashi, S.; Mayer, P.; Ofial, A. R.; Mayr, H. Eur. J. Org. Chem. 2013, 3369. doi:10.1002/ejoc.201300213
See for an interesting comparison of the nucleophilicity of DBU; other N-heterocyclic compounds.
Return to citation in text: [1] -
No reaction occurred when A was exposed to DBU under an argon atmosphere. When the experiment was conducted under an air atmosphere, pyrroline B or pyrrole 5a were not observed but degradation occurred after 24 h. These experiments point to the deconjugation of the acrylate moiety as the first step.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-168-i7.svg?max-width=637&scale=1.18182)
Return to citation in text: [1] -
While the photolytic deconjugation of 4a cannot be completely ruled out, the deconjugation of this compound under daylight was not observed.
Return to citation in text: [1] -
Nguyen, T. B.; Ermolenko, L.; Al-Mourabit, A. J. Am. Chem. Soc. 2013, 135, 118. doi:10.1021/ja311780a
See for a recent elegant application of the sulfur redox ability for the oxidation of methylpyridines.
Return to citation in text: [1] -
Lee, W. Y.; Park, C. H.; Kim, S. J. Am. Chem. Soc. 1993, 115, 1184. doi:10.1021/ja00056a074
Return to citation in text: [1] -
Jiang, H.; Chen, H.; Wang, A.; Liu, X. Chem. Commun. 2010, 46, 7259. doi:10.1039/C0CC00841A
See for an example of Pd-catalyzed acetoxylation of pyridinemethylene position.
Return to citation in text: [1] -
Yamamoto, Y.; Gridnev, I. D.; Patil, N. T.; Jin, T. Chem. Commun. 2009, 5075. doi:10.1039/B909978F
See for a key review on the synthetic innovations available by the activation of the triple bond with Brønsted acids.
Return to citation in text: [1]
| 1. | Li, Q.; Jiang, J.; Fan, A.; Cui, Y.; Jia, Y. Org. Lett. 2011, 13, 312. doi:10.1021/ol1027877 |
| 8. | Mederski, W. W. K. R.; Lefort, M.; Germann, M.; Kux, D. Tetrahedron 1999, 55, 12757. doi:10.1016/S0040-4020(99)00752-8 |
| 9. | Cristau, H.-J.; Cellier, P. P.; Spindler, J.-F.; Taillefer, M. Chem.–Eur. J. 2004, 10, 5607. doi:10.1002/chem.200400582 |
| 10. |
Ley, S. V.; Thomas, A. W. Angew. Chem., Int. Ed. 2003, 42, 5400. doi:10.1002/anie.200300594
See for a review on the copper promoted N-arylation of azoles. |
| 31. |
Yamamoto, Y.; Gridnev, I. D.; Patil, N. T.; Jin, T. Chem. Commun. 2009, 5075. doi:10.1039/B909978F
See for a key review on the synthetic innovations available by the activation of the triple bond with Brønsted acids. |
| 6. |
Yoshihara, T.; Druzhinin, S. I.; Zachariasse, K. A. J. Am. Chem. Soc. 2004, 126, 8535. doi:10.1021/ja049809s
See for some recent applications. |
| 7. |
Delcamp, J. H.; Yella, A.; Holcombe, T. W.; Nazeeruddin, M. K.; Grätzel, M. Angew. Chem., Int. Ed. 2013, 52, 376. doi:10.1002/anie.201205007
See for some recent applications. |
| 13. | Jean, A.; Blanchet, J.; Rouden, J.; Maddaluno, J.; De Paolis, M. Chem. Commun. 2013, 49, 1651. doi:10.1039/c2cc38954a |
| 5. | Patil, N. T.; Yamamoto, Y. ARKIVOC 2007, No. (x), 121. doi:10.3998/ark.5550190.0008.a11 |
| 29. | Lee, W. Y.; Park, C. H.; Kim, S. J. Am. Chem. Soc. 1993, 115, 1184. doi:10.1021/ja00056a074 |
| 2. | Murugesan, D.; Mital, A.; Kaiser, M.; Shackleford, D. M.; Morizzi, J.; Katneni, K.; Campbell, M.; Hudson, A.; Charman, S. A.; Yeates, C.; Gilbert, I. H. J. Med. Chem. 2013, 56, 2975. doi:10.1021/jm400009c |
| 3. | He, R.; Zeng, L.-F.; He, Y.; Wu, L.; Gunawan, A. M.; Zhang, Z.-Y. Chem. Commun. 2013, 49, 2064. doi:10.1039/c3cc38961h |
| 4. | Martinez, G. R.; Hirschfeld, D. R.; Maloney, P. J.; Yang, D. S.; Rosenkranz, R. P.; Walker, K. A. M. J. Med. Chem. 1989, 32, 890. doi:10.1021/jm00124a027 |
| 30. |
Jiang, H.; Chen, H.; Wang, A.; Liu, X. Chem. Commun. 2010, 46, 7259. doi:10.1039/C0CC00841A
See for an example of Pd-catalyzed acetoxylation of pyridinemethylene position. |
| 25. |
Maji, B.; Baidya, M.; Ammer, J.; Kobayashi, S.; Mayer, P.; Ofial, A. R.; Mayr, H. Eur. J. Org. Chem. 2013, 3369. doi:10.1002/ejoc.201300213
See for an interesting comparison of the nucleophilicity of DBU; other N-heterocyclic compounds. |
| 27. | While the photolytic deconjugation of 4a cannot be completely ruled out, the deconjugation of this compound under daylight was not observed. |
| 14. |
Hoekman, S.; Verkade, J. M. M.; Rutjes, F. P. J. T. In Enantioselective Organocatalyzed Reactions II; Mahrwald, R., Ed.; Springer: Dordrecht, The Netherlands, 2011; pp 343 ff. doi:10.1007/978-90-481-3867-8_5
See for a review on the field. |
| 15. |
Kano, T.; Song, S.; Kubota, Y.; Maruoka, K. Angew. Chem., Int. Ed. 2012, 51, 1191. doi:10.1002/anie.201107375
See for selected contributions. |
| 16. |
Moteki, S. A.; Han, J.; Arimitsu, S.; Akakura, M.; Nakayama, K.; Maruoka, K. Angew. Chem., Int. Ed. 2012, 51, 1187. doi:10.1002/anie.201107239
See for selected contributions. |
| 17. |
Kano, T.; Sakamoto, R.; Akakura, M.; Maruoka, K. J. Am. Chem. Soc. 2012, 134, 7516. doi:10.1021/ja301120z
See for selected contributions. |
| 18. |
Kumar, I.; Mir, N. A.; Gupta, V. K.; Rajnikant. Chem. Commun. 2012, 48, 6975. doi:10.1039/C2CC33103A
See for selected contributions. |
| 19. |
Monaco, M. R.; Renzi, P.; Scarpino Schietroma, D. M.; Bella, M. Org. Lett. 2011, 13, 4546. doi:10.1021/ol2017406
See for selected contributions. |
| 20. |
Yang, J. W.; Chandler, C.; Stadler, M.; Kampen, D.; List, B. Nature 2008, 452, 453. doi:10.1038/nature06740
See for selected contributions. |
| 21. |
Córdova, A.; Watanabe, S.-I.; Tanaka, F.; Notz, W.; Barbas, C. F., III. J. Am. Chem. Soc. 2002, 124, 1866. doi:10.1021/ja017833p
See for selected contributions. |
| 22. |
Pouliquen, M.; Blanchet, J.; Lasne, M.-C.; Rouden, J. Org. Lett. 2008, 10, 1029. doi:10.1021/ol8000975
See for the use of 3-aminopyrrolidine based catalysts to promote asymmetric coupling. |
| 23. |
Kano, T.; Hato, Y.; Yamamoto, A.; Maruoka, K. Tetrahedron 2008, 64, 1197. doi:10.1016/j.tet.2007.11.086
See for the use of 3-aminopyrrolidine based catalysts to promote asymmetric coupling. |
| 24. |
Zhang, H.; Mitsumori, S.; Utsumi, N.; Imai, M.; Garcia-Delgado, N.; Mifsud, M.; Albertshofer, K.; Cheong, P. H.-Y.; Houk, K. N.; Tanaka, F.; Barbas, C. F., III. J. Am. Chem. Soc. 2008, 130, 875. doi:10.1021/ja074907+
See for the use of 3-aminopyrrolidine based catalysts to promote asymmetric coupling. |
| 28. |
Nguyen, T. B.; Ermolenko, L.; Al-Mourabit, A. J. Am. Chem. Soc. 2013, 135, 118. doi:10.1021/ja311780a
See for a recent elegant application of the sulfur redox ability for the oxidation of methylpyridines. |
| 13. | Jean, A.; Blanchet, J.; Rouden, J.; Maddaluno, J.; De Paolis, M. Chem. Commun. 2013, 49, 1651. doi:10.1039/c2cc38954a |
| 11. | Zhang, Y.-Q.; Zhu, D.-Y.; Li, B.-S.; Tu, Y.-Q.; Liu, J.-X.; Lu, Y.; Wang, S.-H. J. Org. Chem. 2012, 77, 4167. doi:10.1021/jo300374v |
| 12. | Kumar, I.; Mir, N. A.; Ramaraju, P.; Wakhloo, B. P. RSC Adv. 2012, 2, 8922. doi:10.1039/c2ra21258g |
| 26. |
No reaction occurred when A was exposed to DBU under an argon atmosphere. When the experiment was conducted under an air atmosphere, pyrroline B or pyrrole 5a were not observed but degradation occurred after 24 h. These experiments point to the deconjugation of the acrylate moiety as the first step.
|
© 2013 Jean et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)