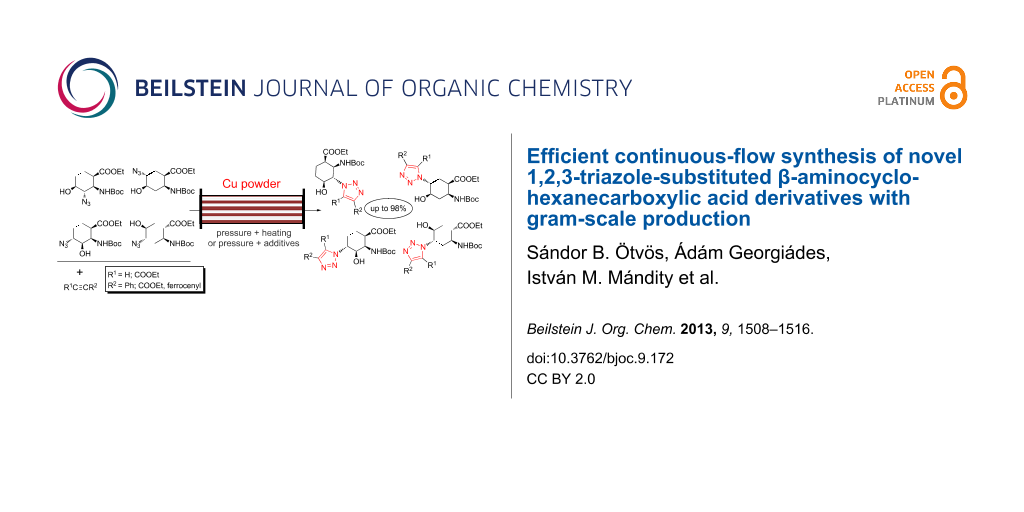
Abstract
The preparation of novel multi-substituted 1,2,3-triazole-modified β-aminocyclohexanecarboxylic acid derivatives in a simple and efficient continuous-flow procedure is reported. The 1,3-dipolar cycloaddition reactions were performed with copper powder as a readily accessible Cu(I) source. Initially, high reaction rates were achieved under high-pressure/high-temperature conditions. Subsequently, the reaction temperature was lowered to room temperature by the joint use of both basic and acidic additives to improve the safety of the synthesis, as azides were to be handled as unstable reactants. Scale-up experiments were also performed, which led to the achievement of gram-scale production in a safe and straightforward way. The obtained 1,2,3-triazole-substituted β-aminocyclohexanecarboxylates can be regarded as interesting precursors for drugs with possible biological effects.

Graphical Abstract
Introduction
In recent years, triazole-containing compounds have become potential targets for drug discovery [1,2]. A large number of 1,2,3-triazoles exhibit various biological effects [3], e.g., antiviral (1), antibacterial (2), antifungal (3) and anticancer (4) activities [4-7] (Figure 1). The 1,2,3-triazole skeleton is frequently used as a pharmacophore for the modification of known pharmaceuticals. Triazole analogues of several bioactive compounds have recently been reported. Examples are those of the well-known highly functionalized antiviral cyclic amino acid derivatives oseltamivir and zanamivir (5 and 6 in Figure 1) [8,9]. The 1,2,3-triazole moiety is a constituent part of many modified nucleosides or carbanucleosides with antiviral, anti-HIV or cytostatic activities [10-12]. However, the scope of triazole chemistry is not confined to drug discovery. There are an increasing number of applications in numerous other areas of modern chemical sciences, such as bioconjugation [13], supramolecular chemistry, [14] and polymer sciences [15].
![[1860-5397-9-172-1]](/bjoc/content/figures/1860-5397-9-172-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of 1,2,3-triazoles with various biological activities.
Figure 1: Examples of 1,2,3-triazoles with various biological activities.
Probably the most useful and powerful procedure for the synthesis of 1,2,3-triazoles is the Huisgen 1,3-dipolar cycloaddition of organic azides with acetylenes [16]. The classical Huisgen reaction, thermally induced, gives an approximate 1:1 mixture of 1,4- and 1,5-disubstituted 1,2,3-triazole isomers (Scheme 1) [17]. However, when Cu(I) catalysis is applied, the reaction becomes regioselective, exclusively yielding the 1,4-regioisomer within a relatively short reaction time [18-20]. Recently, Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) has become the basis of the so-called click chemistry concept due to its wide applicability and efficiency.
![[1860-5397-9-172-i1]](/bjoc/content/inline/1860-5397-9-172-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: 1,3-Dipolar azide–alkyne cycloadditions.
Scheme 1: 1,3-Dipolar azide–alkyne cycloadditions.
Over the past twenty years, alicyclic β-amino acids have attracted great interest among synthetic chemists, thanks to their massive pharmacological potential [21,22]. For example, cispentacin ((1R,2S)-2-aminocyclopentanecarboxylic acid, 7) is a widely investigated naturally occurring carbocyclic β-amino acid with strong antifungal properties against Candida species (Figure 2) [23]. Its synthetic 4-methylene derivative icofungipen (8), also an antifungal agent, is now proceeding through clinical development for the oral treatment of yeast infections (Figure 2) [24]. Certain multi-substituted cyclohexane amino acid derivatives, such as oryzoxymycin (9) and tilidine (10), are also well-known bioactive agents with anticancer, antibacterial, antiviral or analgesic effects (Figure 2) [25,26]. The alicyclic β-amino acids are key intermediates for the synthesis of a series of pharmaceutically relevant products [27], such as amino esters, amino alcohols, azides and heterocycles. Moreover, they are frequently used as building blocks for the synthesis of new peptides and foldamers with possible biological effects [28].
![[1860-5397-9-172-2]](/bjoc/content/figures/1860-5397-9-172-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Selected bioactive alicyclic β-amino acids.
Figure 2: Selected bioactive alicyclic β-amino acids.
Modern continuous-flow (CF) technologies offer many advantages over classical batch-based procedures [29-32], including efficient mixing quality [33], excellent heat and mass transfer [34], shorter reaction times [35-37], reduced reagent consumption [38-40], improved safety [41,42], and operational simplicity [43]. Furthermore, CF methodologies provide opportunities for a simple and rapid scale-up [44,45] and automation [46,47] of chemical processes. They also tend to be environmentally benign technologies [48]. In consequence of these benefits, flow chemistry-based techniques have exerted a significant impact on modern synthetic chemistry, ranging from laboratory-based experiments to industrial-scale production.
Here, we describe a safe and efficient CF synthesis of a series of novel 1,2,3-triazole-modified β-aminocyclohexanecarboxylic acid derivatives as potential biologically active compounds. Gram-scale production is also reported, which predicts a possible usefulness for the pharmaceutical industry.
Results and Discussion
Several approaches are to be found in the literature for the Cu(I)-catalysed flow synthesis of triazoles. Heterogeneous Cu(I) sources are most popular, such as copper-in-charcoal (Cu/C) [49,50], solid supported Cu(I) species [51-54], and heated copper wirings [55-58], but a homogeneous technique has also recently emerged [59]. The main driving forces behind these CF methodologies are the safety aspects associated with the handling of azides and the inherent scalability of flow processing. Moreover, when organic azides are formed in situ, operational safety can be further improved [55,57]. We envisioned that it would be simplest to make use of copper powder as a catalytic source [60]. Similarly to cases when heated rings of copper wire are employed, a copper surface acts as a source of active copper species. Copper is constantly oxidized when exposed to air, and non-self-protecting layers of different oxides, including Cu2O, are formed on its surface [61], which can promote CuAAC. Thus, we utilized copper powder in a stainless steel column, which served as a catalyst bed later on. The catalyst bed was placed into a stainless steel block with a Peltier heating system, which could heat the column up to 100 °C. A backpressure regulator was also integrated to ensure pressures up to 100 bar. The mixture of the reactants was pumped through the system continuously by means of an HPLC pump. This experimental setup is practical and cheap, as it does not require costly catalysts or special apparatus. At the same time this setup is safe, even with unstable reactants such as azides (Figure 3).
![[1860-5397-9-172-3]](/bjoc/content/figures/1860-5397-9-172-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Experimental setup for the CF reactions.
Figure 3: Experimental setup for the CF reactions.
To maximize the CF triazole synthesis reaction rates, it appeared easiest to use high-temperature conditions initially. The application of elevated pressure in CuAAC is also beneficial, as it can promote the product formation in accordance with Le Chatelier’s principle [60] and also prevents the solvent from boiling over when high temperature is used. Thus, 100 °C and 100 bar were selected as conditions A for the CF synthesis. However, when azides are reacted, it is important to minimize the explosion hazard. Accordingly, we attempted to improve the rates of the reaction in the presence of additives, without the use of high temperature [60]. Amines are known to accelerate CuAAC, in particular by coordinating to catalytically active Cu(I) species and promoting their liberation from the copper matrix [62,63]. It was recently shown that the use of certain acids as additives is also beneficial, as this can further accelerate the formation of the triazole product [64-66] and also prevents the accumulation of unwanted byproducts, such as diacetylenes, bistriazoles, etc. [67]. At the same time, byproduct formation is catalysed by a base, and the joint utilization of a basic and an acidic additive is therefore favourable. This buffer system gives rise to a high reactivity in CuAAC, even at room temperature (rt), but without byproduct formation [60,67]. This system thus greatly improves the safety relative to the high-temperature conditions. The literature data led us to select N,N-diisopropylethylamine (DIEA) as a base and HOAc as an acid [67], which were used jointly as additives, each in 0.04 equivalents, at 100 bar and rt as conditions B [60].
As starting materials for the CF CuAAC reactions, azido-substituted β-aminocyclohexanecarboxylates 11–14 were prepared previously by a diastereoselective epoxidation of the corresponding 2-aminocyclohexenecarboxylates, followed by a regioselective oxirane ring opening with NaN3 [68]. Three different alkynes (phenylacetylene, diethyl acetylenedicarboxylate and ethynyl ferrocene) were employed as dipolarophiles to yield a library of novel 1,2,3-triazole-modified cyclic β-amino acid derivatives. Compounds 11–14 were racemates, the structures in Table 1 show their relative stereochemistry. The CF syntheses were carried out under both conditions A and B in order to obtain a clear comparison between the performances of the two approaches. CH2Cl2 was used as a solvent, and the starting azides were used in a concentration of 0.085 M. A higher concentration of the starting azides led to the precipitation of the triazole product and a blockage in the CF reactor. Aliquots of 2.5 mL of a reaction mixture containing 1 equivalent of the azide and 1.5 equivalents of the acetylene were pumped through the reactor in each run with a a flow rate of 0.5 mL min–1. At this flow rate the residence time on the catalyst bed was as low as 1.5 min and it took only 5 min of process time to pump the 2.5 mL aliquots through the system. This resulted in around 100 mg of crude product, depending on the conversion and the molecular masses of the reactants.
Table 1: CF synthesis of 1,2,3-triazole-substituted alicyclic β-amino acid derivatives.
| Entry | Azidea (1 equivalent) | Acetylene (1.5 equivalents) | Product | Yieldb (%) | |
|---|---|---|---|---|---|
| Ac | Bd | ||||
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-172-i3.svg?max-width=637&scale=1.0)
11 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-172-i4.svg?max-width=637&scale=1.0)
|
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-172-i5.svg?max-width=637&scale=1.0)
15 |
61 | 96 |
| 2 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-172-i6.svg?max-width=637&scale=1.0)
12 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-172-i7.svg?max-width=637&scale=1.0)
|
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-172-i8.svg?max-width=637&scale=1.0)
16 |
47 | 97 |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-172-i9.svg?max-width=637&scale=1.0)
13 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-172-i10.svg?max-width=637&scale=1.0)
|
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-172-i11.svg?max-width=637&scale=1.0)
17 |
33 |
76
(98)e |
| 4 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-172-i12.svg?max-width=637&scale=1.0)
14 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-172-i13.svg?max-width=637&scale=1.0)
|
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-172-i14.svg?max-width=637&scale=1.0)
18 |
53 |
89
(98)e |
| 5 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-172-i15.svg?max-width=637&scale=1.0)
11 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-172-i16.svg?max-width=637&scale=1.0)
|
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-172-i17.svg?max-width=637&scale=1.0)
19 |
98 | 97 |
| 6 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-172-i18.svg?max-width=637&scale=1.0)
12 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-172-i19.svg?max-width=637&scale=1.0)
|
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-172-i20.svg?max-width=637&scale=1.0)
20 |
97 | 98 |
| 7 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-172-i21.svg?max-width=637&scale=1.0)
13 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-172-i22.svg?max-width=637&scale=1.0)
|
![[Graphic 21]](/bjoc/content/inline/1860-5397-9-172-i23.svg?max-width=637&scale=1.0)
21 |
97 | 96 |
| 8 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-9-172-i24.svg?max-width=637&scale=1.0)
14 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-9-172-i25.svg?max-width=637&scale=1.0)
|
![[Graphic 24]](/bjoc/content/inline/1860-5397-9-172-i26.svg?max-width=637&scale=1.0)
22 |
97 | 98 |
| 9 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-9-172-i27.svg?max-width=637&scale=1.0)
11 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-9-172-i28.svg?max-width=637&scale=1.0)
|
![[Graphic 27]](/bjoc/content/inline/1860-5397-9-172-i29.svg?max-width=637&scale=1.0)
23 |
95 | 97 |
| 10 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-9-172-i30.svg?max-width=637&scale=1.0)
12 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-9-172-i31.svg?max-width=637&scale=1.0)
|
![[Graphic 30]](/bjoc/content/inline/1860-5397-9-172-i32.svg?max-width=637&scale=1.0)
24 |
91 | 98 |
| 11 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-9-172-i33.svg?max-width=637&scale=1.0)
13 |
![[Graphic 32]](/bjoc/content/inline/1860-5397-9-172-i34.svg?max-width=637&scale=1.0)
|
![[Graphic 33]](/bjoc/content/inline/1860-5397-9-172-i35.svg?max-width=637&scale=1.0)
25 |
96 | 93 |
| 12 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-9-172-i36.svg?max-width=637&scale=1.0)
14 |
![[Graphic 35]](/bjoc/content/inline/1860-5397-9-172-i37.svg?max-width=637&scale=1.0)
|
![[Graphic 36]](/bjoc/content/inline/1860-5397-9-172-i38.svg?max-width=637&scale=1.0)
26 |
75 | 97 |
acazide = 0.085 M. bYield of isolated product. cConditions A: CH2Cl2 as solvent, 100 bar, 100 °C, flow rate 0.5 mL min–1, without any additives. dConditions B: CH2Cl2 as solvent, 100 bar, rt, flow rate 0.5 mL min–1, with 0.04 equivalents of DIEA + 0.04 equivalents of HOAc. eAchieved under the following conditions: CH2Cl2 as solvent, 100 bar, 100 °C, flow rate 0.5 mL min–1, with 0.04 equivalents of DIEA + 0.04 equivalents of HOAc.
In the Cu(I)-catalysed reactions between phenylacetylene and the azido-substituted β-amino acid derivatives 11–14, 1,4-disubstituted 1,2,3-triazole isomers (15–18) were regioselectively formed. The high-pressure/high-temperature conditions A led to only medium yields (Table 1, entries 1–4), but under conditions B the yields of triazoles 15 and 16 were excellent, and those of triazoles 17 and 18 were high (76% and 89%, respectively; Table 1, entries 1–4). When the CF reactions of azides 13 and 14 with phenylacetylene were repeated under high-pressure/high-temperature conditions with the simultaneous use of additives (100 bar, 100 °C, 0.04 equivalents each of DIEA and HOAc; further conditions were not modified), triazoles 17 and 18 were obtained in very high yields (98% in both cases; Table 1, entries 3 and 4).
1,4,5-Trisubstituted 1,2,3-triazoles are of notable importance in drug discovery. For example, several 1,2,3-triazole-4,5-dicarboxylates display significant antituberculotic activity in vitro [69]. Thus, a nonterminal alkyne, diethyl acetylenedicarboxylate, was subjected to CF CuAAC with the azido-functionalized β-amino acid derivatives 11–14 as reaction partners. 1,4,5-Trisubstituted 1,2,3-triazole dicarboxylates 19–22 were obtained in excellent yields (>96%) under both conditions A and B (Table 1, entries 5–8). In this set of CF syntheses, no significant difference was observed between the performances of the two methods.
Ferrocene-triazole conjugates play a crucial role in the labelling and detection of various systems, such as biomolecules, polymers, nanomaterials and supramolecular assemblies [70]. They also have potential applications in medicinal chemistry and drug discovery as biosensing probes, in immunoassays and in host–guest chemistry [71]. Ferrocene-substituted amino acids have been of significant importance in the investigation of the secondary structures of different peptides and foldamers [72]. Thus, conjugates of the azido-functionalized β-amino acid derivatives 11–14 were prepared with ethynylferrocene as a dipolarophile. Both conditions A and B afforded ferrocenyltriazoles 23–25 in excellent yields (>91%; Table 1, entries 9–11). However, in the case of ferrocenyltriazole 26 the high-pressure/high-temperature conditions A led to a yield of only 75%, whereas the use of additives at rt (conditions B) proved more efficient, with a yield of 97% (Table 1, entry 12). Triazoles 23–26 were obtained selectively as 1,4-disubstituted regioisomers.
To understand the differences between the results obtained with the three different dipholarophiles, it must be taken into account that the carboxylate groups of diethyl acetylenedicarboxylate and the aromatic system of the ferrocenyl group as ligands can probably coordinate copper from its matrix. Therefore, the concentration of the catalytically active Cu(I) species is increased as compared to the reactions with phenylacetylene [73-75]. Accordingly, the yields were usually higher in the reactions with diethyl acetylenedicarboxylate and ethynylferrocene than with phenylacetylene (Table 1, entries 5–12 versus entries 1–4). These differences can mainly be observed between the results obtained under conditions A. This is because the base, as an additive, evolves the same effect and improves the reactivity through the CuAAC, thus in the case of conditions B (the use of additives) the influence of the alkyne is practically masked.
The presence of trace amounts of copper in the chromatographically purified triazole products was determined by means of inductively coupled plasma mass spectrometry (ICP–MS). The analytical data in Table 2 show that the contents of copper impurities in the products were appropriately low, i.e., amounts of 3.9–9.1 µg g–1 were detected. It should be noted that the samples obtained with the joint use of DIEA + HOAc (conditions B) contained more copper than those obtained under conditions A (high-temperature/high-pressure without additives). The levels of copper contamination detected in our triazole products compare well with literature results relating to CF [50] and conventional batch experiments [76].
Table 2: Copper contents in the triazole products after column chromatographic purification on silica gel.
| Entry | Product | Copper content (µg g–1)a | |
|---|---|---|---|
| Ab | Bc | ||
| 1 | 15 | 4.6 (±0.5) | 8.4 (±0.6) |
| 2 | 16 | 4.2 (±0.3) | 7.7 (±0.6) |
| 3 | 17 | 3.9 (±0.5) | 8.0 (±0.4) |
| 4 | 18 | 4.7 (±0.6) | 8.2 (±0.7) |
| 5 | 19 | 5.2 (±0.4) | 7.9 (±0.4) |
| 6 | 20 | 5.1 (±0.3) | 7.5 (±0.6) |
| 7 | 21 | 4.8 (±0.6) | 7.7 (±0.7) |
| 8 | 22 | 5.3 (±0.3) | 8.2 (±0.6) |
| 9 | 23 | 6.1 (±0.5) | 8.6 (±0.5) |
| 10 | 24 | 4.8 (±0.4) | 7.7 (±0.8) |
| 11 | 25 | 5.4 (±0.3) | 9.1 (±0.4) |
| 12 | 26 | 4.9 (±0.6) | 7.8 (±0.7) |
aDetermined by ICP–MS. bConditions A: CH2Cl2 as solvent, 100 bar, 100 °C, flow rate 0.5 mL min–1, without any additives. cConditions B: CH2Cl2 as solvent, 100 bar, rt, flow rate 0.5 mL min–1, with 0.04 equivalents of DIEA + 0.04 equivalents of HOAc.
In conventional batch-based chemistry, the scale-up of chemical reactions can be a challenge because the output depends on the batch size. The situation becomes even more complicated when unstable reactants such as azides are handled on a large scale. However, the scalability of CF procedures is a straightforward function of time and the flow rate, and the risks associated with the accumulation of hazardous species are minimized, because the solution of the reactants is eluting continuously from the active zone of the reactor [33,34,44,45,60]. The CF CuAAC between azide 14 and diethyl acetylenedicarboxylate was scaled up in a simple, safe and efficient manner to achieve gram-scale production (Scheme 2). Methods A and B proved equally efficient in the small-scale CF syntheses of triazole 22 (Table 1, entry 8). However, we performed the large-scale experiment at 100 bar and rt in the presence of the additives (conditions B) so as to ensure maximum safety throughout the procedure. A CH2Cl2 solution of the reaction mixture containing 1 equivalent of the azide (cazide = 0.085 M), 1.5 equivalents of the acetylene and 0.04 equivalents of each additive was pumped continuously through the system at a flow rate of 0.5 mL min–1. During the whole scale-up procedure, the same portion of copper powder was used in the catalyst bed. The solution of the crude product was collected for 100 min, and after purification 2.06 g of triazole 22 was obtained, which is equivalent to a yield of 96%.
![[1860-5397-9-172-i2]](/bjoc/content/inline/1860-5397-9-172-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Gramm-scale CF synthesis of triazole 22 under conditions B.
Scheme 2: Gramm-scale CF synthesis of triazole 22 under conditions B.
Conclusion
Twelve highly functionalized 1,2,3-triazole-substituted β-aminocyclohexanecarboxylic acid racemates were successfully prepared in CF mode as a small library of novel compounds with possible biological effects. The CF syntheses were first performed under high-pressure/high-temperature conditions with copper powder as a readily accessible Cu(I) source. Subsequently, to moderate the harsh reaction conditions, the reaction temperature could be lowered to rt in the presence of additives. The joint use of a base and an acid dramatically improved the reactivity in the CuAAC, while it completely eliminated unwanted byproduct formation. These conditions ensured enhanced safety and typically higher yields than those attained under the harsh reaction conditions. Simple, efficient and safe gram-scale production was also implemented in a short processing time, which can be important for potential industrial applications.
Experimental
General Information
The reagents and materials were of the highest commercially available purity grade and were used without any further purification. Flash column chromatography was performed on Merck silica gel 60, particle sizes ranged from 63 to 200 μm, and analytical thin-layer chromatography (TLC) on Merck silica gel 60 F254 plates. Compounds were visualized with UV light or KMnO4. 1H and 13C NMR spectra were recorded on a Bruker Avance DRX 500 spectrometer, in CDCl3 as a solvent, with TMS as internal standard, and at 500.1 and 125.0 MHz, respectively. Microanalyses were performed on a Perkin-Elmer 2400 elemental analyser.
Determination of the copper contents of the triazole products
Copper concentrations were determined by ICP–MS by using an Agilent 7700x instrument equipped with a collision cell. The determination was carried out on the isotope 63Cu, with He as collision gas. The standard solutions for external calibration were prepared from a stock solution (Certipur, Merck) by dilution with doubly deionized water (Millipore MillQ, Merck). All glassware and plastic utensils used during the determination were precleaned by soaking in solutions of trace-metal-grade nitric acid and hydrochloric acid (Suprapur, Merck), followed by rinsing with copious amounts of doubly deionized water.
General procedure for the CF reactions
An H-Cube® system was used as a CF reactor in the “no H2” mode. For the CF reactions, the catalyst bed (internal dimensions: 70 mm × 4 mm) was filled with ~900 mg of copper powder with an average particle size of 200 µm. 70 mg (0.21 mmol, 1 equivalent) of the corresponding azide and 0.32 mmol (1.5 equivalents) of the alkyne, and (only in method B) 1.5 µL (0.0084 mmol, 0.04 equivalents) of DIEA and 0.5 µL (0.0084 mmol, 0.04 equivalents) of HOAc were dissolved in 2.5 mL of CH2Cl2. The solution was homogenized by sonication, and then pumped through the CF reactor under the appropriate conditions. Between two reactions in the CF reactor, the catalyst bed was washed at rt for 5 min with CH2Cl2 at a flow rate of 1 mL min–1. The crude product was checked by TLC with a mixture of n-hexane/EtOAc as an eluent, and the solvent was next evaporated off under vacuum. Column chromatographic purification was carried out on silica gel with a mixture of n-hexane/EtOAc as an eluent. The 1,2,3-triazole-modified compounds were characterized by elemental analysis and NMR experiments. For detailed analytical data see Supporting Information File 1.
Measurement of the residence time on the catalyst bed
To determine the residence time, a CH2Cl2 solution of a blue ink was pumped through the catalyst bed. The time that elapsed between the first contact of the ink with the bed and the moment when the blue colour appeared at the column outlet was measured.
Supporting Information
| Supporting Information File 1: Detailed analytical data of the prepared compounds and a collection of NMR spectra. | ||
| Format: PDF | Size: 1.3 MB | Download |
Acknowledgements
This research was partly realized within the scope of TÁMOP 4.2.4. A/2-11-1-2012-0001 „National Excellence Program – Elaborating and operating an inland student and researcher personal support system convergence program”. The project was subsidized by the European Union and co-financed by the European Social Fund. We are grateful to the Hungarian Research Foundation (OTKA Nos. NK81371, PD103994 and K100530) and TÁMOP 4.2.2/B-10/1-2010-0012. IMM acknowledges the award of a János Bolyai scholarship from the Hungarian Academy of Sciences.
References
-
Moorhouse, A. D.; Moses, J. E. ChemMedChem 2008, 3, 715–723. doi:10.1002/cmdc.200700334
Return to citation in text: [1] -
Hou, J. L.; Liu, X. F.; Shen, J.; Zhao, G. L.; Wang, P. G. Expert. Opin. Drug Discov. 2012, 7, 489–501. doi:10.1517/17460441.2012.682725
Return to citation in text: [1] -
Agalave, S. G.; Maujan, S. R.; Pore, V. S. Chem.–Asian J. 2011, 6, 2696–2718. doi:10.1002/asia.201100432
Return to citation in text: [1] -
Jordão, A. K.; Ferreira, V. F.; Lima, E. S.; de Souza, M. C. B. V.; Carlos, E. C. L.; Castro, H. C.; Geraldo, R. B.; Rodrigues, C. R.; Almeida, M. C. B.; Cunha, A. C. Bioorg. Med. Chem. 2009, 17, 3713–3719. doi:10.1016/j.bmc.2009.03.053
Return to citation in text: [1] -
Vijaya Raghava Reddy, L.; Venkat Reddy, P.; Mishra, N. N.; Shukla, P. K.; Yadav, G.; Srivastava, R.; Shaw, A. K. Carbohydr. Res. 2010, 345, 1515–1521. doi:10.1016/j.carres.2010.03.031
Return to citation in text: [1] -
Aher, N. G.; Pore, V. S.; Mishra, N. N.; Kumar, A.; Shukla, P. K.; Sharma, A.; Bhat, M. K. Bioorg. Med. Chem. Lett. 2009, 19, 759–763. doi:10.1016/j.bmcl.2008.12.026
Return to citation in text: [1] -
Soltis, M. J.; Yeh, H. J.; Cole, K. A.; Whittaker, N.; Wersto, R. P.; Kohn, E. C. Drug Metab. Dispos. 1996, 24, 799–806.
Return to citation in text: [1] -
Cho, J. H.; Bernard, D. L.; Sidwell, R. W.; Kern, E. R.; Chu, C. K. J. Med. Chem. 2006, 49, 1140–1148. doi:10.1021/jm0509750
Return to citation in text: [1] -
Li, J.; Zheng, M.; Tang, W.; He, P.-L.; Zhu, W.; Li, T.; Zuo, J.-P.; Liu, H.; Jiang, H. Bioorg. Med. Chem. Lett. 2006, 16, 5009–5013. doi:10.1016/j.bmcl.2006.07.047
Return to citation in text: [1] -
Xia, Y.; Liu, Y.; Wan, J.; Wang, M.; Rocchi, P.; Qu, F.; Iovanna, J. L.; Peng, L. J. Med. Chem. 2009, 52, 6083–6096. doi:10.1021/jm900960v
Return to citation in text: [1] -
Pérez-Castro, I.; Caamaño, O.; Fernández, F.; García, M. D.; López, C.; De Clercq, E. Org. Biomol. Chem. 2007, 5, 3805–3813. doi:10.1039/b710348d
Return to citation in text: [1] -
Kiss, L.; Forro, E.; Fulop, F. Lett. Org. Chem. 2011, 8, 220–228. doi:10.2174/157017811795038359
Return to citation in text: [1] -
El-Sagheer, A. H.; Brown, T. Chem. Soc. Rev. 2010, 39, 1388–1405. doi:10.1039/b901971p
Return to citation in text: [1] -
Fahrenbach, A. C.; Stoddart, J. F. Chem.–Asian J. 2011, 6, 2660–2669. doi:10.1002/asia.201100457
Return to citation in text: [1] -
Kempe, K.; Krieg, A.; Becer, C. R.; Schubert, U. S. Chem. Soc. Rev. 2012, 41, 176–191. doi:10.1039/c1cs15107j
Return to citation in text: [1] -
Huisgen, R. In 1,3-Dipolar Cycloadditional Chemistry; Padwa, A., Ed.; Wiley: New York, 1984; pp 1–176.
Return to citation in text: [1] -
Huisgen, R.; Szeimis, G.; Möbius, L. Chem. Ber. 1967, 100, 2494–2507. doi:10.1002/cber.19671000806
Return to citation in text: [1] -
Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4
Return to citation in text: [1] -
Wang, Q.; Chan, T. R.; Hilgraf, R.; Fokin, V. V.; Sharpless, K. B.; Finn, M. G. J. Am. Chem. Soc. 2003, 125, 3192–3193. doi:10.1021/ja021381e
Return to citation in text: [1] -
Tornoe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057–3064. doi:10.1021/jo011148j
Return to citation in text: [1] -
Kuhl, A.; Hahn, M. G.; Dumić, M.; Mittendorf, J. Amino Acids 2005, 29, 89–100. doi:10.1007/s00726-005-0212-y
Return to citation in text: [1] -
Weiner, B.; Szymański, W.; Janssen, D. B.; Minnaard, A. J.; Feringa, B. L. Chem. Soc. Rev. 2010, 39, 1656–1691. doi:10.1039/b919599h
Return to citation in text: [1] -
Konishi, M.; Nishio, M.; Saitoh, K.; Miyaki, T.; Oki, T.; Kawaguchi, H. J. Antibiot. 1989, 42, 1749–1755. doi:10.7164/antibiotics.42.1749
Return to citation in text: [1] -
Mittendorf, J.; Kunisch, F.; Matzke, M.; Militzer, H.-C.; Schmidt, A.; Schönfeld, W. Bioorg. Med. Chem. Lett. 2003, 13, 433–436. doi:10.1016/S0960-894X(02)00958-7
Return to citation in text: [1] -
Bunnage, M. E.; Ganesh, T.; Masesane, I. B.; Orton, D.; Steel, P. G. Org. Lett. 2003, 5, 239–242. doi:10.1021/ol0269704
Return to citation in text: [1] -
Palko, M.; Kiss, L.; Fulop, F. Curr. Med. Chem. 2005, 12, 3063–3083. doi:10.2174/092986705774933443
Return to citation in text: [1] -
Kiss, L.; Forro, E.; Fulop, F. In Amino Acids, Peptides and Proteins in Organic Chemistry; Hughes, A. B., Ed.; Wiley-VCH, 2009; pp 367–409.
Return to citation in text: [1] -
Martinek, T. A.; Fülöp, F. Chem. Soc. Rev. 2012, 41, 687–702. doi:10.1039/c1cs15097a
Return to citation in text: [1] -
Wegner, J.; Ceylan, S.; Kirschning, A. Adv. Synth. Catal. 2012, 354, 17–57. doi:10.1002/adsc.201100584
Return to citation in text: [1] -
Yoshida, J.-i.; Kim, H.; Nagaki, A. ChemSusChem 2011, 4, 331–340. doi:10.1002/cssc.201000271
Return to citation in text: [1] -
Geyer, K.; Gustafsson, T.; Seeberger, P. H. Synlett 2009, 2382–2391. doi:10.1055/s-0029-1217828
Return to citation in text: [1] -
Mak, X. Y.; Laurino, P.; Seeberger, P. H. Beilstein J. Org. Chem. 2009, 5, No. 19. doi:10.3762/bjoc.5.19
Return to citation in text: [1] -
Nagy, K. D.; Shen, B.; Jamison, T. F.; Jensen, K. F. Org. Process Res. Dev. 2012, 16, 976–981. doi:10.1021/op200349f
Return to citation in text: [1] [2] -
Hartman, R. L.; McMullen, J. P.; Jensen, K. F. Angew. Chem., Int. Ed. 2011, 50, 7502–7519. doi:10.1002/anie.201004637
Return to citation in text: [1] [2] -
Ötvös, S. B.; Mándity, I. M.; Fülöp, F. ChemSusChem 2012, 5, 266–269. doi:10.1002/cssc.201100332
Return to citation in text: [1] -
Ötvös, S. B.; Mándity, I. M.; Fülöp, F. J. Catal. 2012, 295, 179–185. doi:10.1016/j.jcat.2012.08.006
Return to citation in text: [1] -
Yoshida, J.-i.; Nagaki, A.; Yamada, T. Chem.–Eur. J. 2008, 14, 7450–7459. doi:10.1002/chem.200800582
Return to citation in text: [1] -
Ueno, M.; Suzuki, T.; Naito, T.; Oyamada, H.; Kobayashi, S. Chem. Commun. 2008, 1647–1649. doi:10.1039/b715259k
Return to citation in text: [1] -
Rasheed, M.; Wirth, T. Angew. Chem., Int. Ed. 2011, 50, 357–358. doi:10.1002/anie.201006107
Return to citation in text: [1] -
Rueping, M.; Bootwicha, T.; Sugiono, E. Beilstein J. Org. Chem. 2012, 8, 300–307. doi:10.3762/bjoc.8.32
Return to citation in text: [1] -
Ötvös, S. B.; Mándity, I. M.; Fülöp, F. Mol. Diversity 2011, 15, 605–611. doi:10.1007/s11030-010-9276-z
Return to citation in text: [1] -
Brandt, J. C.; Wirth, T. Beilstein J. Org. Chem. 2009, 5, No. 30. doi:10.3762/bjoc.5.30
Return to citation in text: [1] -
Bryan, M. C.; Wernick, D.; Hein, C. D.; Petersen, J. V.; Eschelbach, J. W.; Doherty, E. M. Beilstein J. Org. Chem. 2011, 7, 1141–1149. doi:10.3762/bjoc.7.132
Return to citation in text: [1] -
He, P.; Haswell, S. J.; Fletcher, P. D. I.; Kelly, S. M.; Mansfield, A. Beilstein J. Org. Chem. 2011, 7, 1150–1157. doi:10.3762/bjoc.7.133
Return to citation in text: [1] [2] -
Kockmann, N.; Gottsponer, M.; Roberge, D. M. Chem. Eng. J. 2011, 167, 718–726. doi:10.1016/j.cej.2010.08.089
Return to citation in text: [1] [2] -
Smith, C. J.; Nikbin, N.; Ley, S. V.; Lange, H.; Baxendale, I. R. Org. Biomol. Chem. 2011, 9, 1938–1947. doi:10.1039/c0ob00815j
Return to citation in text: [1] -
Baumann, M.; Baxendale, I. R.; Ley, S. V.; Smith, C. D.; Tranmer, G. K. Org. Lett. 2006, 8, 5231–5234. doi:10.1021/ol061975c
Return to citation in text: [1] -
Wiles, C.; Watts, P. Green Chem. 2012, 14, 38–54. doi:10.1039/c1gc16022b
Return to citation in text: [1] -
Lipshutz, B. H.; Taft, B. R. Angew. Chem., Int. Ed. 2006, 45, 8235–8238. doi:10.1002/anie.200603726
Return to citation in text: [1] -
Fuchs, M.; Goessler, W.; Pilger, C.; Kappe, C. O. Adv. Synth. Catal. 2010, 352, 323–328. doi:10.1002/adsc.200900726
Return to citation in text: [1] [2] -
Girard, C.; Önen, E.; Aufort, M.; Beauvière, S.; Samson, E.; Herscovici, J. Org. Lett. 2006, 8, 1689–1692. doi:10.1021/ol060283l
Return to citation in text: [1] -
Smith, C. D.; Baxendale, I. R.; Lanners, S.; Hayward, J. J.; Smith, S. C.; Ley, S. V. Org. Biomol. Chem. 2007, 5, 1559–1561. doi:10.1039/b702995k
Return to citation in text: [1] -
Özçubukçu, S.; Ozkal, E.; Jimeno, C.; Pericàs, M. A. Org. Lett. 2009, 11, 4680–4683. doi:10.1021/ol9018776
Return to citation in text: [1] -
Ozkal, E.; Özçubukçu, S.; Jimeno, C.; Pericàs, M. A. Catal. Sci. Technol. 2012, 2, 195–200. doi:10.1039/c1cy00297j
Return to citation in text: [1] -
Bogdan, A. R.; Sach, N. W. Adv. Synth. Catal. 2009, 351, 849–854. doi:10.1002/adsc.200800758
Return to citation in text: [1] [2] -
Bogdan, A. R.; James, K. Chem.–Eur. J. 2010, 16, 14506–14512. doi:10.1002/chem.201002215
Return to citation in text: [1] -
Ceylan, S.; Klande, T.; Vogt, C.; Friese, C.; Kirschning, A. Synlett 2010, 2009–2013. doi:10.1055/s-0030-1258487
Return to citation in text: [1] [2] -
Kupracz, L.; Hartwig, J.; Wegner, J.; Ceylan, S.; Kirschning, A. Beilstein J. Org. Chem. 2011, 7, 1441–1448. doi:10.3762/bjoc.7.168
Return to citation in text: [1] -
Varas, A. C.; Noël, T.; Wang, Q.; Hessel, V. ChemSusChem 2012, 5, 1703–1707. doi:10.1002/cssc.201200323
Return to citation in text: [1] -
Ötvös, S. B.; Mándity, I. M.; Kiss, L.; Fülöp, F. Chem.–Asian J. 2013, 8, 800–808. doi:10.1002/asia.201201125
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Boggio, J. E. J. Chem. Phys. 1979, 70, 5054–5058. doi:10.1063/1.437347
Return to citation in text: [1] -
Meldal, M.; Tornøe, C. W. Chem. Rev. 2008, 108, 2952–3015. doi:10.1021/cr0783479
Return to citation in text: [1] -
Rodionov, V. O.; Fokin, V. V.; Finn, M. G. Angew. Chem., Int. Ed. 2005, 44, 2210–2215. doi:10.1002/anie.200461496
Return to citation in text: [1] -
Nolte, C.; Mayer, P.; Straub, B. F. Angew. Chem., Int. Ed. 2007, 46, 2101–2103. doi:10.1002/anie.200604444
Return to citation in text: [1] -
Shao, C.; Cheng, G.; Su, D.; Xu, J.; Wang, X.; Hu, Y. Adv. Synth. Catal. 2010, 352, 1587–1592. doi:10.1002/adsc.200900768
Return to citation in text: [1] -
Shao, C.; Wang, X.; Xu, J.; Zhao, J.; Zhang, Q.; Hu, Y. J. Org. Chem. 2010, 75, 7002–7005. doi:10.1021/jo101495k
Return to citation in text: [1] -
Shao, C.; Wang, X.; Zhang, Q.; Luo, S.; Zhao, J.; Hu, Y. J. Org. Chem. 2011, 76, 6832–6836. doi:10.1021/jo200869a
Return to citation in text: [1] [2] [3] -
Kiss, L.; Forró, E.; Fülöp, F. Tetrahedron 2012, 68, 4438–4443. doi:10.1016/j.tet.2011.12.065
Return to citation in text: [1] -
Shanmugavelan, P.; Nagarajan, S.; Sathishkumar, M.; Ponnuswamy, A.; Yogeeswari, P.; Sriram, D. Bioorg. Med. Chem. Lett. 2011, 21, 7273–7276. doi:10.1016/j.bmcl.2011.10.048
Return to citation in text: [1] -
Ganesh, V.; Sudhir, V. S.; Kundu, T.; Chandrasekaran, S. Chem.–Asian J. 2011, 6, 2670–2694. doi:10.1002/asia.201100408
Return to citation in text: [1] -
Fouda, M. F. R.; Abd-Elzaher, M. M.; Abdelsamaia, R. A.; Labib, A. A. Appl. Organomet. Chem. 2007, 21, 613–625. doi:10.1002/aoc.1202
Return to citation in text: [1] -
van Staveren, D. R.; Metzler-Nolte, N. Chem. Rev. 2004, 104, 5931–5986. doi:10.1021/cr0101510
Return to citation in text: [1] -
Nakamura, E.; Mori, S. Angew. Chem., Int. Ed. 2000, 39, 3750–3771. doi:10.1002/1521-3773(20001103)39:21<3750::AID-ANIE3750>3.0.CO;2-L
Return to citation in text: [1] -
Grodzicki, A.; Łakomska, I.; Piszczek, P.; Szymańska, I.; Szłyk, E. Coord. Chem. Rev. 2005, 249, 2232–2258. doi:10.1016/j.ccr.2005.05.026
Return to citation in text: [1] -
Liang, L.; Astruc, D. Coord. Chem. Rev. 2011, 255, 2933–2945. doi:10.1016/j.ccr.2011.06.028
Return to citation in text: [1] -
Kovács, S.; Zih-Perényi, K.; Révész, A.; Novák, Z. Synthesis 2012, 44, 3722–3730. doi:10.1055/s-0032-1317697
Return to citation in text: [1]
| 49. | Lipshutz, B. H.; Taft, B. R. Angew. Chem., Int. Ed. 2006, 45, 8235–8238. doi:10.1002/anie.200603726 |
| 50. | Fuchs, M.; Goessler, W.; Pilger, C.; Kappe, C. O. Adv. Synth. Catal. 2010, 352, 323–328. doi:10.1002/adsc.200900726 |
| 51. | Girard, C.; Önen, E.; Aufort, M.; Beauvière, S.; Samson, E.; Herscovici, J. Org. Lett. 2006, 8, 1689–1692. doi:10.1021/ol060283l |
| 52. | Smith, C. D.; Baxendale, I. R.; Lanners, S.; Hayward, J. J.; Smith, S. C.; Ley, S. V. Org. Biomol. Chem. 2007, 5, 1559–1561. doi:10.1039/b702995k |
| 53. | Özçubukçu, S.; Ozkal, E.; Jimeno, C.; Pericàs, M. A. Org. Lett. 2009, 11, 4680–4683. doi:10.1021/ol9018776 |
| 54. | Ozkal, E.; Özçubukçu, S.; Jimeno, C.; Pericàs, M. A. Catal. Sci. Technol. 2012, 2, 195–200. doi:10.1039/c1cy00297j |
| 55. | Bogdan, A. R.; Sach, N. W. Adv. Synth. Catal. 2009, 351, 849–854. doi:10.1002/adsc.200800758 |
| 56. | Bogdan, A. R.; James, K. Chem.–Eur. J. 2010, 16, 14506–14512. doi:10.1002/chem.201002215 |
| 57. | Ceylan, S.; Klande, T.; Vogt, C.; Friese, C.; Kirschning, A. Synlett 2010, 2009–2013. doi:10.1055/s-0030-1258487 |
| 58. | Kupracz, L.; Hartwig, J.; Wegner, J.; Ceylan, S.; Kirschning, A. Beilstein J. Org. Chem. 2011, 7, 1441–1448. doi:10.3762/bjoc.7.168 |
| 62. | Meldal, M.; Tornøe, C. W. Chem. Rev. 2008, 108, 2952–3015. doi:10.1021/cr0783479 |
| 63. | Rodionov, V. O.; Fokin, V. V.; Finn, M. G. Angew. Chem., Int. Ed. 2005, 44, 2210–2215. doi:10.1002/anie.200461496 |
| 64. | Nolte, C.; Mayer, P.; Straub, B. F. Angew. Chem., Int. Ed. 2007, 46, 2101–2103. doi:10.1002/anie.200604444 |
| 65. | Shao, C.; Cheng, G.; Su, D.; Xu, J.; Wang, X.; Hu, Y. Adv. Synth. Catal. 2010, 352, 1587–1592. doi:10.1002/adsc.200900768 |
| 66. | Shao, C.; Wang, X.; Xu, J.; Zhao, J.; Zhang, Q.; Hu, Y. J. Org. Chem. 2010, 75, 7002–7005. doi:10.1021/jo101495k |
| 60. | Ötvös, S. B.; Mándity, I. M.; Kiss, L.; Fülöp, F. Chem.–Asian J. 2013, 8, 800–808. doi:10.1002/asia.201201125 |
| 60. | Ötvös, S. B.; Mándity, I. M.; Kiss, L.; Fülöp, F. Chem.–Asian J. 2013, 8, 800–808. doi:10.1002/asia.201201125 |
| 60. | Ötvös, S. B.; Mándity, I. M.; Kiss, L.; Fülöp, F. Chem.–Asian J. 2013, 8, 800–808. doi:10.1002/asia.201201125 |
| 59. | Varas, A. C.; Noël, T.; Wang, Q.; Hessel, V. ChemSusChem 2012, 5, 1703–1707. doi:10.1002/cssc.201200323 |
| 55. | Bogdan, A. R.; Sach, N. W. Adv. Synth. Catal. 2009, 351, 849–854. doi:10.1002/adsc.200800758 |
| 57. | Ceylan, S.; Klande, T.; Vogt, C.; Friese, C.; Kirschning, A. Synlett 2010, 2009–2013. doi:10.1055/s-0030-1258487 |
| 67. | Shao, C.; Wang, X.; Zhang, Q.; Luo, S.; Zhao, J.; Hu, Y. J. Org. Chem. 2011, 76, 6832–6836. doi:10.1021/jo200869a |
| 60. | Ötvös, S. B.; Mándity, I. M.; Kiss, L.; Fülöp, F. Chem.–Asian J. 2013, 8, 800–808. doi:10.1002/asia.201201125 |
| 67. | Shao, C.; Wang, X.; Zhang, Q.; Luo, S.; Zhao, J.; Hu, Y. J. Org. Chem. 2011, 76, 6832–6836. doi:10.1021/jo200869a |
| 67. | Shao, C.; Wang, X.; Zhang, Q.; Luo, S.; Zhao, J.; Hu, Y. J. Org. Chem. 2011, 76, 6832–6836. doi:10.1021/jo200869a |
| 73. | Nakamura, E.; Mori, S. Angew. Chem., Int. Ed. 2000, 39, 3750–3771. doi:10.1002/1521-3773(20001103)39:21<3750::AID-ANIE3750>3.0.CO;2-L |
| 74. | Grodzicki, A.; Łakomska, I.; Piszczek, P.; Szymańska, I.; Szłyk, E. Coord. Chem. Rev. 2005, 249, 2232–2258. doi:10.1016/j.ccr.2005.05.026 |
| 75. | Liang, L.; Astruc, D. Coord. Chem. Rev. 2011, 255, 2933–2945. doi:10.1016/j.ccr.2011.06.028 |
| 50. | Fuchs, M.; Goessler, W.; Pilger, C.; Kappe, C. O. Adv. Synth. Catal. 2010, 352, 323–328. doi:10.1002/adsc.200900726 |
| 71. | Fouda, M. F. R.; Abd-Elzaher, M. M.; Abdelsamaia, R. A.; Labib, A. A. Appl. Organomet. Chem. 2007, 21, 613–625. doi:10.1002/aoc.1202 |
| 72. | van Staveren, D. R.; Metzler-Nolte, N. Chem. Rev. 2004, 104, 5931–5986. doi:10.1021/cr0101510 |
| 69. | Shanmugavelan, P.; Nagarajan, S.; Sathishkumar, M.; Ponnuswamy, A.; Yogeeswari, P.; Sriram, D. Bioorg. Med. Chem. Lett. 2011, 21, 7273–7276. doi:10.1016/j.bmcl.2011.10.048 |
| 70. | Ganesh, V.; Sudhir, V. S.; Kundu, T.; Chandrasekaran, S. Chem.–Asian J. 2011, 6, 2670–2694. doi:10.1002/asia.201100408 |
| 60. | Ötvös, S. B.; Mándity, I. M.; Kiss, L.; Fülöp, F. Chem.–Asian J. 2013, 8, 800–808. doi:10.1002/asia.201201125 |
| 68. | Kiss, L.; Forró, E.; Fülöp, F. Tetrahedron 2012, 68, 4438–4443. doi:10.1016/j.tet.2011.12.065 |
| 33. | Nagy, K. D.; Shen, B.; Jamison, T. F.; Jensen, K. F. Org. Process Res. Dev. 2012, 16, 976–981. doi:10.1021/op200349f |
| 34. | Hartman, R. L.; McMullen, J. P.; Jensen, K. F. Angew. Chem., Int. Ed. 2011, 50, 7502–7519. doi:10.1002/anie.201004637 |
| 44. | He, P.; Haswell, S. J.; Fletcher, P. D. I.; Kelly, S. M.; Mansfield, A. Beilstein J. Org. Chem. 2011, 7, 1150–1157. doi:10.3762/bjoc.7.133 |
| 45. | Kockmann, N.; Gottsponer, M.; Roberge, D. M. Chem. Eng. J. 2011, 167, 718–726. doi:10.1016/j.cej.2010.08.089 |
| 60. | Ötvös, S. B.; Mándity, I. M.; Kiss, L.; Fülöp, F. Chem.–Asian J. 2013, 8, 800–808. doi:10.1002/asia.201201125 |
| 76. | Kovács, S.; Zih-Perényi, K.; Révész, A.; Novák, Z. Synthesis 2012, 44, 3722–3730. doi:10.1055/s-0032-1317697 |
| 1. | Moorhouse, A. D.; Moses, J. E. ChemMedChem 2008, 3, 715–723. doi:10.1002/cmdc.200700334 |
| 2. | Hou, J. L.; Liu, X. F.; Shen, J.; Zhao, G. L.; Wang, P. G. Expert. Opin. Drug Discov. 2012, 7, 489–501. doi:10.1517/17460441.2012.682725 |
| 10. | Xia, Y.; Liu, Y.; Wan, J.; Wang, M.; Rocchi, P.; Qu, F.; Iovanna, J. L.; Peng, L. J. Med. Chem. 2009, 52, 6083–6096. doi:10.1021/jm900960v |
| 11. | Pérez-Castro, I.; Caamaño, O.; Fernández, F.; García, M. D.; López, C.; De Clercq, E. Org. Biomol. Chem. 2007, 5, 3805–3813. doi:10.1039/b710348d |
| 12. | Kiss, L.; Forro, E.; Fulop, F. Lett. Org. Chem. 2011, 8, 220–228. doi:10.2174/157017811795038359 |
| 25. | Bunnage, M. E.; Ganesh, T.; Masesane, I. B.; Orton, D.; Steel, P. G. Org. Lett. 2003, 5, 239–242. doi:10.1021/ol0269704 |
| 26. | Palko, M.; Kiss, L.; Fulop, F. Curr. Med. Chem. 2005, 12, 3063–3083. doi:10.2174/092986705774933443 |
| 8. | Cho, J. H.; Bernard, D. L.; Sidwell, R. W.; Kern, E. R.; Chu, C. K. J. Med. Chem. 2006, 49, 1140–1148. doi:10.1021/jm0509750 |
| 9. | Li, J.; Zheng, M.; Tang, W.; He, P.-L.; Zhu, W.; Li, T.; Zuo, J.-P.; Liu, H.; Jiang, H. Bioorg. Med. Chem. Lett. 2006, 16, 5009–5013. doi:10.1016/j.bmcl.2006.07.047 |
| 27. | Kiss, L.; Forro, E.; Fulop, F. In Amino Acids, Peptides and Proteins in Organic Chemistry; Hughes, A. B., Ed.; Wiley-VCH, 2009; pp 367–409. |
| 4. | Jordão, A. K.; Ferreira, V. F.; Lima, E. S.; de Souza, M. C. B. V.; Carlos, E. C. L.; Castro, H. C.; Geraldo, R. B.; Rodrigues, C. R.; Almeida, M. C. B.; Cunha, A. C. Bioorg. Med. Chem. 2009, 17, 3713–3719. doi:10.1016/j.bmc.2009.03.053 |
| 5. | Vijaya Raghava Reddy, L.; Venkat Reddy, P.; Mishra, N. N.; Shukla, P. K.; Yadav, G.; Srivastava, R.; Shaw, A. K. Carbohydr. Res. 2010, 345, 1515–1521. doi:10.1016/j.carres.2010.03.031 |
| 6. | Aher, N. G.; Pore, V. S.; Mishra, N. N.; Kumar, A.; Shukla, P. K.; Sharma, A.; Bhat, M. K. Bioorg. Med. Chem. Lett. 2009, 19, 759–763. doi:10.1016/j.bmcl.2008.12.026 |
| 7. | Soltis, M. J.; Yeh, H. J.; Cole, K. A.; Whittaker, N.; Wersto, R. P.; Kohn, E. C. Drug Metab. Dispos. 1996, 24, 799–806. |
| 23. | Konishi, M.; Nishio, M.; Saitoh, K.; Miyaki, T.; Oki, T.; Kawaguchi, H. J. Antibiot. 1989, 42, 1749–1755. doi:10.7164/antibiotics.42.1749 |
| 3. | Agalave, S. G.; Maujan, S. R.; Pore, V. S. Chem.–Asian J. 2011, 6, 2696–2718. doi:10.1002/asia.201100432 |
| 24. | Mittendorf, J.; Kunisch, F.; Matzke, M.; Militzer, H.-C.; Schmidt, A.; Schönfeld, W. Bioorg. Med. Chem. Lett. 2003, 13, 433–436. doi:10.1016/S0960-894X(02)00958-7 |
| 16. | Huisgen, R. In 1,3-Dipolar Cycloadditional Chemistry; Padwa, A., Ed.; Wiley: New York, 1984; pp 1–176. |
| 18. | Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4 |
| 19. | Wang, Q.; Chan, T. R.; Hilgraf, R.; Fokin, V. V.; Sharpless, K. B.; Finn, M. G. J. Am. Chem. Soc. 2003, 125, 3192–3193. doi:10.1021/ja021381e |
| 20. | Tornoe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057–3064. doi:10.1021/jo011148j |
| 15. | Kempe, K.; Krieg, A.; Becer, C. R.; Schubert, U. S. Chem. Soc. Rev. 2012, 41, 176–191. doi:10.1039/c1cs15107j |
| 21. | Kuhl, A.; Hahn, M. G.; Dumić, M.; Mittendorf, J. Amino Acids 2005, 29, 89–100. doi:10.1007/s00726-005-0212-y |
| 22. | Weiner, B.; Szymański, W.; Janssen, D. B.; Minnaard, A. J.; Feringa, B. L. Chem. Soc. Rev. 2010, 39, 1656–1691. doi:10.1039/b919599h |
| 14. | Fahrenbach, A. C.; Stoddart, J. F. Chem.–Asian J. 2011, 6, 2660–2669. doi:10.1002/asia.201100457 |
| 13. | El-Sagheer, A. H.; Brown, T. Chem. Soc. Rev. 2010, 39, 1388–1405. doi:10.1039/b901971p |
| 17. | Huisgen, R.; Szeimis, G.; Möbius, L. Chem. Ber. 1967, 100, 2494–2507. doi:10.1002/cber.19671000806 |
| 33. | Nagy, K. D.; Shen, B.; Jamison, T. F.; Jensen, K. F. Org. Process Res. Dev. 2012, 16, 976–981. doi:10.1021/op200349f |
| 28. | Martinek, T. A.; Fülöp, F. Chem. Soc. Rev. 2012, 41, 687–702. doi:10.1039/c1cs15097a |
| 29. | Wegner, J.; Ceylan, S.; Kirschning, A. Adv. Synth. Catal. 2012, 354, 17–57. doi:10.1002/adsc.201100584 |
| 30. | Yoshida, J.-i.; Kim, H.; Nagaki, A. ChemSusChem 2011, 4, 331–340. doi:10.1002/cssc.201000271 |
| 31. | Geyer, K.; Gustafsson, T.; Seeberger, P. H. Synlett 2009, 2382–2391. doi:10.1055/s-0029-1217828 |
| 32. | Mak, X. Y.; Laurino, P.; Seeberger, P. H. Beilstein J. Org. Chem. 2009, 5, No. 19. doi:10.3762/bjoc.5.19 |
| 46. | Smith, C. J.; Nikbin, N.; Ley, S. V.; Lange, H.; Baxendale, I. R. Org. Biomol. Chem. 2011, 9, 1938–1947. doi:10.1039/c0ob00815j |
| 47. | Baumann, M.; Baxendale, I. R.; Ley, S. V.; Smith, C. D.; Tranmer, G. K. Org. Lett. 2006, 8, 5231–5234. doi:10.1021/ol061975c |
| 43. | Bryan, M. C.; Wernick, D.; Hein, C. D.; Petersen, J. V.; Eschelbach, J. W.; Doherty, E. M. Beilstein J. Org. Chem. 2011, 7, 1141–1149. doi:10.3762/bjoc.7.132 |
| 44. | He, P.; Haswell, S. J.; Fletcher, P. D. I.; Kelly, S. M.; Mansfield, A. Beilstein J. Org. Chem. 2011, 7, 1150–1157. doi:10.3762/bjoc.7.133 |
| 45. | Kockmann, N.; Gottsponer, M.; Roberge, D. M. Chem. Eng. J. 2011, 167, 718–726. doi:10.1016/j.cej.2010.08.089 |
| 38. | Ueno, M.; Suzuki, T.; Naito, T.; Oyamada, H.; Kobayashi, S. Chem. Commun. 2008, 1647–1649. doi:10.1039/b715259k |
| 39. | Rasheed, M.; Wirth, T. Angew. Chem., Int. Ed. 2011, 50, 357–358. doi:10.1002/anie.201006107 |
| 40. | Rueping, M.; Bootwicha, T.; Sugiono, E. Beilstein J. Org. Chem. 2012, 8, 300–307. doi:10.3762/bjoc.8.32 |
| 41. | Ötvös, S. B.; Mándity, I. M.; Fülöp, F. Mol. Diversity 2011, 15, 605–611. doi:10.1007/s11030-010-9276-z |
| 42. | Brandt, J. C.; Wirth, T. Beilstein J. Org. Chem. 2009, 5, No. 30. doi:10.3762/bjoc.5.30 |
| 34. | Hartman, R. L.; McMullen, J. P.; Jensen, K. F. Angew. Chem., Int. Ed. 2011, 50, 7502–7519. doi:10.1002/anie.201004637 |
| 35. | Ötvös, S. B.; Mándity, I. M.; Fülöp, F. ChemSusChem 2012, 5, 266–269. doi:10.1002/cssc.201100332 |
| 36. | Ötvös, S. B.; Mándity, I. M.; Fülöp, F. J. Catal. 2012, 295, 179–185. doi:10.1016/j.jcat.2012.08.006 |
| 37. | Yoshida, J.-i.; Nagaki, A.; Yamada, T. Chem.–Eur. J. 2008, 14, 7450–7459. doi:10.1002/chem.200800582 |
© 2013 Ötvös et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)