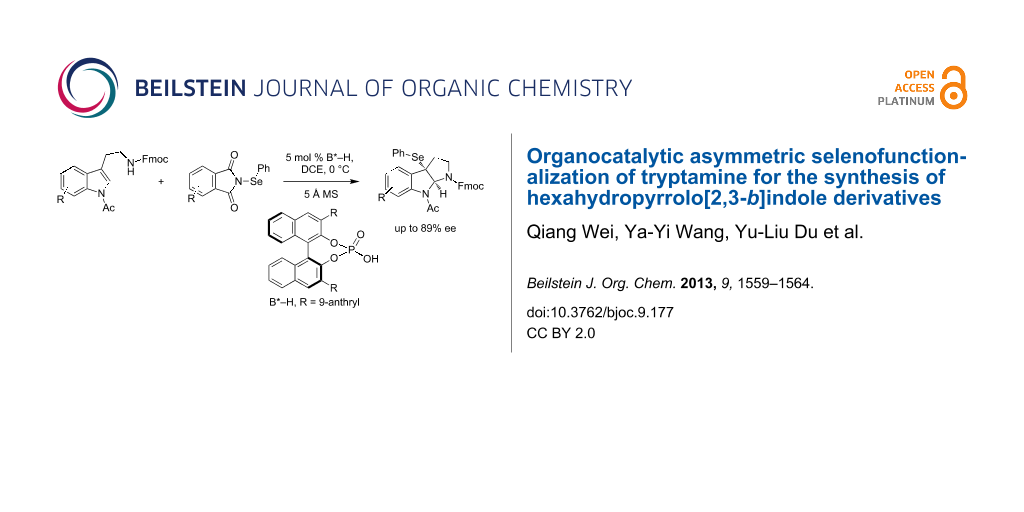
Abstract
A chiral phosphoric acid-catalyzed selenofunctionalization of tryptamine derivatives provides access to 3a-(phenylselenyl)-1,2,3,3a,8,8a-hexahydropyrrolo[2,3-b]indole derivatives in high yields and with synthetically useful levels of enantioselectivity (up to 89% ee).

Graphical Abstract
Introduction
Selenofunctionalization of carbon–carbon double bonds provides practicable opportunities for rapid construction of molecule complexity [1-8], because the versatile carbon–selenium bond could either stabilize carbanions [9,10], serve as a radical precursor [11-13], or undergo a syn-selective oxidative elimination via the selenoxide [14,15]. A widely accepted mechanism suggests that a key discrete seleniranium ion intermediate is initially formed, and then trapped by internal amine through nucleophilic attack to furnish the product. So far, in addition to the chiral-substrate-induced strategy [16], chiral selenylating agents [17-24] are commonly designed for asymmetric selenofunctionalization of carbon–carbon double bonds. In 2010, Denmark and co-workers reported a Lewis base catalyzed asymmetric selenoetherification of olefins, whereas the enantioselectivity was not quite synthetically attractive (up to 70% ee) [25]. As chiral 3-substituted hexahydropyrroloindoline is a key structural moiety prevalent in a large number of bioactive indole alkaloids [26,27], direct access to which by selenofunctionalization has been considered to be promising but challenging. Danishefsky found that the treatment of bis(Cbz)tryptamine with N-phenylselenophthalimide (N-PSP) in the presence of a catalytic amount of p-toluenesulfonic acid (PTSA) was able to afford a racemic selenofunctionalization product in 84% yield [28]. These leading findings indicate that either Lewis base or Brønsted acid shows catalytic activity for the selenofunctionalization reaction. Since chiral phosphoric acids have been shown to be Brønsted acid/Lewis base bifunctional organocatalysts [29-33], we ask whether the chiral BINOL-based phosphoric acids are able to catalyze the selenofunctionalization of tryptamine derivatives.
Results and Discussion
Initially, we investigated a reaction of bis(Cbz)tryptamine reagent 1a with N-phenylselenophthalimide (N-PSP) (2a) by using phosphoric acids 3 (Table 1, Figure 1) as catalysts for the validation of our hypothesis. Encouragingly, the reaction proceeded smoothly in the presence of 10 mol % of the phosphoric acids evaluated under the assistance of 5 Å molecular sieves. Apparently, the stereoselectivity depended on the N-protecting group of tryptamine 1. When nitrogen atoms of the tryptamine were both protected with Cbz, a very poor enantioselectivity was observed regardless of the catalysts used (Table 1, entries 1–3). Notably, only the substrate bearing an electron-withdrawing N-protecting group at the indole nitrogen (R2) underwent a smooth reaction to afford the desired product. When the R2 was replaced with a methyl group, the N-PSP directly underwent a coupling reaction with tryptamine derivatives 1 at the 2-position in 76% yield [34], indicating that the electronically rich substitution inhibited the desired selenofunctionalization reaction. Other selenofunctionalization reagents, such as 2b and 2c could also participate in the reaction under similar conditions, but both showed lower reactivity than 2a. Interestingly, the selenofunctionalization reagent 2d, which was the best substrate in the reaction developed by Denmark [25], however, was completely unreactive in our case. After the optimal R2 protecting group and the phenylseleno reagent were determined, we focused on the evaluation of the N-protecting group of the tryptamine (R1) to improve the enantioselectivity. The Fmoc group was found to be better than any other substituents that were screened (Table 1, entries 10 and 11 versus 12). The optimization of reaction parameters including solvents and temperature found dichloroethane (DCE) to be the best solvent in terms of enantioselectivity, and the best results could be accessed by conducting at 0 °C (Table 1, entries 13 and 14).
Table 1: Optimization of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-177-i3.svg?max-width=637&scale=1.0)
|
||||||
| entry | 3 | R1 | R2 | 2 | yield (%)b | ee (%)c |
|---|---|---|---|---|---|---|
| 1 | 3a | Cbz | Cbz | 2a | 56 | 10 |
| 2 | 3b | Cbz | Cbz | 2a | 46 | 6 |
| 3 | 3c | Cbz | Cbz | 2a | 36 | 0 |
| 4 | 3a | Cbz | Boc | 2a | 49 | 6 |
| 5 | 3a | Cbz | Me | 2a | – | – |
| 6 | 3a | Cbz | Ac | 2a | 67 | 24 |
| 7 | 3a | Cbz | Ac | 2b | 63 | 25 |
| 8 | 3a | Cbz | Ac | 2c | 37 | 5 |
| 9 | 3b | Cbz | Ac | 2d | – | – |
| 10 | 3b | Cbz | Ac | 2a | 65 | 48 |
| 11 | 3b | CO2Et | Ac | 2a | 61 | 27 |
| 12 | 3b | Fmoc | Ac | 2a | 60 | 63 |
| 13d | 3b | Fmoc | Ac | 2a | 75 | 77 |
| 14d,e | 3b | Fmoc | Ac | 2a | 78 | 86 |
aThe reaction was performed in 0.1 mmol scale in DCM (1 mL) with 5 Å MS (100 mg). bIsolated yield. cThe ee was determined by HPLC. dIn DCE. eThe temperature was 0 °C.
![[1860-5397-9-177-1]](/bjoc/content/figures/1860-5397-9-177-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Catalysts and seleno reagents evaluated in this study.
Figure 1: Catalysts and seleno reagents evaluated in this study.
After optimizing the reaction conditions, a variety of tryptamine analogues were synthesized for this chiral phosphoric acid-catalyzed asymmetric selenofunctionalization. As shown in Figure 2, no matter what the chemical and electronic feature of the substituents on the benzene moiety of either substrates or N-PSP, various tryptamine analogues could be smoothly transformed into the corresponding products in satisfactory yields (65–85%) and with good enantioselectivities (71–89%; Figure 2, 4b–4i). In addition, the products were solid and easy to recrystallize to enhance the optical purity. After a single recrystallization from methanol, the optical purity of some products, such as 4e, was enhanced to >99% ee. Importantly, the configuration could be assigned by X-ray crystallography. The crystal structure of 4a (>99%) indicated that the configuration of the stereogenic centers was assigned to be (3aR,8aS) (Figure 3) [35].
![[1860-5397-9-177-2]](/bjoc/content/figures/1860-5397-9-177-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Generality for substitution at the indoline moiety. The reaction was performed in 0.1 mmol scale in DCE (1 mL) with 5 Å MS (100 mg) in 0 °C and the ratio of 2/1 is 3:1. The reaction was performed for 1–3 days. The given yields are isolated yields, and the ee’s were determined by HPLC.
Figure 2: Generality for substitution at the indoline moiety. The reaction was performed in 0.1 mmol scale in...
![[1860-5397-9-177-3]](/bjoc/content/figures/1860-5397-9-177-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: X-ray crystallography of 4a catalyzed by (S)-3b.
Figure 3: X-ray crystallography of 4a catalyzed by (S)-3b.
On the basis of experimental observations, we proposed a reaction mechanism (Scheme 1). The phosphoric acid acts as a bifunctional catalyst and simultaneously activates both the tryptamine derivative and N-PSP by hydrogen-bonding interaction. Then, the asymmetric selenofunctionalization occurred at the 3-substituted tryptamine and subsequently the proton of the phosphoric acid protonates the phthalimide anion to release phthalimide. Finally, the amide on the side chain of the tryptamine derivatives attacks the resultant iminium cation leading to the formation of the product in an enantioselective manner.
![[1860-5397-9-177-i1]](/bjoc/content/inline/1860-5397-9-177-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The plausible reaction mechanism.
Scheme 1: The plausible reaction mechanism.
Finally, we demonstrate the synthetic application of this reaction in the construction of the 3a-(phenylselenyl)bispyrrolidino[2,3-b]indoline core structure (Scheme 2). Under the optimized reaction conditions, the enantioselective substitution reaction gave 4a in 78% yield and 86% ee. However, when the reaction was scaled up to 20 mmol, both the yield and the stereoselectivity were significantly sacrificed. To our delight, the reaction running on a similar scale could give 4a in 80% yield and 82% ee by tuning the stoichiometry of the phenylseleno reagent (2a) to 1.5 equiv by using 5 mol % of catalyst (R)-3b. After a single recrystallization from methanol, the product 4a was obtained in 50% yield and with 97% ee. The oxidative deselenation of 4a with MCPBA afforded the corresponding alcohol 5 in 95% yield. The stereochemistry of the alcohol was found to be identical to that of the parent selenide, as demonstrated previously [36].
![[1860-5397-9-177-i2]](/bjoc/content/inline/1860-5397-9-177-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scale up of the protocol and synthetic application.
Scheme 2: Scale up of the protocol and synthetic application.
Conclusion
In summary, we have developed a reaction for the enantioselective selenofunctionalization of tryptamine derivatives with N-phenylselenophthalimide (N-PSP) catalyzed by chiral phosphoric acids (up to 89% ee). In this context, we used this protocol to prepare the key chiral precursor of the (+)-alline.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data for new compounds. In addition, confirmatory crystallographic data are included. | ||
| Format: PDF | Size: 1.3 MB | Download |
References
-
Nicolaou, K. C.; Petasis, N. A. Selenium in Natural Product Synthesis; CIS: Philadelphia, PA, U.S.A., 1984.
Return to citation in text: [1] -
Paulmier, C. Selenium Reagents and Intermediates in Organic Synthesis; Pergamon Press: Oxford, 1986.
Return to citation in text: [1] -
Liotta, D., Ed. Organoselenium Chemistry; Wiley & Sons: New York, NY, USA, 1987; pp 1–163.
Return to citation in text: [1] -
Back, T. G. Preparative uses of organoselenium and organotellurium compounds. In The Chemistry of Organic Selenium and Tellurium Compounds; Patai, S., Ed.; Wiley & Sons: New York, NY, USA, 1987; Vol. 2, pp 91–215.
Return to citation in text: [1] -
Back, T. G., Ed. Organoselenium Chemistry A Practical Approach; Oxford University Press: Oxford, 1999.
Return to citation in text: [1] -
Tiecco, M. Electrophilic Selenium, Selenocyclizations. In Organoselenium Chemistry; Wirth, T., Ed.; Topics in Current Chemistry, Vol. 208; Springer Verlag: Heidelberg, 2000; pp 7–54. doi:10.1007/3-540-48171-0_2
Return to citation in text: [1] -
Petragnani, N.; Stefani, H. A.; Valduga, C. J. Tetrahedron 2001, 57, 1411–1448. doi:10.1016/S0040-4020(00)01033-4
Return to citation in text: [1] -
Ranganathan, S.; Muraleedharan, K. M.; Vaish, N. K.; Jayaraman, N. Tetrahedron 2004, 60, 5273–5308. doi:10.1016/j.tet.2004.04.014
Return to citation in text: [1] -
Petragnani, N.; Ferraz, H. M. C. Synthesis 1978, 476–478. doi:10.1055/s-1978-24790
Return to citation in text: [1] -
Reich, H. J. Acc. Chem. Res. 1979, 12, 22–30. doi:10.1021/ar50133a004
Return to citation in text: [1] -
Clive, D. L. J.; Chittattu, G. J.; Farina, V.; Kiel, W. A.; Menchen, S. M.; Russell, C. G.; Singh, A.; Wong, C. K.; Curtis, N. J. J. Am. Chem. Soc. 1980, 102, 4438–4447. doi:10.1021/ja00533a024
Return to citation in text: [1] -
Kraus, G. A.; Taschner, M. J. J. Org. Chem. 1980, 45, 1175–1176. doi:10.1021/jo01294a058
Return to citation in text: [1] -
Bachi, M. D.; Hoornaert, C. Tetrahedron Lett. 1981, 22, 2693–2694. doi:10.1016/S0040-4039(01)92972-7
Return to citation in text: [1] -
Jones, D. N.; Mundy, D.; Whitehouse, R. D. J. Chem. Soc. D 1970, 86–87. doi:10.1039/C29700000086
Return to citation in text: [1] -
Reich, H. J.; Wollowitz, S.; Trend, J. E.; Chow, F.; Wendelborn, D. F. J. Org. Chem. 1978, 43, 1697–1705. doi:10.1021/jo00403a016
Return to citation in text: [1] -
Rodriguez-Escrich, C.; Olivella, A.; Urpí, F.; Vilarrasa, J. Org. Lett. 2007, 9, 989–992. doi:10.1021/ol063035y
Return to citation in text: [1] -
Deziel, R.; Goulet, S.; Grenier, L.; Bordeleau, J.; Bernier, J. J. Org. Chem. 1993, 58, 3619–3621. doi:10.1021/jo00066a010
Return to citation in text: [1] -
Deziel, R.; Malenfant, E. J. Org. Chem. 1995, 60, 4660–4662. doi:10.1021/jo00119a055
Return to citation in text: [1] -
Déziel, R.; Malenfant, E.; Thibault, C.; Fréchette, S.; Gravel, M. Tetrahedron Lett. 1997, 38, 4753–4756. doi:10.1016/S0040-4039(97)01030-7
Return to citation in text: [1] -
Fragale, G.; Wirth, T.; Neuberger, M. Chem. Commun. 1998, 1867–1868. doi:10.1039/a804264k
Return to citation in text: [1] -
Wirth, T. Tetrahedron 1999, 55, 1–28. doi:10.1016/S0040-4020(98)00946-6
Return to citation in text: [1] -
Wirth, T. Angew. Chem., Int. Ed. 2000, 39, 3740–3749. doi:10.1002/1521-3773(20001103)39:21<3740::AID-ANIE3740>3.0.CO;2-N
Return to citation in text: [1] -
Tiecco, M.; Testaferri, L.; Marini, F.; Bagnoli, L.; Santi, C.; Temperini, A.; Sternativo, S.; Tomassini, C. Phosphorus, Sulfur Silicon Relat. Elem. 2005, 180, 729–740. doi:10.1080/10426500590907462
Return to citation in text: [1] -
Browne, D. M.; Wirth, T. Curr. Org. Chem. 2006, 10, 1893–1903. doi:10.2174/138527206778521213
Return to citation in text: [1] -
Denmark, S. E.; Kalyani, D.; Collins, W. R. J. Am. Chem. Soc. 2010, 132, 15752–15765. doi:10.1021/ja106837b
Return to citation in text: [1] [2] -
Hino, T.; Nakagawa, M. Alkaloids 1998, 34, 1–75.
Return to citation in text: [1] -
Anthoni, U.; Christophersen, C.; Nielsen, P. H. Naturally occurring cyclotryptophans and cyclotryptamines. In Alkaloids: Chemical and Biological Perspectives; Pelletier, S. W., Ed.; Pergamon Press: New York, NY, USA, 1999; Vol. 13, pp 163–236. doi:10.1016/S0735-8210(99)80025-9
Return to citation in text: [1] -
Depew, K. M.; Marsden, S. P.; Zatorska, D.; Zatorski, A.; Bornmann, W. G.; Danishefsky, S. J. J. Am. Chem. Soc. 1999, 121, 11953–11963. doi:10.1021/ja991558d
Return to citation in text: [1] -
Akiyama, T. Chem. Rev. 2007, 107, 5744–5748. doi:10.1021/cr068374j
Return to citation in text: [1] -
Terada, M. Chem. Commun. 2008, 4097–4112. doi:10.1039/b807577h
Return to citation in text: [1] -
Terada, M. Synthesis 2010, 1929–1982. doi:10.1055/s-0029-1218801
Return to citation in text: [1] -
Yu, J.; Shi, F.; Gong, L.-Z. Acc. Chem. Res. 2011, 44, 1156–1171. doi:10.1021/ar2000343
Return to citation in text: [1] -
Huang, D.; Wang, H.; Xue, F.; Guan, H.; Li, L.; Peng, X.; Shi, Y. Org. Lett. 2011, 13, 6350–6353. doi:10.1021/ol202527g
Return to citation in text: [1] -
Zhao, X.; Yu, Z.-K.; Xu, T.-Y.; Wu, P.; Yu, H.-F. Org. Lett. 2007, 9, 5263–5266. doi:10.1021/ol7024795
Return to citation in text: [1] -
CCDC: 844280.
Return to citation in text: [1] -
Hewitt, P. R.; Cleator, E.; Ley, S. V. Org. Biomol. Chem. 2004, 2, 2415–2417. doi:10.1039/b410180d
Return to citation in text: [1]
| 1. | Nicolaou, K. C.; Petasis, N. A. Selenium in Natural Product Synthesis; CIS: Philadelphia, PA, U.S.A., 1984. |
| 2. | Paulmier, C. Selenium Reagents and Intermediates in Organic Synthesis; Pergamon Press: Oxford, 1986. |
| 3. | Liotta, D., Ed. Organoselenium Chemistry; Wiley & Sons: New York, NY, USA, 1987; pp 1–163. |
| 4. | Back, T. G. Preparative uses of organoselenium and organotellurium compounds. In The Chemistry of Organic Selenium and Tellurium Compounds; Patai, S., Ed.; Wiley & Sons: New York, NY, USA, 1987; Vol. 2, pp 91–215. |
| 5. | Back, T. G., Ed. Organoselenium Chemistry A Practical Approach; Oxford University Press: Oxford, 1999. |
| 6. | Tiecco, M. Electrophilic Selenium, Selenocyclizations. In Organoselenium Chemistry; Wirth, T., Ed.; Topics in Current Chemistry, Vol. 208; Springer Verlag: Heidelberg, 2000; pp 7–54. doi:10.1007/3-540-48171-0_2 |
| 7. | Petragnani, N.; Stefani, H. A.; Valduga, C. J. Tetrahedron 2001, 57, 1411–1448. doi:10.1016/S0040-4020(00)01033-4 |
| 8. | Ranganathan, S.; Muraleedharan, K. M.; Vaish, N. K.; Jayaraman, N. Tetrahedron 2004, 60, 5273–5308. doi:10.1016/j.tet.2004.04.014 |
| 16. | Rodriguez-Escrich, C.; Olivella, A.; Urpí, F.; Vilarrasa, J. Org. Lett. 2007, 9, 989–992. doi:10.1021/ol063035y |
| 14. | Jones, D. N.; Mundy, D.; Whitehouse, R. D. J. Chem. Soc. D 1970, 86–87. doi:10.1039/C29700000086 |
| 15. | Reich, H. J.; Wollowitz, S.; Trend, J. E.; Chow, F.; Wendelborn, D. F. J. Org. Chem. 1978, 43, 1697–1705. doi:10.1021/jo00403a016 |
| 11. | Clive, D. L. J.; Chittattu, G. J.; Farina, V.; Kiel, W. A.; Menchen, S. M.; Russell, C. G.; Singh, A.; Wong, C. K.; Curtis, N. J. J. Am. Chem. Soc. 1980, 102, 4438–4447. doi:10.1021/ja00533a024 |
| 12. | Kraus, G. A.; Taschner, M. J. J. Org. Chem. 1980, 45, 1175–1176. doi:10.1021/jo01294a058 |
| 13. | Bachi, M. D.; Hoornaert, C. Tetrahedron Lett. 1981, 22, 2693–2694. doi:10.1016/S0040-4039(01)92972-7 |
| 9. | Petragnani, N.; Ferraz, H. M. C. Synthesis 1978, 476–478. doi:10.1055/s-1978-24790 |
| 10. | Reich, H. J. Acc. Chem. Res. 1979, 12, 22–30. doi:10.1021/ar50133a004 |
| 36. | Hewitt, P. R.; Cleator, E.; Ley, S. V. Org. Biomol. Chem. 2004, 2, 2415–2417. doi:10.1039/b410180d |
| 28. | Depew, K. M.; Marsden, S. P.; Zatorska, D.; Zatorski, A.; Bornmann, W. G.; Danishefsky, S. J. J. Am. Chem. Soc. 1999, 121, 11953–11963. doi:10.1021/ja991558d |
| 34. | Zhao, X.; Yu, Z.-K.; Xu, T.-Y.; Wu, P.; Yu, H.-F. Org. Lett. 2007, 9, 5263–5266. doi:10.1021/ol7024795 |
| 26. | Hino, T.; Nakagawa, M. Alkaloids 1998, 34, 1–75. |
| 27. | Anthoni, U.; Christophersen, C.; Nielsen, P. H. Naturally occurring cyclotryptophans and cyclotryptamines. In Alkaloids: Chemical and Biological Perspectives; Pelletier, S. W., Ed.; Pergamon Press: New York, NY, USA, 1999; Vol. 13, pp 163–236. doi:10.1016/S0735-8210(99)80025-9 |
| 25. | Denmark, S. E.; Kalyani, D.; Collins, W. R. J. Am. Chem. Soc. 2010, 132, 15752–15765. doi:10.1021/ja106837b |
| 25. | Denmark, S. E.; Kalyani, D.; Collins, W. R. J. Am. Chem. Soc. 2010, 132, 15752–15765. doi:10.1021/ja106837b |
| 17. | Deziel, R.; Goulet, S.; Grenier, L.; Bordeleau, J.; Bernier, J. J. Org. Chem. 1993, 58, 3619–3621. doi:10.1021/jo00066a010 |
| 18. | Deziel, R.; Malenfant, E. J. Org. Chem. 1995, 60, 4660–4662. doi:10.1021/jo00119a055 |
| 19. | Déziel, R.; Malenfant, E.; Thibault, C.; Fréchette, S.; Gravel, M. Tetrahedron Lett. 1997, 38, 4753–4756. doi:10.1016/S0040-4039(97)01030-7 |
| 20. | Fragale, G.; Wirth, T.; Neuberger, M. Chem. Commun. 1998, 1867–1868. doi:10.1039/a804264k |
| 21. | Wirth, T. Tetrahedron 1999, 55, 1–28. doi:10.1016/S0040-4020(98)00946-6 |
| 22. | Wirth, T. Angew. Chem., Int. Ed. 2000, 39, 3740–3749. doi:10.1002/1521-3773(20001103)39:21<3740::AID-ANIE3740>3.0.CO;2-N |
| 23. | Tiecco, M.; Testaferri, L.; Marini, F.; Bagnoli, L.; Santi, C.; Temperini, A.; Sternativo, S.; Tomassini, C. Phosphorus, Sulfur Silicon Relat. Elem. 2005, 180, 729–740. doi:10.1080/10426500590907462 |
| 24. | Browne, D. M.; Wirth, T. Curr. Org. Chem. 2006, 10, 1893–1903. doi:10.2174/138527206778521213 |
| 29. | Akiyama, T. Chem. Rev. 2007, 107, 5744–5748. doi:10.1021/cr068374j |
| 30. | Terada, M. Chem. Commun. 2008, 4097–4112. doi:10.1039/b807577h |
| 31. | Terada, M. Synthesis 2010, 1929–1982. doi:10.1055/s-0029-1218801 |
| 32. | Yu, J.; Shi, F.; Gong, L.-Z. Acc. Chem. Res. 2011, 44, 1156–1171. doi:10.1021/ar2000343 |
| 33. | Huang, D.; Wang, H.; Xue, F.; Guan, H.; Li, L.; Peng, X.; Shi, Y. Org. Lett. 2011, 13, 6350–6353. doi:10.1021/ol202527g |
© 2013 Wei et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)