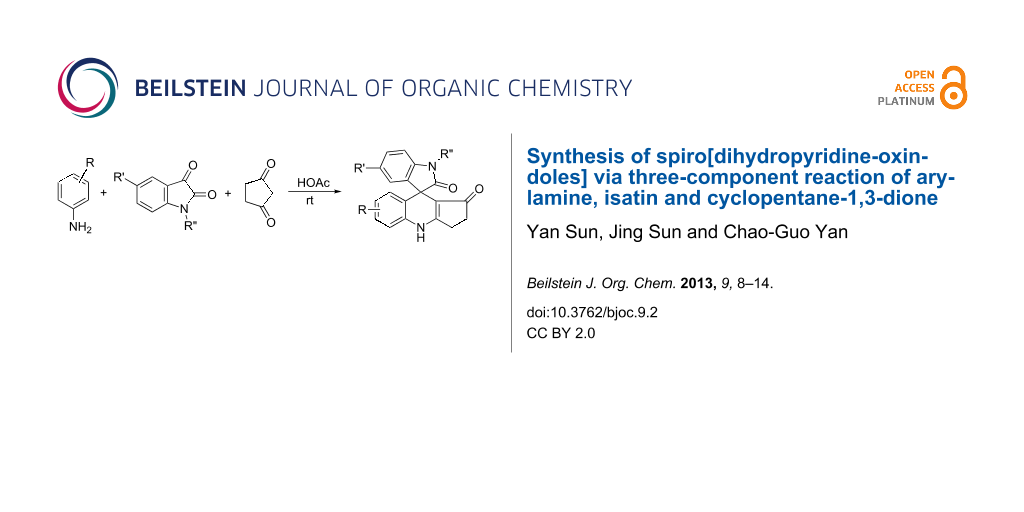
Abstract
A fast and convenient protocol for the synthesis of novel spiro[dihydropyridine-oxindole] derivatives in satisfactory yields was developed by the three-component reactions of arylamine, isatin and cyclopentane-1,3-dione in acetic acid at room temperature. On the other hand the condensation of isatin with two equivalents of cyclopentane-1,3-dione gave 3,3-bis(2-hydroxy-5-oxo-cyclopent-1-enyl)oxindole in high yields. The reaction mechanism and substrate scope of this novel reaction is briefly discussed.

Graphical Abstract
Introduction
The spirooxindole is among the most important class of naturally occurring substances, characterized by highly pronounced biological properties, and is also the core structure of many synthetic pharmaceuticals [1,2]. The various biological activities of spirooxindole derivatives have attracted much attention from organic chemists, and as a consequence, a number of methods have been reported for the preparation of spirooxindole-fused heterocycles [3-6]. Isatin and its derivatives may be the most useful starting materials or precursors in the synthesis of a wide number of spirocyclic oxindoles [7,8]. Due to its simple process, easy operation, efficiency and high atomic economy, the multicomponent reaction based on isatin and its derivatives have become an efficient method for the synthesis of various spirooxindoles in recent years [9,10]. It is known that the multicomponent reactions of isatins with in situ formed azomethine ylides have become the efficient synthetic procedure for constructing versatile spirooxindole systems [11-14]. Considering the above reports, and as part of our program aimed at developing new multicomponent reactions for the construction of complex heterocyclic compounds, we wish in this work to report the efficient synthesis of unprecedented cyclopentyl-fused spiro[dihydropyridine-oxindoles] by the three-component reaction of arylamine, isatin and cyclopentane-1,3-dione.
Results and Discussion
Recently we found that the four-component reactions of arylamine, acetylenedicarboxylate, isatin and dimedone in acetic acid resulted in the novel functionalized tetrahydrospiro[indoline-3,2’-quinoline] derivatives in moderate yields (Scheme 1a) [15]. In order to explore the generality of this four-component reaction, the reactivity of the other cyclic 1,3-diketones was also investigated. In an exploratory experiment, the four-component reaction of p-methoxyaniline, dimethyl acetylenedicarboxylate, 1-benzyl-5-methylisatin and cyclopentane-1,3-dione in acetic acid was carried out at room temperature. After workup, we were a little surprised to find that there was no unit of acetylenedicarboxylate in the obtained product (Scheme 1b). In another experiment, the three-component reaction of isatin, cyclopentane-1,3-dione and β-enamino ester, which was prepared in situ from the addition of p-methoxyaniline to dimethyl acetylenedicarboxylate in acetic acid according to our previously established procedure [15], also gave this new kind of spiro product in nearly the same yield. This result clearly indicated that the three-component reaction of arylamine, isatin and clopentane-1,3-dione gave the final spiro product, while the acetylenedicarboxylate could not take part in the reaction.
![[1860-5397-9-2-i1]](/bjoc/content/inline/1860-5397-9-2-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The four-component reactions containing dimedone (a) and cyclopentane-1,3-dione (b).
Scheme 1: The four-component reactions containing dimedone (a) and cyclopentane-1,3-dione (b).
Attention was therefore turned to evaluate the generality of the three-component reaction of arylamine, isatin and cyclopentane-1,3-dione. Under similar conditions various arylamines and isatins with different substituents reacted with cyclopentane-1,3-dione in acetic acid at room temperature for 8–10 hours to afford the corresponding spiro[dihydropyridine-oxindole] compounds 1a–1k in good yields. The results are shown in Table 1. From these results we could see that only anilines with electron-donating alkyl and alkoxy groups reacted smoothly. When anilines with electron-withdrawing p-chloro, p-bromo, m-nitro, or p-nitro groups were used in this three-component reaction, no expected spiro[dihydropyridine-oxindole] could be separated from the reaction system. On the other hand, the reaction of α-naphthylamine also gave good yields of spiro compound 1l (Table 1, entry 12).
Table 1: Synthesis of spiro[dihydropyridine-oxindole] from three-component reactions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-2-i4.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Compound | R | R’ | R” | Yield (%) |
|---|---|---|---|---|---|
| 1 | 1a | p-CH3O | CH3 | CH2Ph | 88 |
| 2 | 1b | p-CH3 | CH3 | CH2Ph | 85 |
| 3 | 1c | p-CH3CH2O | CH3 | CH2Ph | 60 |
| 4 | 1d | p-(CH3)2CH | CH3 | CH2Ph | 60 |
| 5 | 1e | p-(CH3)3C | CH3 | CH2Ph | 65 |
| 6 | 1f | p-CH3 | H | CH2Ph | 74 |
| 7 | 1g | p-CH3O | Cl | n-C4H9 | 52 |
| 8 | 1h | p-CH3 | CH3 | n-C4H9 | 82 |
| 9 | 1i | p-CH3 | Cl | n-C4H9 | 56 |
| 10 | 1j | p-CH3 | F | n-C4H9 | 58 |
| 11 | 1k | p-OH | CH3 | n-C4H9 | 79 |
| 12 | 1l | α-naphthyl | CH3 | CH2Ph | 67 |
The structures of spiro compounds 1a–1l were fully characterized by 1H and 13C NMR, HRMS, and IR spectra and were further confirmed by a single-crystal X-ray diffraction study performed for the compound 1b (Figure 1).
![[1860-5397-9-2-1]](/bjoc/content/figures/1860-5397-9-2-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Molecular structure of spiro[dihydropyridine-oxindole] 1b.
Figure 1: Molecular structure of spiro[dihydropyridine-oxindole] 1b.
It should be pointed out that the structure of the obtained spiro compounds is very interesting, in which the oxindole was connected to the ortho-position of the amino group of arylamine. It is well known that the one-pot reactions of arylamine, isatin and cyclic 1,3-ketone under different catalytic conditions usually gave a kind of spiro[pyridine-oxindole] (I in Figure 2) as the main product, in which the arylamine only provided the amino group to form the pyridyl ring [16-21]. There are only very few papers describing that either 2-naphthylamine [22-25], functionalized 5-aminopyrazoles [26-28], or 2-aminobenzothiazoles [29] reacted with isatin and cyclic 1,3-dicarbonyl compounds to give the similar spiro[dihydropyridine-oxindole] (II, III in Figure 2), in which both the amino group and the aryl ring were involved in the construction of the pyridyl ring. In these cases only some special amines such as naphthylamine or heterocyclic amines were employed. It is well known that the reactivity at the α-position of 2-naphthylamine and the heterocyclic amine is much higher than that at the ortho-position of aniline. To the best of our knowledge, this new reaction provided the first example of normally substituted aniline showing this kind of reaction pattern.
![[1860-5397-9-2-2]](/bjoc/content/figures/1860-5397-9-2-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The two kinds of spiro compounds from reactions of isatins with arylamines and cyclic 1,3-diketones.
Figure 2: The two kinds of spiro compounds from reactions of isatins with arylamines and cyclic 1,3-diketones....
Encouraged by this success, we extended this three-component reaction to other isatins. When isatins without N-substituent were used under similar conditions, the reaction successfully resulted in the expected spiro[dihydropyridine-oxindole] 2a–2g in lower yields (Table 2) and byproducts 3a–3d, which obviously came from the condensation of isatins with two molar cyclopentane-1,3-diones. It is mentioned in Table 1 that isatins with an N-substituent afforded solely spiro[dihydropyridine-oxindole] 1a–1l in satisfactory yields. Trying to increase the yields of spiro compounds and decrease the yields of condensation products was not successful. Both the spiro compounds 2a–2g and condensation products 3a–3d have low solubility in common organic solvents, such as chloroform, ethanol and THF, and could be partially dissolved in DMF. The structures of them were successfully established by spectroscopic methods and single crystal determination of compound 2f (Figure 3).
Table 2: Synthesis of spiro[dihydropyridine-oxindole] from three-component reactions.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-2-i5.svg?max-width=637&scale=1.0)
|
||||||
| Entry | R | R’ | Compound 2 | Yield (%) | Compound 3 | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | p-CH3O | H | 2a | 35 | 3a | 20 |
| 2 | p-CH3O | CH3 | 2b | 36 | 3b | 18 |
| 3 | p-CH3O | Cl | 2c | 35 | 3c | 16 |
| 4 | p-CH3O | F | 2d | 30 | 3d | 23 |
| 5 | p-CH3 | H | 2e | 26 | 3a | 25 |
| 6 | p-CH3 | CH3 | 2f | 35 | 3b | 15 |
| 7 | p-CH3 | Cl | 2g | 32 | 3c | 20 |
![[1860-5397-9-2-3]](/bjoc/content/figures/1860-5397-9-2-3.svg?scale=1.0&max-width=1024&background=FFFFFF)
Figure 3: Molecular structure of spiro[dihydropyridine-oxindole] 2f.
Figure 3: Molecular structure of spiro[dihydropyridine-oxindole] 2f.
A literature survey showed that even though there are a lot of reports about the condensation of isatins with cyclic 1,3-dicarbonyl compounds [30-33], the condensation reaction of isatin with cyclopentane-1,3-dione seemed still not to have been investigated and the 3,3-bis(2-hydroxy-5-oxo-cyclopent-1-enyl)oxindoles 3a–3d have not been prepared until now. Thus, the direct condensation of isatins with two molar cyclopentane-1,3-dione was carried out in acetic acid and the desired condensation products 3a–3d were obtained in high yields (Scheme 2). Then full characterization data were provided for them, and the single-crystal structure of compound 3d was determined (Figure 4). 1H NMR spectra of 3,3-bis(2-hydroxy-5-oxo-cyclopent-1-enyl)oxindoles 3a–3d showed a broad signal of two hydroxy groups at about 11.55 ppm, which clearly indicates cyclopentane-1,3-dione units exist in enol form. In the crystal structure of 3d it is clearly seen that there is one carbonyl group and one enol group in each cyclopentane-1,3-dione moiety.
![[1860-5397-9-2-i2]](/bjoc/content/inline/1860-5397-9-2-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Condensation reactions of isatins with cyclopentane-1,3-dione.
Scheme 2: Condensation reactions of isatins with cyclopentane-1,3-dione.
![[1860-5397-9-2-4]](/bjoc/content/figures/1860-5397-9-2-4.svg?scale=1.0&max-width=1024&background=FFFFFF)
Figure 4: Molecular structure of compound 3d.
Figure 4: Molecular structure of compound 3d.
Although at present the exact mechanism of this three-component reaction is not very clear, a plausible reaction mechanism for the formation of spiro[dihydropyridine-oxindoles] is presented based on the similar multicomponent reactions of isatins [22-29]. Firstly, the reaction of isatin with one equivalent of cyclopentane-1,3-dione in acetic acid forms an aldol adduct (A in Scheme 3). Secondly, a carbonium ion intermediate B was formed by acidic dehydration of adduct A. Then further reaction of carbonium ion B with a second equivalent of cyclopentane-1,3-dione gave the double condensation product 3. On the other hand the carbonyl ion B reacted with the arylamine to give intermediate C. Then the intramolecular dehydration of C resulted in the final spiro compound 1 or 2. In this reaction process the reactivity of arylamine played an important factor. The inactive arylamine bearing electron-withdrawing groups could not react with carbonium ion B, thus could not give the expected spiro compound.
![[1860-5397-9-2-i3]](/bjoc/content/inline/1860-5397-9-2-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed reaction mechanism for the three-component reaction.
Scheme 3: Proposed reaction mechanism for the three-component reaction.
Conclusion
In conclusion, we have described a one-pot three-component reaction of arylamine, isatin and cyclopentane-1,3-dione and found an efficient procedure for the synthesis of a new type of polysubstituted spiro[dihydropyridine-oxindoles]. The reaction mechanism and substrate scope of this novel reaction were briefly discussed. Prominent among the advantages of this new method are operational simplicity, good yields of products in short reaction times, and easy workup procedures. Further expansion of the reaction scope and synthetic applications of this methodology are in progress in our laboratory.
Experimental
Reagents and apparatus: All reagents and solvents were commercially available with analytical grade and used as received. Evaporative removal of organic solvents was carried out with a rotary evaporator in conjunction with a water jet pump. Melting points were taken on a hot-plate microscope apparatus and were uncorrected. 1H and 13C NMR spectra were recorded on a Bruker AV-600 instrument. IR spectra were obtained on a Bruker Tensor27 spectrometer (KBr disc). HRMS were measured on an AB 5800 MALDI–TOF/TOF instrument. X-ray data were collected on a Bruker Smart APEX-2 diffractometer.
Typical procedure for the synthesis of spiro[dihydropyridine-oxindoles] 1a–1l from the three-component reaction of arylamine, cyclopentane-1,3-dione and isatin: A mixture of arylamine (2.0 mmol), isatin (2.0 mmol) and cyclopentane-1,3-dione (2.0 mmol, 0.196 g) in 10.0 mL acetic acid was stirred at room temperature for about 9–12 hours. The resulting precipitates were collected by filtration and washed with cold ethanol to give pure product for analysis. 1a: white solid, 88%; mp >300 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.29 (s, 1H, NH), 7.51 (d, J = 7.8 Hz, 2H, ArH), 7.33 (t, J = 7.8 Hz, 2H, ArH), 7.27 (d, J = 7.2 Hz, 1H, ArH), 7.00 (d, J = 8.4 Hz, 1H, ArH), 6.94 (d, J = 7.8 Hz, 1H, ArH), 6.84–6.82 (m, 1H, ArH), 6.75 (d, J = 7.8 Hz, 1H, ArH), 6.70 (brs, 1H, ArH), 5.90 (d, J = 8.4 Hz, 1H, ArH), 4.99–4.91 (m, 2H, CH2), 3.47 (s, 3H, OCH3), 2.78–2.77 (m, 2H, CH2), 2.26–2.25 (m, 2H, CH2), 2.13 (s, 3H, CH3); 13C NMR (150 MHz, DMSO-d6) δ 198.3, 177.6, 165.9, 155.4, 139.5, 137.1, 136.6, 131.6, 130.3, 128.4, 128.1, 127.3, 127.2, 124.5, 124.4, 117.6, 113.9, 112.5, 108.6, 108.0, 56.0, 55.1, 50.5, 43.1, 32.8, 24.3, 20.5, 18.5; IR (KBr) ν: 3237, 3112, 3060, 2917, 1686, 1603, 1540, 1494, 1378, 1336, 1288, 1229, 1161, 1114, 1037, 859, 818 cm−1; HRMS–ESI (m/z): [M − H]− calcd. for C28H23N2O3: 435.1714; found, 435.1711.
Typical procedure for the synthesis of spiro[dihydropyridine-oxindoles] 2a–2g from three-component reaction of arylamine, cyclopentane-1,3-dione and isatin: A mixture of arylamine (2.0 mmol), isatin (2.0 mmol) and cyclopentane-1,3-dione (2.0 mmol, 0.196 g) in 10.0 mL acetic acid was stirred at room temperature for about 9–12 hours. The resulting precipitates were collected by filtration and washed with cold ethanol to give the product. Recrystallization in DMF resulted in pure product for analysis.
2a: white solid, 35%; mp >300 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.41 (s, 1H, NH), 10.21 (s, 1H, NH), 7.15–7.13 (m, 1H, ArH), 6.97 (d, J = 8.4 Hz, 1H, ArH), 6.87 (d, J = 7.8 Hz, 1H, ArH), 6.84–6.82 (m, 2H, ArH), 6.80 (d, J = 6.6 Hz, 1H, ArH), 6.61 (s, 1H, ArH), 5.99 (d, J = 3.0 Hz, 1H, ArH), 3.54 (s, 3H, OCH3), 2.74–2.72 (m, 2H, CH2), 2.22–2.20 (m, 2H, CH2); 13C NMR (150 MHz, DMSO-d6) δ 198.2, 179.2, 165.7, 155.3, 141.4, 137.9, 130.3, 127.7, 124.6, 123.9, 121.8, 117.4, 113.3, 112.9, 109.2, 108.1, 55.2, 50.9, 32.8, 24.2; IR (KBr) ν: 3203, 3108, 2966, 2925, 2833, 1703, 1661, 1590, 1538, 1494, 1388, 1330, 1288, 1251, 1194, 1162, 1124, 1039, 911, 857, 805 cm−1; HRMS–ESI (m/z): [M − H]− calcd. for C20H15N2O3: 331.1088; found, 331.1088.
Typical procedure for the synthesis of condensation products 3a–3c from condensation reaction of cyclopentane-1,3-dione and isatin: A mixture of isatin (2.0 mmol) and cyclopentane-1,3-dione (2.0 mmol, 0.196 g) in 10.0 mL acetic acid was stirred at room temperature overnight. Then, a second portion of cyclopentane-1,3-dione (2.0 mmol, 0.196 g) was added to the system, and the solution was stirred at room temperature for 12 hours. The resulting precipitates were collected by filtration and washed with cold ethanol to give the pure product for analysis.
3a: white solid, 76%; mp 239–241 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.57 (brs, 2H, OH), 10.42 (s, 1H, NH), 7.23 (d, J = 6.6 Hz, 1H, ArH), 7.02 (brs, 1H, ArH), 6.75 (brs, 1H, ArH), 6.68 (d, J = 7.2 Hz, 1H, ArH), 2.28 (brs, 8H, CH2); 13C NMR (150 MHz, DMSO-d6) δ 179.0, 141.6, 132.7, 132.6, 126.8, 126.7, 125.7, 120.6, 112.4, 108.5, 43.3, 30.4, 30.3, 30.2, 30.1, 30.0, 29.9, 29.8, 29.7; IR (KBr) ν: 3196, 2925, 1676, 1612, 1573, 1478, 1435, 1385, 1362, 1306, 1232, 1110, 1069, 1040, 881 cm−1; HRMS–ESI (m/z): [M − H]− calcd. for C18H14NO5: 324.0877; found, 324.0874.
Crystallographic data (CIF) of all new compounds are available free of charge via the Internet. Single crystal data for compound 1b (CCDC 869516), 2f (CCDC 869519) and 3d (CCDC 881763) have been deposited in the Cambridge Crystallographic Data Centre. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk./data_request/cif
Supporting Information
| Supporting Information File 1: Spectroscopic and analytical data. | ||
| Format: PDF | Size: 1.3 MB | Download |
References
-
Tan, B.; Hernández-Torres, G.; Barbas, C. F., III. J. Am. Chem. Soc. 2011, 133, 12354–12357. doi:10.1021/ja203812h
Return to citation in text: [1] -
Bazgir, A.; Noroozi Tisseh, Z.; Mirzaei, P. Tetrahedron Lett. 2008, 49, 5165–5168. doi:10.1016/j.tetlet.2008.06.077
Return to citation in text: [1] -
Jadidi, K.; Ghahremanzadeh, R.; Bazgir, A. Tetrahedron 2009, 65, 2005–2009. doi:10.1016/j.tet.2009.01.013
Return to citation in text: [1] -
Badillo, J. J.; Arevalo, G. E.; Fettinger, J. C.; Franz, A. K. Org. Lett. 2011, 13, 418–421. doi:10.1021/ol1027305
Return to citation in text: [1] -
Esmaeili, A. A.; Vesalipoor, H.; Hosseinabadi, R.; Zavareh, A. F.; Naseri, N. A.; Ghiamati, E. A. Tetrahedron Lett. 2011, 52, 4865–4867. doi:10.1016/j.tetlet.2011.07.039
Return to citation in text: [1] -
Kiruthika, S. E.; Lakshmi, N. V.; Banu, B. R.; Perumal, P. T. Tetrahedron Lett. 2011, 52, 6508–6511. doi:10.1016/j.tetlet.2011.09.119
Return to citation in text: [1] -
Shanmugam, P.; Viswambharan, B.; Madhavan, S. Org. Lett. 2007, 9, 4095–4098. doi:10.1021/ol701533d
Return to citation in text: [1] -
Deng, H.-P.; Wei, Y.; Shi, M. Org. Lett. 2011, 13, 3348–3351. doi:10.1021/ol201094f
Return to citation in text: [1] -
Viswambharan, B.; Selvakumar, K.; Madhavan, S.; Shanmugam, P. Org. Lett. 2010, 12, 2108–2111. doi:10.1021/ol100591r
Return to citation in text: [1] -
Quiroga, J.; Portillo, S.; Pérez, A.; Gálvez, J.; Abonia, R.; Insuasty, B. Tetrahedron Lett. 2011, 52, 2664–2666. doi:10.1016/j.tetlet.2011.03.067
Return to citation in text: [1] -
Shakibaei, G. I.; Feiz, A.; Khavasi, H. R.; Soorki, A. A.; Bazgir, A. ACS Comb. Sci. 2011, 13, 96–99. doi:10.1021/co1000053
Return to citation in text: [1] -
Mohammadi, A. A.; Dabiri, M.; Qaraat, H. Tetrahedron 2009, 65, 3804–3808. doi:10.1016/j.tet.2009.02.037
Return to citation in text: [1] -
Basavaiah, D.; Reddy, K. R. Org. Lett. 2007, 9, 57–60. doi:10.1021/ol062561m
Return to citation in text: [1] -
Liu, H.; Dou, D.; Shi, D. J. Comb. Chem. 2010, 12, 633–637. doi:10.1021/cc100035q
Return to citation in text: [1] -
Sun, J.; Sun, Y.; Gao, H.; Yan, C.-G. Eur. J. Org. Chem. 2012, 1976–1983. doi:10.1002/ejoc.201101737
Return to citation in text: [1] [2] -
Joshi, K. C.; Jain, R.; Arora, S. J. Fluorine Chem. 1989, 42, 149–162. doi:10.1016/S0022-1139(00)82745-1
Return to citation in text: [1] -
Dabiri, M.; Bahramnejad, M.; Baghbanzadeh, M. Tetrahedron 2009, 65, 9443–9447. doi:10.1016/j.tet.2009.08.070
Return to citation in text: [1] -
Ghahremanzadeh, R.; Ahadi, S.; Shakibaei, G. I.; Bazgir, A. Tetrahedron Lett. 2010, 51, 499–502. doi:10.1016/j.tetlet.2009.11.041
Return to citation in text: [1] -
Ghahremanzadeh, R.; Shakibaei, G. I.; Ahadi, S.; Bazgir, A. J. Comb. Chem. 2010, 12, 191–194. doi:10.1021/cc900130a
Return to citation in text: [1] -
Jadidi, K.; Ghahremanzadeh, R.; Bazgir, A. J. Comb. Chem. 2009, 11, 341–344. doi:10.1021/cc800167h
Return to citation in text: [1] -
Kefayati, H.; Narchin, F.; Rad-Moghadam, K. Tetrahedron Lett. 2012, 53, 4573–4575. doi:10.1016/j.tetlet.2012.06.070
Return to citation in text: [1] -
Ghahremanzadeh, R.; Fereshtehnejad, F.; Bazgir, A. J. Heterocycl. Chem. 2010, 47, 1031–1034. doi:10.1002/jhet.412
Return to citation in text: [1] [2] -
Rad-Moghadam, K.; Youseftabar-Miri, L. Synlett 2010, 1969–1973. doi:10.1055/s-0030-1258506
Return to citation in text: [1] [2] -
Lu, G.-P.; Cai, C. J. Chem. Res. 2011, 35, 547–551. doi:10.3184/174751911X13157531279974
Return to citation in text: [1] [2] -
Rad-Moghadam, K.; Youseftabar-Miri, L. J. Fluorine Chem. 2012, 135, 213–219. doi:10.1016/j.jfluchem.2011.11.007
Return to citation in text: [1] [2] -
Ghahremanzadeh, R.; Sayyafi, M.; Ahadi, S.; Bazgir, A. J. Comb. Chem. 2009, 11, 393–396. doi:10.1021/cc8001958
Return to citation in text: [1] [2] -
Bazgir, A.; Ahadi, S.; Ghahremanzadeh, R.; Khavasi, H. R.; Mirzaei, P. Ultrason. Sonochem. 2010, 17, 447–462. doi:10.1016/j.ultsonch.2009.09.009
Return to citation in text: [1] [2] -
Khorrami, A. R.; Faraji, F.; Bazgir, A. Ultrason. Sonochem. 2010, 17, 587–591. doi:10.1016/j.ultsonch.2009.11.017
Return to citation in text: [1] [2] -
Arya, K. A.; Kumar, M. Mol. Diversity 2011, 15, 781–789. doi:10.1007/s11030-011-9309-2
Return to citation in text: [1] [2] -
Stefanović, G.; Pavičić-Woss, M.; Lorenc, L.; Mihailović, M. L. Tetrahedron 1959, 6, 97–102. doi:10.1016/0040-4020(59)85001-8
Return to citation in text: [1] -
Joshi, K. C.; Jain, R.; Arora, S. Indian J. Heterocycl. Chem. 1991, 1, 95–97.
Return to citation in text: [1] -
Dabiri, M.; Tisseh, Z. N.; Bahramnejad, M.; Bazgir, A. Ultrason. Sonochem. 2011, 18, 1153–1159. doi:10.1016/j.ultsonch.2010.12.004
Return to citation in text: [1] -
Ghahremanzadeh, R.; Fereshtehnejad, F.; Mirzaei, P.; Bazgir, A. Ultrason. Sonochem. 2011, 18, 415–418. doi:10.1016/j.ultsonch.2010.07.010
Return to citation in text: [1]
| 1. | Tan, B.; Hernández-Torres, G.; Barbas, C. F., III. J. Am. Chem. Soc. 2011, 133, 12354–12357. doi:10.1021/ja203812h |
| 2. | Bazgir, A.; Noroozi Tisseh, Z.; Mirzaei, P. Tetrahedron Lett. 2008, 49, 5165–5168. doi:10.1016/j.tetlet.2008.06.077 |
| 11. | Shakibaei, G. I.; Feiz, A.; Khavasi, H. R.; Soorki, A. A.; Bazgir, A. ACS Comb. Sci. 2011, 13, 96–99. doi:10.1021/co1000053 |
| 12. | Mohammadi, A. A.; Dabiri, M.; Qaraat, H. Tetrahedron 2009, 65, 3804–3808. doi:10.1016/j.tet.2009.02.037 |
| 13. | Basavaiah, D.; Reddy, K. R. Org. Lett. 2007, 9, 57–60. doi:10.1021/ol062561m |
| 14. | Liu, H.; Dou, D.; Shi, D. J. Comb. Chem. 2010, 12, 633–637. doi:10.1021/cc100035q |
| 9. | Viswambharan, B.; Selvakumar, K.; Madhavan, S.; Shanmugam, P. Org. Lett. 2010, 12, 2108–2111. doi:10.1021/ol100591r |
| 10. | Quiroga, J.; Portillo, S.; Pérez, A.; Gálvez, J.; Abonia, R.; Insuasty, B. Tetrahedron Lett. 2011, 52, 2664–2666. doi:10.1016/j.tetlet.2011.03.067 |
| 7. | Shanmugam, P.; Viswambharan, B.; Madhavan, S. Org. Lett. 2007, 9, 4095–4098. doi:10.1021/ol701533d |
| 8. | Deng, H.-P.; Wei, Y.; Shi, M. Org. Lett. 2011, 13, 3348–3351. doi:10.1021/ol201094f |
| 22. | Ghahremanzadeh, R.; Fereshtehnejad, F.; Bazgir, A. J. Heterocycl. Chem. 2010, 47, 1031–1034. doi:10.1002/jhet.412 |
| 23. | Rad-Moghadam, K.; Youseftabar-Miri, L. Synlett 2010, 1969–1973. doi:10.1055/s-0030-1258506 |
| 24. | Lu, G.-P.; Cai, C. J. Chem. Res. 2011, 35, 547–551. doi:10.3184/174751911X13157531279974 |
| 25. | Rad-Moghadam, K.; Youseftabar-Miri, L. J. Fluorine Chem. 2012, 135, 213–219. doi:10.1016/j.jfluchem.2011.11.007 |
| 26. | Ghahremanzadeh, R.; Sayyafi, M.; Ahadi, S.; Bazgir, A. J. Comb. Chem. 2009, 11, 393–396. doi:10.1021/cc8001958 |
| 27. | Bazgir, A.; Ahadi, S.; Ghahremanzadeh, R.; Khavasi, H. R.; Mirzaei, P. Ultrason. Sonochem. 2010, 17, 447–462. doi:10.1016/j.ultsonch.2009.09.009 |
| 28. | Khorrami, A. R.; Faraji, F.; Bazgir, A. Ultrason. Sonochem. 2010, 17, 587–591. doi:10.1016/j.ultsonch.2009.11.017 |
| 29. | Arya, K. A.; Kumar, M. Mol. Diversity 2011, 15, 781–789. doi:10.1007/s11030-011-9309-2 |
| 3. | Jadidi, K.; Ghahremanzadeh, R.; Bazgir, A. Tetrahedron 2009, 65, 2005–2009. doi:10.1016/j.tet.2009.01.013 |
| 4. | Badillo, J. J.; Arevalo, G. E.; Fettinger, J. C.; Franz, A. K. Org. Lett. 2011, 13, 418–421. doi:10.1021/ol1027305 |
| 5. | Esmaeili, A. A.; Vesalipoor, H.; Hosseinabadi, R.; Zavareh, A. F.; Naseri, N. A.; Ghiamati, E. A. Tetrahedron Lett. 2011, 52, 4865–4867. doi:10.1016/j.tetlet.2011.07.039 |
| 6. | Kiruthika, S. E.; Lakshmi, N. V.; Banu, B. R.; Perumal, P. T. Tetrahedron Lett. 2011, 52, 6508–6511. doi:10.1016/j.tetlet.2011.09.119 |
| 22. | Ghahremanzadeh, R.; Fereshtehnejad, F.; Bazgir, A. J. Heterocycl. Chem. 2010, 47, 1031–1034. doi:10.1002/jhet.412 |
| 23. | Rad-Moghadam, K.; Youseftabar-Miri, L. Synlett 2010, 1969–1973. doi:10.1055/s-0030-1258506 |
| 24. | Lu, G.-P.; Cai, C. J. Chem. Res. 2011, 35, 547–551. doi:10.3184/174751911X13157531279974 |
| 25. | Rad-Moghadam, K.; Youseftabar-Miri, L. J. Fluorine Chem. 2012, 135, 213–219. doi:10.1016/j.jfluchem.2011.11.007 |
| 29. | Arya, K. A.; Kumar, M. Mol. Diversity 2011, 15, 781–789. doi:10.1007/s11030-011-9309-2 |
| 16. | Joshi, K. C.; Jain, R.; Arora, S. J. Fluorine Chem. 1989, 42, 149–162. doi:10.1016/S0022-1139(00)82745-1 |
| 17. | Dabiri, M.; Bahramnejad, M.; Baghbanzadeh, M. Tetrahedron 2009, 65, 9443–9447. doi:10.1016/j.tet.2009.08.070 |
| 18. | Ghahremanzadeh, R.; Ahadi, S.; Shakibaei, G. I.; Bazgir, A. Tetrahedron Lett. 2010, 51, 499–502. doi:10.1016/j.tetlet.2009.11.041 |
| 19. | Ghahremanzadeh, R.; Shakibaei, G. I.; Ahadi, S.; Bazgir, A. J. Comb. Chem. 2010, 12, 191–194. doi:10.1021/cc900130a |
| 20. | Jadidi, K.; Ghahremanzadeh, R.; Bazgir, A. J. Comb. Chem. 2009, 11, 341–344. doi:10.1021/cc800167h |
| 21. | Kefayati, H.; Narchin, F.; Rad-Moghadam, K. Tetrahedron Lett. 2012, 53, 4573–4575. doi:10.1016/j.tetlet.2012.06.070 |
| 30. | Stefanović, G.; Pavičić-Woss, M.; Lorenc, L.; Mihailović, M. L. Tetrahedron 1959, 6, 97–102. doi:10.1016/0040-4020(59)85001-8 |
| 31. | Joshi, K. C.; Jain, R.; Arora, S. Indian J. Heterocycl. Chem. 1991, 1, 95–97. |
| 32. | Dabiri, M.; Tisseh, Z. N.; Bahramnejad, M.; Bazgir, A. Ultrason. Sonochem. 2011, 18, 1153–1159. doi:10.1016/j.ultsonch.2010.12.004 |
| 33. | Ghahremanzadeh, R.; Fereshtehnejad, F.; Mirzaei, P.; Bazgir, A. Ultrason. Sonochem. 2011, 18, 415–418. doi:10.1016/j.ultsonch.2010.07.010 |
| 15. | Sun, J.; Sun, Y.; Gao, H.; Yan, C.-G. Eur. J. Org. Chem. 2012, 1976–1983. doi:10.1002/ejoc.201101737 |
| 15. | Sun, J.; Sun, Y.; Gao, H.; Yan, C.-G. Eur. J. Org. Chem. 2012, 1976–1983. doi:10.1002/ejoc.201101737 |
| 26. | Ghahremanzadeh, R.; Sayyafi, M.; Ahadi, S.; Bazgir, A. J. Comb. Chem. 2009, 11, 393–396. doi:10.1021/cc8001958 |
| 27. | Bazgir, A.; Ahadi, S.; Ghahremanzadeh, R.; Khavasi, H. R.; Mirzaei, P. Ultrason. Sonochem. 2010, 17, 447–462. doi:10.1016/j.ultsonch.2009.09.009 |
| 28. | Khorrami, A. R.; Faraji, F.; Bazgir, A. Ultrason. Sonochem. 2010, 17, 587–591. doi:10.1016/j.ultsonch.2009.11.017 |
© 2013 Sun et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)