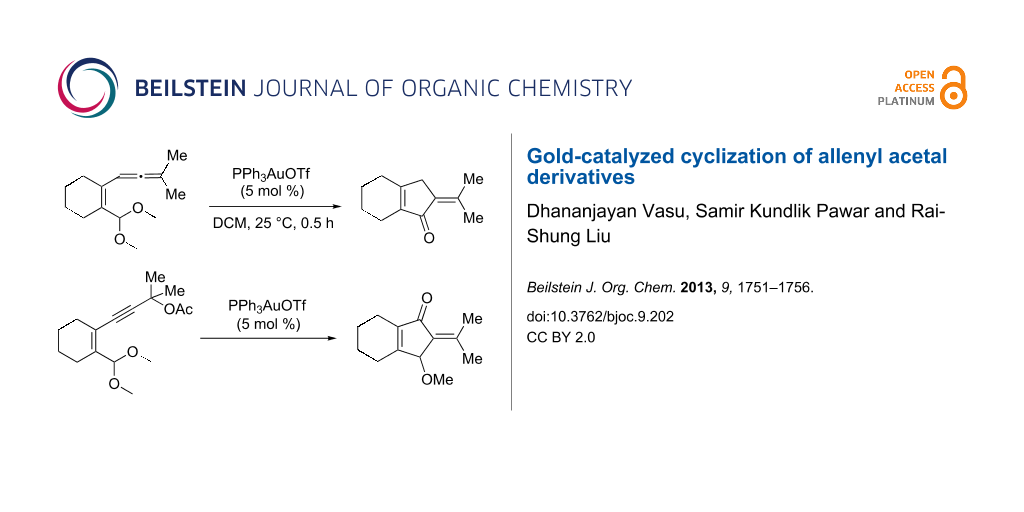
Abstract
The gold-catalyzed transformation of allenyl acetals into 5-alkylidenecyclopent-2-en-1-ones is described. The outcome of our deuterium labeling experiments supports a 1,4-hydride shift of the resulting allyl cationic intermediates because a complete deuterium transfer is observed. We tested the reaction on various acetal substrates bearing a propargyl acetate, giving 4-methoxy-5-alkylidenecyclopent-2-en-1-ones 4 via a degradation of the acetate group at the allyl cation intermediate.

Graphical Abstract
Introduction
Gold-catalyzed cyclization/cycloaddition reactions [1-5] are useful synthetic methods to construct complicated carbo- and oxacyclic frameworks. Such cascade reactions have been well studied on various difunctionalized molecules including oxoalkynes [6-13], oxoallenes [14], oxoalkenes [15] and allenyl acetals [16-18]. In this cascade sequence, two new rings and three chemical bonds are generated in a one-pot procedure. We previously reported gold-catalyzed reactions of allenyl acetals with suitable dipolarophiles such as 1,3-diones to chemoselectively produce the cycloaddition product 2 [17] (Scheme 1). Similar reactions with nitrones stereoselectively delivered distinct formal cycloadducts 3 [18]. We postulate that compounds 2 arise from the attack of 1,3-diones at initially generated allyl cation intermediates I. In the case of electrophilic nitrones, allyl cations I release a proton to form reactive 1-methoxyfulvenes II to achieve a [3 + 2]-nitrone cycloaddition. The versatility of cationic intermediates I encourages us to understand their behavior in the absence of a dipolarophile. This work reports gold-catalyzed intramolecular cyclizations of these allenyl acetals [19].
![[1860-5397-9-202-i1]](/bjoc/content/inline/1860-5397-9-202-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reported cascade reactions on allenyl acetals.
Scheme 1: Reported cascade reactions on allenyl acetals.
Results and Discussion
We first tested the intramolecular cyclizations of allenyl acetal 1a with PPh3AuCl/AgSbF6 (5 mol %), which was shown to be an active catalyst in the two cascade reactions, as depicted in Scheme 1 [17,18]. As shown in Table 1, the treatment of compound 1a with this gold catalyst (5 mol %) in dichloromethane (DCM, 28 °C, 0.5 h) afforded 5-isopropylidenecyclopent-2-en-1-one derivative 4a in 65% yield (Table 1, entry 1). With a change of the counter anion as in PPh3AuCl/AgOTf, the product yield increased to 89% (Table 1, entry 2). PPh3AuCl/AgNTf2 was also active to give the same product in 83% yield (Table 1, entry 3). Under the same conditions, AgOTf alone gave the desired 4a in 48% yield (Table 1, entry 4). AuCl3 and PtCl2 enabled a complete consumption of the starting material 1a, but the yields of compound 4a were 51% and 30%, respectively (Table 1, entries 5 and 6).
Table 1: Catalyst screening over various acid catalysts.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-202-i5.svg?max-width=637&scale=1.0)
|
|||
| Entrya | Catalyst | Time (h) | Yield (%)b |
|---|---|---|---|
| 1 | PPh3AuCl/AgSbF6 | 0.5 | 65 |
| 2 | PPh3AuCl/AgOTf | 0.5 | 89 |
| 3 | PPh3AuCl/AgNTf2 | 0.5 | 83 |
| 4 | AgOTf | 2.0 | 48 |
| 5 | AuCl3/CO | 1.5 | 51 |
| 6 | PtCl2/CO | 1.5 | 30 |
a[1a] = 0.1 M. bIsolated yields.
Table 2 shows the substrate scope including additional allenyl acetals 1b–1h. The reactions were catalyzed by PPh3AuCl/AgNTf2 (5 mol %) in DCM. As shown in entries 1–3, this cyclization was applicable to allenyl acetals 1b–1d bearing a cyclopentyl bridge. The resulting products 4b–4d were produced with satisfactory yields (68–82%). We also tested the reaction on acyclic allenyl acetal 1e (E/Z = 3:1), and afforded the desired product 4e in 52% yield according to initial E-configured 1e. The structure of compound 4e was determined by 1H NMR NOE spectra. The reaction was still operable with 1f, bearing a 1,2-disubsituted allene, giving the desired 4f in moderate yield (49%). Its E-configuration was determined by NOE measurements, and assignable to other products including 4g and 4h. The reaction worked well with substrates bearing a different trisubstituted allenes, giving the desired cyclopentenone 4g and 4h in 82–83% yields.
Table 2: Gold-catalyzed cyclization of allenyl acetals.
| Entry | Substratesa | Time/min | Product (yield)b |
|---|---|---|---|
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-202-i6.svg?max-width=637&scale=1.0)
1b |
15 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-202-i7.svg?max-width=637&scale=1.0)
4b (82%) |
| 2 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-202-i8.svg?max-width=637&scale=1.0)
1c |
10 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-202-i9.svg?max-width=637&scale=1.0)
4c (68%) |
| 3 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-202-i10.svg?max-width=637&scale=1.0)
1d |
10 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-202-i11.svg?max-width=637&scale=1.0)
4d (70%) |
| 4c |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-202-i12.svg?max-width=637&scale=1.0)
1e |
30 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-202-i13.svg?max-width=637&scale=1.0)
4e (52%) |
| 5 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-202-i14.svg?max-width=637&scale=1.0)
1f |
30 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-202-i15.svg?max-width=637&scale=1.0)
4f (49%) |
| 6 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-202-i16.svg?max-width=637&scale=1.0)
1g |
30 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-202-i17.svg?max-width=637&scale=1.0)
4g (82%) |
| 7 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-202-i18.svg?max-width=637&scale=1.0)
1h |
10 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-202-i19.svg?max-width=637&scale=1.0)
4h (83%) |
a5 mol % AuClPPh3/AgOTf, [1] = 0.1 M, 25 °C, DCM. bIsolated yield. c10 mol % of gold catalyst.
The preceding cyclization is mechanistically interesting because it involves a cleavage of the C–H bond of the acetal group. We prepared d1-1a bearing a deuterium (>98%, Scheme 2, reaction 1) at its acetal group. The resulting product d1-4a has almost one full deuterium (X = 0.98 D) at one of the methylene protons according to DEPT 13C NMR analysis. In the presence of added D2O, undeuterated 1a gave the product without deuterium content (Scheme 2, reaction 2). The results of these labeling experiments reveal a 1,4-hydrogen shift [20-22] in the d1-1a→d1-4a transformation.
![[1860-5397-9-202-i2]](/bjoc/content/inline/1860-5397-9-202-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Gold-catalyzed cyclization of deuterated d1-1a.
Scheme 2: Gold-catalyzed cyclization of deuterated d1-1a.
Scheme 3 shows a plausible mechanism to rationalize the transformation of the allenyl acetal 1e into the observed cyclopentenone 4e. The deuterium labeling experiment of the d1-1a→d1-4a transformation (Scheme 2, reaction 1) indicates that one methylene proton of 4a is derived from the original acetal group. Accordingly, we postulate a 1,4-hydride shift [21,22] for the intermediate transformation B→C. We excluded an alternative route involving the protonation of the fulvene intermediate D because this route would water as a proton source. The formation of the fulvene intermediate D from allyl cation B is assisted by a weak base like nitrone [18]. We envisage that a 1,2-hydrogen shift for the allyl cation B fails to explain a complete deuterium transfer for the d1-1a→d1-4a transformation because its resulting cyclopent-3-en-1-one derivative became isomerized to the final product 4a with a loss of deuterium content.
![[1860-5397-9-202-i3]](/bjoc/content/inline/1860-5397-9-202-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: A plausible reaction mechanism.
Scheme 3: A plausible reaction mechanism.
We also prepared the substrate 5a bearing a propargyl acetate moiety because this functionality can be transferred to the allenyl acetate 5a’ by a gold catalyst [23,24]. As shown in Scheme 4, the treatment of species 5a with PPh3AuOTf (5 mol %) in dichloromethane (28 °C, 5 min) gave 4-methoxy-5-isopropylidenecyclopent-2-en-1-one 6a in 76% yield. The structure of compound 6a was determined by an X-ray diffraction study (crystallographic data are provided in Supporting Information File 1). Formation of this product is postulated to arise from the attack of the methoxy anion at the acetyl group of the corresponding allyl cation E, a process not involving a 1,4-hydride shift. This alternative pathway highlights the diversified mechanism of such oxidative cyclizations.
![[1860-5397-9-202-i4]](/bjoc/content/inline/1860-5397-9-202-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: The reaction of propargyl acetate 5a.
Scheme 4: The reaction of propargyl acetate 5a.
We prepared the additional substrates 5b–5g bearing an acetate group to examine the scope of the reaction, results are shown in Table 3. This gold-catalyzed cyclization was applicable to compound 5b bearing a cyclopentyl bridge, giving the desired 6b in 96% yield. The reaction worked also with 5c and 5d bearing a cyclohexyl bridge, delivering the desired products 6c and 6d in 78% and 72% yields, respectively (Table 3, entries 2 and 3). We tested the reaction with the benzenoid substrates 5e–5g, giving the corresponding enones 6e–6g in 63–78% yields.
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-202-i20.svg?max-width=637&scale=1.0)
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-202-i21.svg?max-width=637&scale=1.0)
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-202-i22.svg?max-width=637&scale=1.0)
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-202-i23.svg?max-width=637&scale=1.0)
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-202-i24.svg?max-width=637&scale=1.0)
![[Graphic 21]](/bjoc/content/inline/1860-5397-9-202-i25.svg?max-width=637&scale=1.0)
![[Graphic 22]](/bjoc/content/inline/1860-5397-9-202-i26.svg?max-width=637&scale=1.0)
![[Graphic 23]](/bjoc/content/inline/1860-5397-9-202-i27.svg?max-width=637&scale=1.0)
![[Graphic 24]](/bjoc/content/inline/1860-5397-9-202-i28.svg?max-width=637&scale=1.0)
![[Graphic 25]](/bjoc/content/inline/1860-5397-9-202-i29.svg?max-width=637&scale=1.0)
![[Graphic 26]](/bjoc/content/inline/1860-5397-9-202-i30.svg?max-width=637&scale=1.0)
![[Graphic 27]](/bjoc/content/inline/1860-5397-9-202-i31.svg?max-width=637&scale=1.0)
Conclusion
In summary, we report a gold-catalyzed transformation of allenyl acetals 1 into 5-alkylidenecyclopent-2-en-1-ones 4. Our deuterium labeling experiments support a 1,4-hydride shift for the resulting allyl cation because of a complete deuterium transfer. This observation excludes the pathway involving the protonation of a 1-methoxyfulvene species. We tested the reactions of acetal substrates 5 bearing a propargyl acetate to afford 4-methoxy-5-alkylidenecyclopent-2-en-1-ones 6. The formation mechanism involves a degradation of the acetate group at the corresponding allyl cation.
Experimental
General procedure for the gold-catalyzed carbocyclization
General procedure for the the gold(I)-catalyzed carbocyclization of vinylallenyl acetal: A two-necked flask was charged with chloro(triphenylphosphine)gold(I) (11.1 mg, 0.022 mmol) and silver triflate (5.8 mg, 0.022 mmol), and to this mixture CH2Cl2 (2.0 mL) was added. The resulting solution was stirred at room temperature for 10 min. To this mixture a solution of vinylallenyl acetal 1a (100 mg, 0.45 mmol) in CH2Cl2 (2.5 mL) was added dropwise, and the mixture was kept stirring at 25 °C for 30 min before it was filtered over a short silica bed. The solvent was evaporated under reduced pressure. The crude product was eluted through a short silica column (3% ethyl acetate in hexane) to afford the desired ketone 4a (70.6 mg, 0.40 mmol, 89%) as a pale yellow oil.
General procedure for the gold(I)-catalyzed carbocyclization of propargylic ester acetals: Chloro(triphenylphosphine)gold(I) (8.0 mg, 0.016 mmol) and silver triflate (4.2 mg, 0.016 mmol) were added to a dried Schlenk tube under an N2 atmosphere, and freshly distilled CH2Cl2 (1.0 mL) was introduced by a syringe. The resulting mixture was stirred at room temperature for 10 minutes before the addition of propargylic ester acetal 5a (100 mg, 0.32 mmol) in CH2Cl2 (2.2 mL). The reaction mixture was stirred for additional 5 minutes at 25 °C. After the completion of reaction, the brown suspension was filtered through a short bed of silica gel. The solvent was removed under reduced pressure. The crude product was purified by flash chromatography to afford the desired ketone 6a (58 mg, 0.25 mmol, 76%) as a dark yellow oil.
Supporting Information
| Supporting Information File 1: Experimental details. | ||
| Format: PDF | Size: 6.1 MB | Download |
References
-
Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395. doi:10.1021/cr050041j
Return to citation in text: [1] -
Sohel, S. M. A.; Liu, R.-S. Chem. Soc. Rev. 2009, 38, 2269. doi:10.1039/b807499m
Return to citation in text: [1] -
López, F.; Mascareñas, J. L. Beilstein J. Org. Chem. 2011, 7, 1075. doi:10.3762/bjoc.7.124
Return to citation in text: [1] -
Aubert, C.; Fensterbank, L.; Garcia, P.; Malacria, M.; Simonneau, A. Chem. Rev. 2011, 111, 1954. doi:10.1021/cr100376w
Return to citation in text: [1] -
Rudolph, M.; Hashmi, A. S. K. Chem. Soc. Rev. 2012, 41, 2448. doi:10.1039/c1cs15279c
Return to citation in text: [1] -
Teng, T.-M.; Das, A.; Huple, D. B.; Liu, R.-S. J. Am. Chem. Soc. 2010, 132, 12565. doi:10.1021/ja106493h
Return to citation in text: [1] -
Liu, F.; Qian, D.; Li, L.; Zhao, X.; Zhang, J. Angew. Chem., Int. Ed. 2010, 49, 6669. doi:10.1002/anie.201003136
Return to citation in text: [1] -
Liu, F.; Yu, Y.; Zhang, J. Angew. Chem., Int. Ed. 2009, 48, 5505. doi:10.1002/anie.200901299
Return to citation in text: [1] -
Asao, N.; Kasahara, T.; Yamamoto, Y. Angew. Chem., Int. Ed. 2003, 42, 3504. doi:10.1002/anie.200351390
Return to citation in text: [1] -
Asao, N.; Aikawa, H.; Yamamoto, Y. J. Am. Chem. Soc. 2004, 126, 7458. doi:10.1021/ja0477367
Return to citation in text: [1] -
Hsu, Y.-C.; Ting, C.-M.; Liu, R.-S. J. Am. Chem. Soc. 2009, 131, 2090. doi:10.1021/ja809560c
Return to citation in text: [1] -
Hashmi, A. S. K.; Littmann, A. Chem.–Asian J. 2012, 7, 1435. doi:10.1002/asia.201200046
Return to citation in text: [1] -
Teng, T.-M.; Liu, R.-S. J. Am. Chem. Soc. 2010, 132, 9298. doi:10.1021/ja1043837
Return to citation in text: [1] -
Lin, C.-C.; Teng, T.-M.; Tsai, C.-C.; Liao, H.-Y.; Liu, R.-S. J. Am. Chem. Soc. 2008, 130, 16417. doi:10.1021/ja806415t
Return to citation in text: [1] -
Lin, C.-C.; Teng, T.-M.; Odedra, A.; Liu, R.-S. J. Am. Chem. Soc. 2007, 129, 3798. doi:10.1021/ja069171f
Return to citation in text: [1] -
Bhunia, S.; Liu, R.-S. J. Am. Chem. Soc. 2008, 130, 16488. doi:10.1021/ja807384a
Return to citation in text: [1] -
Teng, T.-M.; Lin, M.-S.; Vasu, D.; Bhunia, S.; Liu, T.-A.; Liu, R.-S. Chem.–Eur. J. 2010, 16, 4744. doi:10.1002/chem.201000041
Return to citation in text: [1] [2] [3] -
Vasu, D.; Liu, R.-S. Chem.–Eur. J. 2012, 18, 13638. doi:10.1002/chem.201201777
Return to citation in text: [1] [2] [3] [4] -
Nakamura, I.; Mizushima, Y.; Gridnev, I. D.; Yamamoto, Y. J. Am. Chem. Soc. 2005, 127, 9844. doi:10.1021/ja051114j
Return to citation in text: [1] -
Busch-Petersen, J.; Corey, E. J. Org. Lett. 2000, 2, 1641. doi:10.1021/ol005964
Return to citation in text: [1] -
Shintani, R.; Okamoto, K.; Hayashi, T. J. Am. Chem. Soc. 2005, 127, 2872. doi:10.1021/ja042582g
Return to citation in text: [1] [2] -
Bolte, B.; Odabachian, Y.; Gagosz, F. J. Am. Chem. Soc. 2010, 132, 7294. doi:10.1021/ja1020469
Return to citation in text: [1] [2] -
Zhang, L. J. Am. Chem. Soc. 2005, 127, 16804. doi:10.1021/ja056419c
Return to citation in text: [1] -
Marion, N.; Díez-González, S.; de Frémont, P.; Noble, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2006, 45, 3647. doi:10.1002/anie.200600571
Return to citation in text: [1]
| 1. | Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395. doi:10.1021/cr050041j |
| 2. | Sohel, S. M. A.; Liu, R.-S. Chem. Soc. Rev. 2009, 38, 2269. doi:10.1039/b807499m |
| 3. | López, F.; Mascareñas, J. L. Beilstein J. Org. Chem. 2011, 7, 1075. doi:10.3762/bjoc.7.124 |
| 4. | Aubert, C.; Fensterbank, L.; Garcia, P.; Malacria, M.; Simonneau, A. Chem. Rev. 2011, 111, 1954. doi:10.1021/cr100376w |
| 5. | Rudolph, M.; Hashmi, A. S. K. Chem. Soc. Rev. 2012, 41, 2448. doi:10.1039/c1cs15279c |
| 16. | Bhunia, S.; Liu, R.-S. J. Am. Chem. Soc. 2008, 130, 16488. doi:10.1021/ja807384a |
| 17. | Teng, T.-M.; Lin, M.-S.; Vasu, D.; Bhunia, S.; Liu, T.-A.; Liu, R.-S. Chem.–Eur. J. 2010, 16, 4744. doi:10.1002/chem.201000041 |
| 18. | Vasu, D.; Liu, R.-S. Chem.–Eur. J. 2012, 18, 13638. doi:10.1002/chem.201201777 |
| 15. | Lin, C.-C.; Teng, T.-M.; Odedra, A.; Liu, R.-S. J. Am. Chem. Soc. 2007, 129, 3798. doi:10.1021/ja069171f |
| 14. | Lin, C.-C.; Teng, T.-M.; Tsai, C.-C.; Liao, H.-Y.; Liu, R.-S. J. Am. Chem. Soc. 2008, 130, 16417. doi:10.1021/ja806415t |
| 23. | Zhang, L. J. Am. Chem. Soc. 2005, 127, 16804. doi:10.1021/ja056419c |
| 24. | Marion, N.; Díez-González, S.; de Frémont, P.; Noble, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2006, 45, 3647. doi:10.1002/anie.200600571 |
| 6. | Teng, T.-M.; Das, A.; Huple, D. B.; Liu, R.-S. J. Am. Chem. Soc. 2010, 132, 12565. doi:10.1021/ja106493h |
| 7. | Liu, F.; Qian, D.; Li, L.; Zhao, X.; Zhang, J. Angew. Chem., Int. Ed. 2010, 49, 6669. doi:10.1002/anie.201003136 |
| 8. | Liu, F.; Yu, Y.; Zhang, J. Angew. Chem., Int. Ed. 2009, 48, 5505. doi:10.1002/anie.200901299 |
| 9. | Asao, N.; Kasahara, T.; Yamamoto, Y. Angew. Chem., Int. Ed. 2003, 42, 3504. doi:10.1002/anie.200351390 |
| 10. | Asao, N.; Aikawa, H.; Yamamoto, Y. J. Am. Chem. Soc. 2004, 126, 7458. doi:10.1021/ja0477367 |
| 11. | Hsu, Y.-C.; Ting, C.-M.; Liu, R.-S. J. Am. Chem. Soc. 2009, 131, 2090. doi:10.1021/ja809560c |
| 12. | Hashmi, A. S. K.; Littmann, A. Chem.–Asian J. 2012, 7, 1435. doi:10.1002/asia.201200046 |
| 13. | Teng, T.-M.; Liu, R.-S. J. Am. Chem. Soc. 2010, 132, 9298. doi:10.1021/ja1043837 |
| 17. | Teng, T.-M.; Lin, M.-S.; Vasu, D.; Bhunia, S.; Liu, T.-A.; Liu, R.-S. Chem.–Eur. J. 2010, 16, 4744. doi:10.1002/chem.201000041 |
| 18. | Vasu, D.; Liu, R.-S. Chem.–Eur. J. 2012, 18, 13638. doi:10.1002/chem.201201777 |
| 21. | Shintani, R.; Okamoto, K.; Hayashi, T. J. Am. Chem. Soc. 2005, 127, 2872. doi:10.1021/ja042582g |
| 22. | Bolte, B.; Odabachian, Y.; Gagosz, F. J. Am. Chem. Soc. 2010, 132, 7294. doi:10.1021/ja1020469 |
| 19. | Nakamura, I.; Mizushima, Y.; Gridnev, I. D.; Yamamoto, Y. J. Am. Chem. Soc. 2005, 127, 9844. doi:10.1021/ja051114j |
| 18. | Vasu, D.; Liu, R.-S. Chem.–Eur. J. 2012, 18, 13638. doi:10.1002/chem.201201777 |
| 18. | Vasu, D.; Liu, R.-S. Chem.–Eur. J. 2012, 18, 13638. doi:10.1002/chem.201201777 |
| 17. | Teng, T.-M.; Lin, M.-S.; Vasu, D.; Bhunia, S.; Liu, T.-A.; Liu, R.-S. Chem.–Eur. J. 2010, 16, 4744. doi:10.1002/chem.201000041 |
| 20. | Busch-Petersen, J.; Corey, E. J. Org. Lett. 2000, 2, 1641. doi:10.1021/ol005964 |
| 21. | Shintani, R.; Okamoto, K.; Hayashi, T. J. Am. Chem. Soc. 2005, 127, 2872. doi:10.1021/ja042582g |
| 22. | Bolte, B.; Odabachian, Y.; Gagosz, F. J. Am. Chem. Soc. 2010, 132, 7294. doi:10.1021/ja1020469 |
© 2013 Vasu et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)