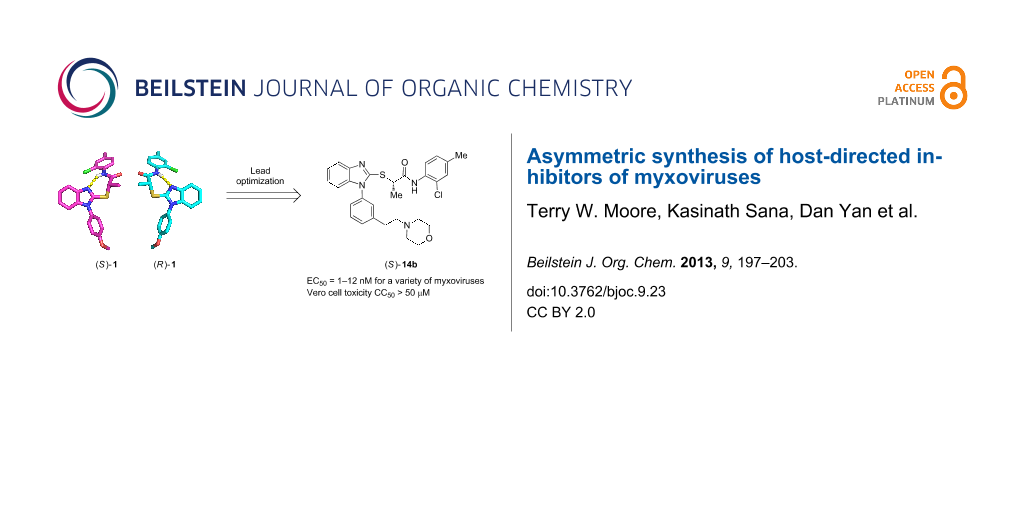
Abstract
High-throughput screening (HTS) previously identified benzimidazole 1 (JMN3-003) as a compound with broad antiviral activity against different influenza viruses and paramyxovirus strains. In pursuit of a lead compound from this series for development, we sought to increase both the potency and the aqueous solubility of 1. Lead optimization has achieved compounds with potent antiviral activity against a panel of myxovirus family members (EC50 values in the low nanomolar range) and much improved aqueous solubilities relative to that of 1. Additionally, we have devised a robust synthetic strategy for preparing 1 and congeners in an enantio-enriched fashion, which has allowed us to demonstrate that the (S)-enantiomers are generally 7- to 110-fold more potent than the corresponding (R)-isomers.

Graphical Abstract
Introduction
Myxoviruses are divided into two evolutionarily distinct yet related families: the orthomyxoviridae, which is composed largely of the influenza viruses, and the paramyxoviridae, which includes respiratory syncytial virus (RSV), measles virus (MeV), human parainfluenza virus (HPIV) and others [1]. Because myxoviruses are responsible for the majority of human morbidity and mortality cases due to viral respiratory illness globally, a therapeutic strategy that targets these viruses could have a substantial impact on human health [2-5]. Although antivirals typically seek to disable viral proteins, cellular host proteins can also be targeted, as is the case with selzentry, which inactivates the coreceptor (CCR5) for human immunodeficiency virus (HIV) entry [6]. The former approach is likely to yield compounds with a narrow spectrum of antiviral activities, and such inhibitors will certainly face the inevitable challenge of resistance [7,8]. Our research group has been actively engaged in the identification of small molecule inhibitors against myxoviruses in recent years [8-12], with a particular focus on the development of agents that target host-cell proteins enabling viral reproduction. Advantages of this strategy include a vastly expanded list of potential targets; a broader spectrum of activity, because many of the relevant host proteins are shared among related viruses; and, in principle, less susceptibility to the development of resistance.
Using high-throughput screening, in combination with counter-screening for detecting a broadened viral target spectrum that extends to other pathogens of the myxovirus families, our research group has been successful in identifying small-molecule antiviral hits resident in host cells [13]. One such molecule recently described is benzimidazole 1 [14] (Figure 1). Although the compound is active in vitro against a number of different para- and orthomyxoviruses, 1 has poor water solubility (<15 μg/mL), which may contribute to its low oral bioavailability [15]. Additionally, it was shown that the methyl group at the stereogenic center alpha to the carbonyl is important for biological activity [8,12,14]. Compound 1 was previously prepared as the racemate, but subsequently separated into enantiomers by chiral HPLC. To enable large-scale preparation of the pure isomers for further pharmacokinetic and animal studies, we present here an asymmetric synthesis of 1 and its congeners with improved aqueous solubility and antiviral potency.
![[1860-5397-9-23-1]](/bjoc/content/figures/1860-5397-9-23-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of first-generation lead compound 1.
Figure 1: Structure of first-generation lead compound 1.
Results and Discussion
Design. We previously reported a series of compounds with antiviral activity against a number of myxoviruses [8,12,14]. Structure–activity relationship (SAR) studies suggested both the 2-chloro-4-methylanilide and the central α-thiopropionamide to be moieties that confer good activity. Relatively unexplored in our previous work was the importance of the p-methoxyphenyl ring as well as the influence of the stereochemistry at the chiral center. In the current work, we examine the replacement of the p-methoxyphenyl ring with basic moieties that may increase aqueous solubility while maintaining activity, and we also developed synthetic routes to produce each enantiomer of these compounds.
Synthesis. The compounds were prepared by modifications of our previously reported routes. Briefly, nitroanilines 7 were obtained by one of two routes: ortho-nitrobenzaldehyde (2) was treated with N-methylpiperazine (3) in the presence of sodium triacetoxyborohydride to give nitrobenzene 4, which was reduced under hydrogenation conditions to provide aniline 5. o-Fluoronitrobenzene (6) was coupled with the previously formed aniline under SNAr conditions to furnish anilino nitrobenzene 7a (Scheme 1). Alternatively, meta- and para-nitrophenylethanols 8 were combined with o-fluoronitrobenzene (6) to deliver o-nitroanilines 9. The hydroxy groups of 9 were activated as the p-nitrobenzenesulfonates 10 and displaced with morpholine to give o-nitroanilines 7b and 7c (Scheme 2). Hydrogenation was used to reduce o-nitroanilines 7 followed by cyclization using thiocarbonyldiimidazole to yield benzothiazoles 11. In the case of the racemates, these were combined with α-bromopropionamide 12 to afford racemic 14 (Scheme 3).
![[1860-5397-9-23-i1]](/bjoc/content/inline/1860-5397-9-23-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of anilino nitrobenzene 7a.
Scheme 1: Synthesis of anilino nitrobenzene 7a.
![[1860-5397-9-23-i2]](/bjoc/content/inline/1860-5397-9-23-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of morpholinyl-o-nitroanilines.
Scheme 2: Preparation of morpholinyl-o-nitroanilines.
![[1860-5397-9-23-i3]](/bjoc/content/inline/1860-5397-9-23-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Asymmetric synthesis of (R)- and (S)-isomers by using two different approaches.
Scheme 3: Asymmetric synthesis of (R)- and (S)-isomers by using two different approaches.
To address the issue of stereochemical control, we developed two strategies for preparing the compounds enantioselectively. The first approach utilized commercially available (R)- or (S)-2-bromopropionic acid (15) from the chiral pool in a two-step sequence (Scheme 4a). The first step involved amide bond formation of 2-bromopropionic acid (15) with 2-chloro-4-methylaniline (16) by using HATU (O-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate), a reagent known to limit racemization in peptide coupling reactions [16]. The enantiomeric excesses of the starting materials were ca. 95%. No erosion of stereochemistry in the products 12 was observed as determined by chiral HPLC. Recrystallization of 12 from ethanol did not increase the enantiomeric excess of the α-bromoamide. Interestingly, the final product can be achieved in both high yield and ee by adding the preformed potassium salt of the 2-mercaptobenzimidazole 11 to (S)- or (R)-α-bromopropionamides 12. Using a substoichiometric amount of potassium carbonate (0.9 equiv) in warm methanol, followed by removal of solvents under vacuum, provided a convenient route to the potassium salt of 11. The salt was suspended in anhydrous DMF and added dropwise to a cold (0 °C) solution of the previously formed bromides, to give the desired products 1 and 14 in high yield (80–90%) with little to no loss in enantiomeric excess. Isolating the potassium salt proved necessary, as attempts at preforming the potassium salt in dry DMF, followed by addition to the chiral α-bromoamide, led to ee’s of ca. 60%. Although both chiral acids are readily available from commercial sources, their enantiomeric excesses seem to be limited to ca. 95%. For these studies, ee’s >90% were sufficient; if material with higher enantiopurity is needed, we note that one recrystallization of (S)-1 from ethyl ether increases the enantiomeric excess from 94% to 97.4%.
![[1860-5397-9-23-i4]](/bjoc/content/inline/1860-5397-9-23-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Preparation of chiral propionamides by HATU-mediated coupling (a) or thionyl chloride-mediated coupling (b).
Scheme 4: Preparation of chiral propionamides by HATU-mediated coupling (a) or thionyl chloride-mediated coup...
In an alternative approach that provides material with high enantiomeric excess (>98% ee), we utilized the Mitsunobu reaction of 2-mercaptobenzimidazoles with an amide obtained from (L)-lactic acid (17). Using one equivalent each of 2-mercaptobenzimidazole and α-hydroxyamide 13 (prepared from thionyl chloride-mediated coupling of (L)-lactic acid (17) and 2-chloro-4-methylaniline (16) [17] (Scheme 4b)) in the presence of a slight excess of triphenylphosphine and diethyl azodicarboxylate, we acquired the desired products 14 in good yield and ee. This methodology is attractive because of the high ee that can be obtained from the inexpensive and readily available (L)-lactic acid (17). However, the enantiomeric (D)-lactic acid is substantially more expensive, and as seen below, the products arising from (L)-lactic acid are less active, making this route less attractive than the route utilizing chiral α-bromopropionamides 12.
Crystal structures of (R)- and (S)-1 obtained by crystallization from ethyl ether allowed us to unambiguously assign the absolute configuration of each enantiomer. Shown in Figure 2 are the (S)-enantiomer (left, magenta) and (R)-enantiomer (right, cyan) of 1. It is interesting to note that the hydrogen-bond formed between the amide N–H and the unsubstituted benzimidazole nitrogen in these crystal structures results in pseudo seven-membered rings. Whether this conformation is biologically relevant is unknown.
![[1860-5397-9-23-2]](/bjoc/content/figures/1860-5397-9-23-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Renderings of crystal structures of (S)- (left, magenta) and (R)- (right, cyan) enantiomers of 1.
Figure 2: Renderings of crystal structures of (S)- (left, magenta) and (R)- (right, cyan) enantiomers of 1.
Structure–activity relationships
A small set of compounds was synthesized based on variations of 1 by replacing the p-methoxyphenyl group with other substituted phenyl rings or heterocyclic rings. The compounds were initially assayed in two screening assays: (1) a measles virus cytopathic effect (CPE) reduction assay [12] and (2) a solubility assay based on laser nephelometry [18]. Unfortunately, compounds with the highest aqueous solubilities (>100 µg/mL) had the poorest antiviral activity (i.e., 18a and 18e, Table 1). To explore the possibility of increasing solubility by salt formation, the L-tartaric acid salt and benzenesulfonic acid salts of the most active compound 18f were synthesized (Figure 3) and subjected to solubility testing. However, these salts failed to improve solubility compared with the parent. Several compounds exhibited moderate antiviral activities but poor to moderate aqueous solubilities (i.e., 1, 18b, 18d, and 18f, Table 1). Since 14a, 14b, and 14c showed good antiviral activities, as well as moderate aqueous solubilities, we decided to examine the broader antiviral activities of these compounds and to determine what, if any, effect the stereocenter present in each of these compounds may cause.
Table 1: In vitro screening of analogues of 1.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-23-i5.svg?max-width=637&scale=1.0)
|
|||||
| Compd. | MeVa (CPE, µM) |
Aqueous solubilityb
(µg/mL at pH 7.4) |
Compd. | MeVa (CPE, µM) |
Aqueous solubilityb
(µg/mL at pH 7.4) |
|---|---|---|---|---|---|
| 1 | 3.1 | <15 | 18f-tartrate | NDc | <15 |
| 18a | >75 | 140 | 18f-benzene sulfonate | ND | <15 |
| 18b | >0.29 | 19 | 18g | 0.05d | <15 |
| 18c | >75 | <15 | 14a | 0.179 | 20 |
| 18d | 4.9 | 22 | 14b | 0.20 | 15 |
| 18e | >75 | 120 | 14c | 0.6 | 25 |
| 18f | 0.27 | <15 | |||
a50% inhibitory concentrations were calculated by using the variable-slope (four parameters) nonlinear regression-fitting algorithm embedded in the Prism 5 software package (GraphPad Software). Values represent averages of four experiments; highest concentration assessed, 75 μM. bDetermined through laser nephelometry; cND. Not determined. dVirus yield reduction assay was used.
![[1860-5397-9-23-3]](/bjoc/content/figures/1860-5397-9-23-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: L-Tartaric acid salt (18f-tartrate) and benzenesulfonic acid salt (18f-benzenesulfonate) of 18f.
Figure 3: L-Tartaric acid salt (18f-tartrate) and benzenesulfonic acid salt (18f-benzenesulfonate) of 18f.
We were motivated by the results of these two assays to more completely characterize the antiviral activities and solubility parameters of the most promising compounds. We assayed the compounds in three additional biological assays: (1) a firefly luciferase minireplicon assay whose output is driven by infection with influenza A/sw/Texas/2009 (WSN); (2) an assay using a renilla luciferase reporter embedded as an additional transcription unit in the genome of a measles virus (MeV) recombinant; and (3) a colorimetric assay that measures reduction of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) as a surrogate for general cytotoxicity, reported here as CC50. Additionally, the aqueous solubilities of the compounds were measured by using laser nephelometry at pH 3.0, 5.0, and 7.4 (Table 2).
Table 2: Antiviral potencies and solubilities for 1 and analogues.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-23-i6.svg?max-width=637&scale=1.0)
|
|||||||
| Compd. | R | EC50 (nM)a | Solubility (µg/mL)b | CC50 (nM)c | |||
|---|---|---|---|---|---|---|---|
| MeV | WSN | pH 3.0 | pH 5.0 | pH 7.4 | |||
|
1
(S)-1 (R)-1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-23-i7.svg?max-width=637&scale=1.0)
|
31
37 880 |
13
5 160 |
<15 | <15 | <15 |
>50000
>50000 >50000 |
|
14b
(S)-14b (R)-14b |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-23-i8.svg?max-width=637&scale=1.0)
|
13
11 210 |
0.3
1 110 |
158 | 33 | 15 |
>50000
>50000 >50000 |
|
14c
(S)-14c (R)-14c |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-23-i9.svg?max-width=637&scale=1.0)
|
110
180 530 |
43
36 120 |
212 | 51 | 25 |
NDd
ND ND |
|
14a
(S)-14ae (R)-14ae |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-23-i10.svg?max-width=637&scale=1.0)
|
41
68 470 |
8
4 120 |
>300 | 92 | 20 |
ND
25000 ND |
a50% effective concentrations were calculated by using the variable-slope (four parameters) nonlinear regression-fitting algorithm embedded in the Prism 5 software package (GraphPad Software). Values represent averages of four experiments; highest concentration assessed 75 μM. bDetermined through laser nephelometry. cDetermined through MTT assay, highest concentration assessed 50 μM. dND: Not determined. eAssayed as a 1:1 mixture of atropisomers. Details of the separation of the atropisomers will be discussed elsewhere.
Analysis of the data reveals several trends. First, with the exception of a single case in which enantiomer potencies are similar (14c, ΔEC50 < 3-fold), six other comparisons reveal the (S)-enantiomer to be more active than the (R)-form by 7 to 110-fold. Although we do not know the identity of the specific biological target(s), the enantio-discrimination implies to us that the molecules bind in a well-defined binding pocket that is able to accommodate the S-enantiomer more readily than the (R)-enantiomer.
We note that the activity trends for the (S)- and (R)-enantiomers against the measles and WSN influenza strain are qualitatively similar. However, for antiviral potency differences between the racemate and the (S)-enantiomer, the attenuation is indistinguishable under the testing conditions. Accordingly, we attribute the assay discrepancy to the inherent variability in the assay system.
While the compounds appear to be active against both influenza and measles virus, they are somewhat more active against the influenza virus strain (WSN) than against the measles virus. Among the compounds surveyed, (S)-14b is the most potent compound, with EC50 values of 1–11 nM against the two viruses. Because the CC50 values of the compounds are greater than 50 µM, the upper limit of the assay, we assume that the compounds are not generally cytotoxic, giving selectivity indices (CC50/EC50) for the active enantiomers of at least 103–104.
We have also assayed the most active compounds in a human parainfluenza viral (HPIV) titer assay based on plaque assay titration. The values for (S)-1, (S)-14a, (S)-14b, and (S)-14c are 80, 13, 80, and 11 nM, respectively. These data further corroborate the broad-spectrum activity of these compounds.
Lastly, the solubilities of the compounds have been improved relative to that of 1. At all pH values examined, the aqueous solubility of 1 was below the limits of detection of the nephelometry assay (i.e., <15 µg/mL). However, compounds bearing basic amine functionalities have improved solubilities relative to 1, particularly at acidic pH values, but also at pH 7.4.
Conclusion
We have extended our previously published work on host-directed inhibitors of myxovirus replication by preparing analogues that positively address the poor aqueous solubility of 1 (JMN3-003) while simultaneously improving its potency. The compounds presented here furnish EC50 values as low as 1 nM with aqueous solubilities ranging from 15–25 µg/mL at pH 7.4 to >300 µg/mL at pH 3.0. Additionally, we have developed two complementary methods for the synthesis of each of the enantiomers of the compounds discussed and have unequivocally demonstrated that the (S)-enantiomer is more active in this series than the (R)-enantiomer. Further work from our laboratories regarding the in vivo efficacy of these compounds is underway.
Supporting Information
Contains detailed synthetic procedures and characterization data for molecules described herein, a more detailed description of the laser nephelometry assay, and data tables for the crystal structures of (S)-1 and (R)-1.
| Supporting Information File 1: Detailed synthetic procedures and characterization data. | ||
| Format: PDF | Size: 2.6 MB | Download |
Acknowledgments
This work was supported, in part, by Public Health Service Grants AI071002 and AI085328 (to R. K. P.) from the NIH/NIAID and by Public Health Service Grant HG003918-02 (to J.P.S.) from the NIH. We also thank Deborah Culver for solubility testing and Dr. John Bacsa for helpful discussions of crystal structures.
References
-
König, R.; Stertz, S.; Zhou, Y.; Inoue, A.; Hoffmann, H.-H.; Bhattacharyya, S.; Alamares, J. G.; Tscherne, D. M.; Ortigoza, M. B.; Liang, Y.; Gao, Q.; Andrews, S. E.; Bandyopadhyay, S.; Jesus, P. D.; Tu, B. P.; Pache, L.; Shih, C.; Orth, A.; Bonamy, G.; Miraglia, L.; Ideker, T.; García-Sastre, A.; Young, J. A. T.; Palese, P.; Shaw, M. L.; Chanda, S. K. Nature 2010, 463, 813–817. doi:10.1038/nature08699
Return to citation in text: [1] -
Martin, P.; Jensen, D. M. J. Gastroenterol. Hepatol. 2008, 23, 844–855. doi:10.1111/j.1440-1746.2008.05398.x
Return to citation in text: [1] -
Dixit, N. M.; Perelson, A. S. Cell. Mol. Life Sci. 2006, 63, 832–842. doi:10.1007/s00018-005-5455-y
Return to citation in text: [1] -
Buckwold, V. E.; Wei, J.; Wenzel-Mathers, M.; Russell, J. Antimicrob. Agents Chemother. 2003, 47, 2293–2298. doi:10.1128/AAC.47.7.2293-2298.2003
Return to citation in text: [1] -
Willis, R. C.; Carson, D. A.; Seegmiller, J. E. Proc. Natl. Acad. Sci. U. S. A. 1978, 75, 3042–3044. doi:10.1073/pnas.75.7.3042
Return to citation in text: [1] -
De Clercq, E. Nat. Rev. Drug Discovery 2002, 1, 13–25. doi:10.1038/nrd703
Return to citation in text: [1] -
Whitley, R. J.; Roizman, B. Lancet 2001, 357, 1513–1518. doi:10.1016/S0140-6736(00)04638-9
Return to citation in text: [1] -
Yoon, J.-J.; Krumm, S. A.; Ndungu, J. M.; Hoffman, V.; Bankamp, B.; Rota, P. A.; Sun, A.; Snyder, J. P.; Plemper, R. K. Antimicrob. Agents Chemother. 2009, 53, 3860–3870. doi:10.1128/AAC.00503-09
Return to citation in text: [1] [2] [3] [4] -
Sun, A.; Yoon, J. J.; Yin, Y.; Prussia, A.; Yang, Y.; Min, J.; Plemper, R. K.; Snyder, J. P. J. Med. Chem. 2008, 51, 3731–3741. doi:10.1021/jm701239a
Return to citation in text: [1] -
White, L. K.; Yoon, J.-J.; Lee, J. K.; Sun, A.; Du, Y.; Fu, H.; Snyder, J. P.; Plemper, R. K. Antimicrob. Agents Chemother. 2007, 51, 2293–2303. doi:10.1128/AAC.00289-07
Return to citation in text: [1] -
Sun, A.; Chandrakumar, N.; Yoon, J.-J.; Plemper, R. K.; Snyder, J. P. Bioorg. Med. Chem. Lett. 2007, 17, 5199–5203. doi:10.1016/j.bmcl.2007.06.084
Return to citation in text: [1] -
Krumm, S. A.; Ndungu, J. M.; Yoon, J.-J.; Dochow, M.; Sun, A.; Natchus, M.; Snyder, J. P.; Plemper, R. K. PLoS One 2011, 6, e20069. doi:10.1371/journal.pone.0020069
Return to citation in text: [1] [2] [3] [4] -
Yoon, J.-J.; Chawla, D.; Paal, T.; Ndungu, M.; Du, Y.; Kurtkaya, S.; Sun, A.; Snyder, J. P.; Plemper, R. K. J. Biomol. Screening 2008, 13, 591–608. doi:10.1177/1087057108321089
Return to citation in text: [1] -
Sun, A.; Ndungu, J. M.; Krumm, S. A.; Yoon, J.-J.; Thepatchri, P.; Natchus, M.; Plemper, R. K.; Snyder, J. P. ACS Med. Chem. Lett. 2011, 2, 798–803. doi:10.1021/ml200125r
Return to citation in text: [1] [2] [3] -
Pharmacokinetics Study of JMN3-003. Unpublished results.
Return to citation in text: [1] -
Carpino, L. A. J. Am. Chem. Soc. 1993, 115, 4397–4398. doi:10.1021/ja00063a082
Return to citation in text: [1] -
Nair, V. P.; Mustafa, S. M.; Mohler, M. L.; Yang, J.; Kirkovsky, L. I.; Dalton, J. T.; Miller, D. D. Tetrahedron Lett. 2005, 46, 4821–4823. doi:10.1016/j.tetlet.2005.04.143
Return to citation in text: [1] -
Hoelke, B.; Gieringer, S.; Arlt, M.; Saal, C. Anal. Chem. 2009, 81, 3165–3172. doi:10.1021/ac9000089
Return to citation in text: [1]
| 1. | König, R.; Stertz, S.; Zhou, Y.; Inoue, A.; Hoffmann, H.-H.; Bhattacharyya, S.; Alamares, J. G.; Tscherne, D. M.; Ortigoza, M. B.; Liang, Y.; Gao, Q.; Andrews, S. E.; Bandyopadhyay, S.; Jesus, P. D.; Tu, B. P.; Pache, L.; Shih, C.; Orth, A.; Bonamy, G.; Miraglia, L.; Ideker, T.; García-Sastre, A.; Young, J. A. T.; Palese, P.; Shaw, M. L.; Chanda, S. K. Nature 2010, 463, 813–817. doi:10.1038/nature08699 |
| 8. | Yoon, J.-J.; Krumm, S. A.; Ndungu, J. M.; Hoffman, V.; Bankamp, B.; Rota, P. A.; Sun, A.; Snyder, J. P.; Plemper, R. K. Antimicrob. Agents Chemother. 2009, 53, 3860–3870. doi:10.1128/AAC.00503-09 |
| 9. | Sun, A.; Yoon, J. J.; Yin, Y.; Prussia, A.; Yang, Y.; Min, J.; Plemper, R. K.; Snyder, J. P. J. Med. Chem. 2008, 51, 3731–3741. doi:10.1021/jm701239a |
| 10. | White, L. K.; Yoon, J.-J.; Lee, J. K.; Sun, A.; Du, Y.; Fu, H.; Snyder, J. P.; Plemper, R. K. Antimicrob. Agents Chemother. 2007, 51, 2293–2303. doi:10.1128/AAC.00289-07 |
| 11. | Sun, A.; Chandrakumar, N.; Yoon, J.-J.; Plemper, R. K.; Snyder, J. P. Bioorg. Med. Chem. Lett. 2007, 17, 5199–5203. doi:10.1016/j.bmcl.2007.06.084 |
| 12. | Krumm, S. A.; Ndungu, J. M.; Yoon, J.-J.; Dochow, M.; Sun, A.; Natchus, M.; Snyder, J. P.; Plemper, R. K. PLoS One 2011, 6, e20069. doi:10.1371/journal.pone.0020069 |
| 7. | Whitley, R. J.; Roizman, B. Lancet 2001, 357, 1513–1518. doi:10.1016/S0140-6736(00)04638-9 |
| 8. | Yoon, J.-J.; Krumm, S. A.; Ndungu, J. M.; Hoffman, V.; Bankamp, B.; Rota, P. A.; Sun, A.; Snyder, J. P.; Plemper, R. K. Antimicrob. Agents Chemother. 2009, 53, 3860–3870. doi:10.1128/AAC.00503-09 |
| 12. | Krumm, S. A.; Ndungu, J. M.; Yoon, J.-J.; Dochow, M.; Sun, A.; Natchus, M.; Snyder, J. P.; Plemper, R. K. PLoS One 2011, 6, e20069. doi:10.1371/journal.pone.0020069 |
| 2. | Martin, P.; Jensen, D. M. J. Gastroenterol. Hepatol. 2008, 23, 844–855. doi:10.1111/j.1440-1746.2008.05398.x |
| 3. | Dixit, N. M.; Perelson, A. S. Cell. Mol. Life Sci. 2006, 63, 832–842. doi:10.1007/s00018-005-5455-y |
| 4. | Buckwold, V. E.; Wei, J.; Wenzel-Mathers, M.; Russell, J. Antimicrob. Agents Chemother. 2003, 47, 2293–2298. doi:10.1128/AAC.47.7.2293-2298.2003 |
| 5. | Willis, R. C.; Carson, D. A.; Seegmiller, J. E. Proc. Natl. Acad. Sci. U. S. A. 1978, 75, 3042–3044. doi:10.1073/pnas.75.7.3042 |
| 18. | Hoelke, B.; Gieringer, S.; Arlt, M.; Saal, C. Anal. Chem. 2009, 81, 3165–3172. doi:10.1021/ac9000089 |
| 8. | Yoon, J.-J.; Krumm, S. A.; Ndungu, J. M.; Hoffman, V.; Bankamp, B.; Rota, P. A.; Sun, A.; Snyder, J. P.; Plemper, R. K. Antimicrob. Agents Chemother. 2009, 53, 3860–3870. doi:10.1128/AAC.00503-09 |
| 12. | Krumm, S. A.; Ndungu, J. M.; Yoon, J.-J.; Dochow, M.; Sun, A.; Natchus, M.; Snyder, J. P.; Plemper, R. K. PLoS One 2011, 6, e20069. doi:10.1371/journal.pone.0020069 |
| 14. | Sun, A.; Ndungu, J. M.; Krumm, S. A.; Yoon, J.-J.; Thepatchri, P.; Natchus, M.; Plemper, R. K.; Snyder, J. P. ACS Med. Chem. Lett. 2011, 2, 798–803. doi:10.1021/ml200125r |
| 16. | Carpino, L. A. J. Am. Chem. Soc. 1993, 115, 4397–4398. doi:10.1021/ja00063a082 |
| 17. | Nair, V. P.; Mustafa, S. M.; Mohler, M. L.; Yang, J.; Kirkovsky, L. I.; Dalton, J. T.; Miller, D. D. Tetrahedron Lett. 2005, 46, 4821–4823. doi:10.1016/j.tetlet.2005.04.143 |
| 14. | Sun, A.; Ndungu, J. M.; Krumm, S. A.; Yoon, J.-J.; Thepatchri, P.; Natchus, M.; Plemper, R. K.; Snyder, J. P. ACS Med. Chem. Lett. 2011, 2, 798–803. doi:10.1021/ml200125r |
| 13. | Yoon, J.-J.; Chawla, D.; Paal, T.; Ndungu, M.; Du, Y.; Kurtkaya, S.; Sun, A.; Snyder, J. P.; Plemper, R. K. J. Biomol. Screening 2008, 13, 591–608. doi:10.1177/1087057108321089 |
| 8. | Yoon, J.-J.; Krumm, S. A.; Ndungu, J. M.; Hoffman, V.; Bankamp, B.; Rota, P. A.; Sun, A.; Snyder, J. P.; Plemper, R. K. Antimicrob. Agents Chemother. 2009, 53, 3860–3870. doi:10.1128/AAC.00503-09 |
| 12. | Krumm, S. A.; Ndungu, J. M.; Yoon, J.-J.; Dochow, M.; Sun, A.; Natchus, M.; Snyder, J. P.; Plemper, R. K. PLoS One 2011, 6, e20069. doi:10.1371/journal.pone.0020069 |
| 14. | Sun, A.; Ndungu, J. M.; Krumm, S. A.; Yoon, J.-J.; Thepatchri, P.; Natchus, M.; Plemper, R. K.; Snyder, J. P. ACS Med. Chem. Lett. 2011, 2, 798–803. doi:10.1021/ml200125r |
© 2013 Moore et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)