Abstract
Amine radical cations are highly useful reactive intermediates in amine synthesis. They have displayed several modes of reactivity leading to some highly sought-after synthetic intermediates including iminium ions, α-amino radicals, and distonic ions. One appealing method to access amine radical cations is through one-electron oxidation of the corresponding amines under visible light photoredox conditions. This approach and subsequent chemistries are emerging as a powerful tool in amine synthesis. This article reviews synthetic applications of amine radical cations produced by visible light photocatalysis.

Graphical Abstract
Introduction
Amine radical cations, which are an odd-electron species, are of great utility in amine syntheses [1-8]. They can be formed by loss of an electron from the corresponding amines. This one-electron oxidation process has been realized by using electrochemistry [9-11], chemical oxidants [12-14], metal-catalyzed oxidation [15-18], UV light-mediated photochemistry [7,19-21], and visible light-mediated photochemistry [22,23]. Recently, the last approach has become a major research focus in organic chemistry. The enthusiasm surrounding this approach is partially driven not only by its green characteristics (i.e. using visible light), but also more importantly by its unique ability to achieve unconventional bond formation.
Like most organic compounds, amines do not absorb visible light efficiently, unless they have a chromophore (e.g., conjugated π-bond systems). Therefore, a photocatalyst is often required to initialize electron-transfer reactions with amines. Some of the frequently used photocatalysts include ruthenium [24-26] and iridium [27,28] polypyridyl complexes as well as organic dyes [29,30] that are absorbed in the visible-light region. They all share one common characteristic: a facile intersystem crossing (ISC) that allows the conversion of the initially formed singlet photoexcited state to the relatively long-lived triplet photoexcited state. The triplet photoexcited state’s long lifetime permits it to engage in single-electron transfer with organic molecules such as amines. The photoexcited state is both more oxidizing and more reducing than the ground state. It can be quenched reductively by accepting an electron from an electron donor or oxidatively by donating an electron to an electron acceptor. Amines are often used as an electron donor to reductively quench the photoexcited state while they are oxidized to amine radical cations. This single-electron transfer process was investigated intensively in the late 1970s and early 1980s because amines were used as a sacrificial electron donor in water splitting [31,32] and carbon dioxide reduction [33,34]. Since 2008, seminal works from MacMillan, Yoon, and Stephenson have reinvigorated the field of visible light photoredox catalysis [35-42]. The use of amines as both the electron donor and the substrate, rather than just the electron donor, has become a major approach to exploit synthetic utility of photogenically produced amine radical cations.
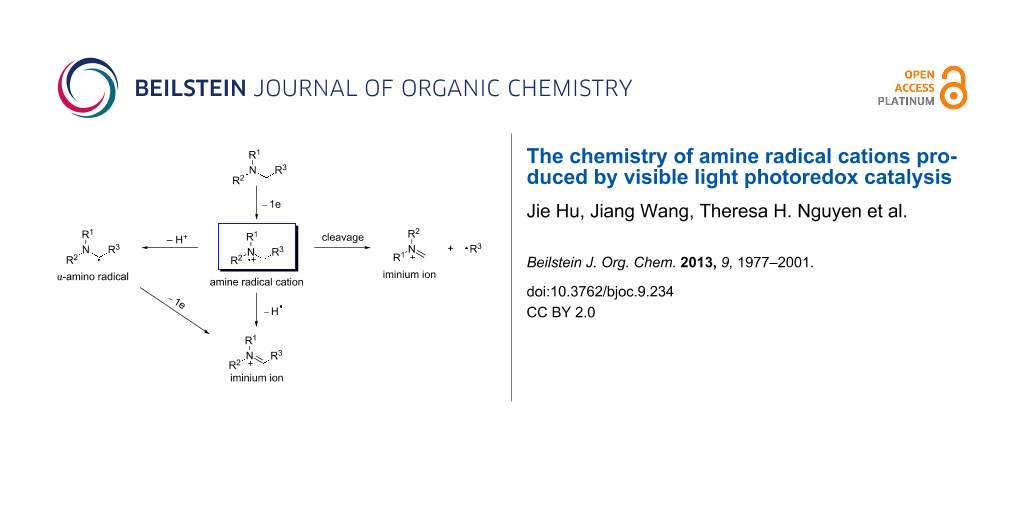
Reductive quenching of the photoexcited state of a photocatalyst (M) by amine 1 is governed by the reduction potentials of the photoexcited state and the amine (Scheme 1). The amine’s reduction potential, which can be readily measured by cyclic voltammetry, should be less positive than that of the photoexcited M. The solvent also has a significant impact on the oxidation and the subsequent reactions [43,44]. A polar solvent is generally favored for electron-transfer reactions involving amine radical cations, but identification of the optimal solvent requires experimentation. Once formed, amine radical cation 2 has been shown to have four modes of reactivity. The first mode is the back electron transfer reaction, which involves amine radical cation 2 giving back one electron to M(n−1). This is a major side reaction competing against the other productive downstream reactions of 2. To circumvent this side reaction, two approaches or a combination thereof can be exploited [45,46]. One approach involves modifying the structure of the ligand on M to retard the back electron transfer. The other involves designing fast and/or irreversible downstream reactions of 2. The second mode involves hydrogen atom abstraction from 2 to produce iminium ion 4, when a good hydrogen atom acceptor is present in the reaction. The use of amine radical cation 2 as the source of a hydrogen radical has been applied to a number of visible light-mediated reductions such as reductive dehalogenation [47-51], reductive radical cyclization [52-54], reduction of activated ketones [49], and reduction of aromatic azides [55]. The third mode involves deprotonation of amine radical cation 2 to form α-amino radical 3, which is converted to iminium ion 4 by another one-electron oxidation. The acidifying effect of one-electron oxidation on the α-C–H bond remains debatable [56-60]. The rate for deprotonation of amine radical cation 2 has been measured experimentally by several groups, and a broad range has been obtained [61,62]. α-Amino radical 3 is strongly reducing [45,63], thus making the second one-electron oxidation facile. The last mode involves cleavage of a C–C bond α to the nitrogen atom, yielding a neutral free radical 6 and iminium ion 5. Iminium ion 4, an excellent electrophile, is amenable to interception by a variety of nucleophiles to directly install a new bond at the position α to the nitrogen atom. In contrast, α-amino radical 3 is nucleophilic. It tends to add to electron-deficient alkenes to form a C–C bond, also at the position α to the nitrogen atom.
![[1860-5397-9-234-i1]](/bjoc/content/inline/1860-5397-9-234-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Amine radical cations’ mode of reactivity.
Scheme 1: Amine radical cations’ mode of reactivity.
This review will summarize the work to date on the use of amine radical cations generated under visible light photoredox conditions as a key intermediate to trigger downstream reactions. The work is grouped based on the key intermediates (iminium ions and α-amino radicals) or processes (cleavage of C–C and N–N bonds) involved. The chemistries that have focused on the use of amines as a sacrificial electron donor only or as a hydrogen radical donor only will not be discussed in the review. These chemistries have been recently reviewed [22,23,35-42]. Photooxidation of amines to amine radical cations can also be achieved using UV light with a sensitizer. This approach and subsequent chemistries are also outside the scope of this review. Interested readers are referred to these reviews [7,19-21].
Review
Iminium ions
Intercepted by carbon nucleophiles
One of the major modes of reactivity for amine radical cations is their conversion to the powerful electrophilic iminium ions, which can be intercepted by a range of pronucleophiles to form a number of important bonds such as C–C, C–N, C–O, and C–P. The chemistry involving iminium ions has seen the most synthetic applications so far.
The Whitten group provided some early studies to establish the conversion of amine radical cations to iminium ions. In 1980, Giannotti and Whitten reported that irradiation of triethylamine with three ruthenium polypyridyl complexes using visible light in the presence of water yielded acetaldehyde, presumably formed by the hydrolysis of iminium ion 12 (Scheme 2) [46]. They proposed that reductive quenching of the photoexcited Ru(II) complex by triethylamine produced Ru(I) and amine radical cation 9. Then amine radical cation 9 can either abstract a hydrogen atom from the solvent (CH3CN) to form carbon radical 10, or lose a proton to another molecule of triethylamine to form α-amino radical 11. Carbon radical 10 is converted to α-amino radical 11 by abstracting a hydrogen atom from a second molecule of triethylamine and CH3CN is ultimately regenerated. Finally, one electron oxidation of α-amino radical 11 furnishes iminium ion 12 that is hydrolyzed to acetaldehyde. Although the authors were not able to detect amine radical cation 12 spectroscopically, they were able to use ESR (electron spin resonance) techniques to detect Ru(I) and α-amino radical 11 with the aid of a spin trap, nitrosodurene.
![[1860-5397-9-234-i2]](/bjoc/content/inline/1860-5397-9-234-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reductive quenching of photoexcited Ru complexes by Et3N.
Scheme 2: Reductive quenching of photoexcited Ru complexes by Et3N.
In 2010, Stephenson and coworkers reported a visible light-mediated aza-Henry reaction that harnesses the synthetic potential of iminium ions. Using only 1 mol % of [Ir(ppy)2(dtbbpy)](PF6) and visible light, a variety of N-aryltetrahydroisoquinolines were oxidatively coupled with nitroalkanes to provide the aza-Henry products in excellent yields (Scheme 3) [64]. They suggested that reductive quenching of the Ir(III) photoexcited state by N-aryltetrahydroisoquinolines 13 leads to the formation of amine radical cation 14 and the powerful reducing agent Ir(II) (Ir(III)/Ir(II), −1.51 V vs SCE). The Ir(II) catalyst then reduces nitromethane or oxygen to a radical anion that may abstract a hydrogen atom from amine radical cation 14 to form the iminium ion 15. Interception of the iminium ion by the anion of nitromethane affords the aza-Henry product 16.
![[1860-5397-9-234-i3]](/bjoc/content/inline/1860-5397-9-234-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Oxygen has been the most often used stoichiometric oxidant in the formation of iminium ions under photoredox conditions. However, this use has some limitations. The catalyst turnover mediated by O2 is often slow, resulting in long reaction time. O2 can also intercept α-amino radicals, one of the key intermediates in the formation of iminium ions, to produce amides and thus compromise the formation of iminium ions [65,66]. The Stephenson group sought an alternative stoichiometric oxidant to overcome the limitations encountered by O2 (Scheme 4) [58]. BrCCl3 was identified as such an alternative and iminium ions were produced cleanly within 3 hours. A broad range of nucleophiles, including nitroalkanes, was shown to add to iminium ions. The authors proposed two possible mechanisms for the formation of iminium ions based on the two divergent pathways for the conversion of amine radical cations to iminium ions. The first mechanism is based on the pathway involving abstraction of a hydrogen atom from amine radical cation 14. The hydrogen atom acceptor is a trichloromethyl radical, which is formed via one-electron reduction of BrCCl3 by Ru(I). The second is centered on the pathway involving deprotonation of amine radical cation 14 followed by one-electron oxidation. BrCCl3 is the one-electron oxidant via electron transfer or atom transfer. The trichloromethyl radical, which is generated by this oxidation, then abstracts a hydrogen atom of another molecule of N-aryltetrahydroisoquinoline to produce the α-amino radical 17, which once again enters the radical chain process with BrCCl3.
![[1860-5397-9-234-i4]](/bjoc/content/inline/1860-5397-9-234-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Formation of iminium ions using BrCCl3 as stoichiometric oxidant.
Scheme 4: Formation of iminium ions using BrCCl3 as stoichiometric oxidant.
König and coworkers showed that the same aza-Henry reaction can be catalyzed by the organic dye Eosin Y to afford the aza-Henry product 18 (Scheme 5) [67]. In addition to nitroalkanes, dialkyl malonates and malononitrile can be used as pronucleophiles to provide β-diester amine 19 and α-aminonitrile 20. The authors proposed a mechanism similar to that proposed by Stephenson and coworkers for the aza-Henry reaction catalyzed by the Ir complex (Scheme 3). The Tan group simultaneously reported that another organic dye, Rose Bengal (RB), can be used in place of Eosin Y to catalyze the aza-Henry reaction [68].
![[1860-5397-9-234-i5]](/bjoc/content/inline/1860-5397-9-234-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Oxidative functionalization of N-aryltetrahydroisoquinolines using Eosin Y.
Scheme 5: Oxidative functionalization of N-aryltetrahydroisoquinolines using Eosin Y.
The Wu group concurrently developed the Eosin Y-catalyzed aza-Henry reaction as reported by König and also performed mechanistic studies on the reaction. Their proposed catalytic cycle for the reaction is detailed in Scheme 6 [69]. Wu and coworkers were able to obtain experimental evidence to lend support to some of the key steps in the catalytic cycle. An oxygen uptake experiment showed that 0.75 equiv of O2 was consumed for the complete conversion of N-phenyltetrahydroisoquinoline 13. This data strongly supports the role of O2 as the stoichiometric oxidant. Flash photolysis studies established that reductive quenching of the triplet excited state of Eosin Y by N-phenyltetrahydroisoquinoline 13 produced the Eosin Y radical anion. An ESR study on the irradiated solution of DMPO (5,5-dimethyl-1-pyrroline-N-oxide), Eosin Y, and N-phenyltetrahydroisoquinoline in air-saturated CH3CN detected the adduct of superoxide to DMPO. In contrast, an ESR study on the same solution but with DMPO being replaced by TEMP (2,3,6,6-tetramethylpiperidine) did not detect TEMPO, the oxidation product of TEMP by singlet oxygen. However, TEMPO was detected in the absence of N-phenyltetrahydroisoquinoline. The results from these ESR studies are consistent with the notion that singlet oxygen is not formed in the presence of N-phenyltetrahydroisoquinoline and the Eosin Y radical anion reduces oxygen to superoxide. Finally, the yield of the product 18 increased when the reaction mixture was kept stirring in the dark after 4 h irradiation. This observation supports the formation of hydroperoxide intermediate 21.
![[1860-5397-9-234-i6]](/bjoc/content/inline/1860-5397-9-234-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthetic and mechanistic studies of Eosin Y-catalyzed aza-Henry reaction.
Scheme 6: Synthetic and mechanistic studies of Eosin Y-catalyzed aza-Henry reaction.
Tan and coworkers employed a cocatalyst system composed of the organic dye Rose Bengal and graphite oxide (GO) for α-cyanation of N-aryltetrahydroisoquinolines (Scheme 7) [70]. The use of GO as carbocatalyst, pioneered by the Bielawski group, has been shown to facilitate a variety of reactions including oxidation, reduction, dehydration, and C–C bond formation [71-74]. GO was found to improve the yields of the α-cyanation reaction, and this was the first example of using GO to promote visible light-mediated reactions. The synergistic effect between carbocatalysis and visible light-mediated photocatalysis has the potential to be further explored in other photocatalyzed reactions.
![[1860-5397-9-234-i7]](/bjoc/content/inline/1860-5397-9-234-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Oxidative functionalization of N-aryltetrahydroisoquinolines using RB and GO.
Scheme 7: Oxidative functionalization of N-aryltetrahydroisoquinolines using RB and GO.
Since visible light photocatalysis is often orthogonal to or compatible with a number of common catalytic processes, merging it with another type of catalysis has become a recent development in the field of visible light photocatalysis. One direct benefit of this dual catalysis approach is to allow expansion of the types of nucleophiles capable of adding to the iminium ions generated under photoredox conditions.
In 2011, Rueping and coworkers described a dual catalytic system combining photoredox and Lewis base catalysis for the functionalization of C–H bonds α to the nitrogen atom of N-aryltetrahydroisoquinoline 13 (Scheme 8) [65]. In the presence of a Lewis base, a ketone is converted to enamine nucleophile 28 in situ, which is then added to photogenically formed iminium ion 27 to yield the Mannich product 23. The Mannich reaction was sluggish without the Lewis base, and a side reaction, formation of the oxidized isoquinoline, became significant. The choice of Lewis base was found to be also crucial for the outcome of the reaction and proline was more effective than pyrrolidine. Additionally, to maximize the yields, the optimal rates for the two catalytic processes need to be similar. Since formation of the iminium ions is much faster than the addition of the enamine nucleophiles, higher yields were realized with slower formation of the iminium ions. This was achieved by use of [Ru(bpy)3](PF6)2 in conjunction with a weak light source (5 W fluorescence bulb).
![[1860-5397-9-234-i8]](/bjoc/content/inline/1860-5397-9-234-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Merging Ru-based photoredox catalysis and Lewis base catalysis for the Mannich reaction.
Scheme 8: Merging Ru-based photoredox catalysis and Lewis base catalysis for the Mannich reaction.
The Che group synthesized a photoactive gold(III) complex that was shown to catalyze α-cyanation of N-aryltetrahydroisoquinolines [75]. Very recently, Zhu and coworkers used an analogous gold(III) complex to catalyze the reactions similar to those reported by the Rueping group (Scheme 9) [76]. A 5 W blue LED was used as the light source. One advantage of using the gold complex over [Ru(bpy)3](PF6)2 is that long-chain aliphatic ketones work much better using the former catalyst. Other types of pronucleophiles such as malonates are also effective in the Mannich reaction.
![[1860-5397-9-234-i9]](/bjoc/content/inline/1860-5397-9-234-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Merging Au-based photoredox catalysis and Lewis base catalysis for the Mannich reaction.
Scheme 9: Merging Au-based photoredox catalysis and Lewis base catalysis for the Mannich reaction.
The Rueping group extended the concept of dual catalysis by merging visible light photocatalysis with a metal-catalyzed process (Scheme 10) [77]. To make this approach work, several hurdles need to be addressed. First, a labile carbon–metal bond is desired in order to have an efficient turnover of the metal. Second, the metal complex needs to be compatible with the strongly reducing intermediates (e.g., superoxide) produced in the photocatalytic cycle. Third, the rates of the two catalytic cycles have to be comparable, as iminium ions are known to be converted to amides by superoxide [65,66]. Rueping and coworkers discovered that using a weak light source (5 W fluorescent bulb), copper acetylide 31, formed in situ by (MeCN)4CuPF6, was added efficiently to the photogenically-produced iminium ion 27a, thus achieving the formation of Csp3–Csp bonds.
![[1860-5397-9-234-i10]](/bjoc/content/inline/1860-5397-9-234-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Merging Ru-based photoredox catalysis and Cu-catalyzed alkynylation reaction.
Scheme 10: Merging Ru-based photoredox catalysis and Cu-catalyzed alkynylation reaction.
Rovis and coworkers recently developed another mode of dual catalysis involving visible light photocatalysis with chiral N-heterocyclic carbene catalysis, which allows catalytic asymmetric α-acylation of N-aryltetrahydroisoquinoline 13 with an aliphatic aldehyde (Scheme 11) [78]. In the presence of a N-heterocyclic carbene (NHC), the aldehyde is converted to a chiral acyl anion or homoenolate equivalent 37, which is then added to the iminium ion 27 to form Csp3–Csp2 bonds asymmetrically. It is interesting to note that the use of m-dinitrobenzene (m-DNB) is critical to achieve the desirable conversion and yield of the expected product 32. m-DNB is proposed to act as an electron acceptor to promote an oxidative quenching cycle of Ru(bpy)32+* to Ru(bpy)33+. N-aryltetrahydroisoquinoline 13 is then oxidized by Ru(bpy)33+. This is in contrast to the majority of reported examples in which the conversion to the iminum ion such as 27 is realized in a reductive quenching cycle of Ru(bpy)32+* to Ru(bpy)31+, where N-aryltetrahydroisoquinoline 13 is oxidized by Ru(bpy)32+* instead.
![[1860-5397-9-234-i11]](/bjoc/content/inline/1860-5397-9-234-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Merging Ru-based photoredox catalysis and NHC catalysis.
Scheme 11: Merging Ru-based photoredox catalysis and NHC catalysis.
Xiao [79] and Rueping [80] independently reported that when tetrahydroisoquinolines (e.g., 41 and 45) were substituted with a methylene group attached to one or two esters, the initially formed iminium ions were readily converted to azomethine ylides. They subsequently underwent 1,3-dipolar cycloaddition with a range of dipolarophiles to form fused pyrrolidines 43 and 47 (Scheme 12). Xiao also showed that the pyrrolidine ring of 43 could be further oxidized to a fused pyrrole 44 under the same photoredox conditions or by treatment with NBS. Both Ru(bpy)3Cl2 and Ir(bpy)(ppy)2 were found to be effective catalysts.
![[1860-5397-9-234-i12]](/bjoc/content/inline/1860-5397-9-234-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: 1,3-Dipolar cycloaddition of photogenically formed azomethine ylides.
Scheme 12: 1,3-Dipolar cycloaddition of photogenically formed azomethine ylides.
A plausible mechanism for the 1,3-dipolar cycloaddition is shown in Scheme 13. The reaction commences with oxidation of tetrahydroisoquinoline 41 to amine radical cation 48 by the photoexcited state of Ru2+. Subsequently, abstraction of a hydrogen atom α to the nitrogen atom of 48 yields iminium ion 49, which is then converted to azomethine ylide 50 by loss of a proton. 1,3-Dipolar cycloaddition of 50 with a dipolarophile 46 furnishes fused pyrrolidine 51 that is further oxidized to pyrrole 52.
![[1860-5397-9-234-i13]](/bjoc/content/inline/1860-5397-9-234-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Plausible mechanism for photoredox 1,3-dipolar cycloaddition.
Scheme 13: Plausible mechanism for photoredox 1,3-dipolar cycloaddition.
The Zhu group discovered that the use of α-ketoester 53 as a pronucleophile to intercept the iminium ion of 13 triggered a new cascade reaction en route to fused isoxazolidine 54 in excellent diastereoselectivity (>20:1, Scheme 14) [81]. Alcohols were found to be the solvent of choice for this reaction. Among the three alcohols screened, iPrOH was more effective than MeOH or EtOH, resulting in a shorter reaction time. The addition of a catalytic amount of TfOH had marginally beneficial effects on the reaction time and yields. Interestingly, depending on the electronic character of the N-aryl group, [Ir(ppy)2(dtbbpy)](BF4) or Ru(bpy)3Cl2 was used to obtain optimal yields. The former catalyst worked better with electron-poor N-aryl groups while the latter was more effective for electron-rich N-aryl groups.
![[1860-5397-9-234-i14]](/bjoc/content/inline/1860-5397-9-234-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Photoredox-catalyzed cascade reaction for the synthesis of fused isoxazolidines.
Scheme 14: Photoredox-catalyzed cascade reaction for the synthesis of fused isoxazolidines.
The authors proposed a possible mechanism that starts with reductive quenching of the photoexcited state of Ru(II) or Ir(III) by N-phenyltetrahydroisoquinoline 13 (Scheme 15). The initially formed amine radical cation 14 is converted to iminium ion 15 by abstraction of a hydrogen atom directly. The addition of the enol form of α-ketoester 59 to 15 furnishes the Mannich adduct 60. A retro-aza-Michael reaction via enol 61 allows cleavage of the C–N bond to yield secondary aniline 62. Aniline 62 is first oxidized to imine 63, which is further oxidized to nitrone 64. Finally, an intramolecular 1,3-dipolar cycloaddition of 64 furnishes isoxazolidine 55.
![[1860-5397-9-234-i15]](/bjoc/content/inline/1860-5397-9-234-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Plausible mechanism for the photoredox-catalyzed cascade reaction.
Scheme 15: Plausible mechanism for the photoredox-catalyzed cascade reaction.
Tetrahydroisoquinolines are arguably the most exploited amines in visible light photoredox catalysis. However, efforts towards expanding the scope of amines have been recently reported. Li [82] and Rueping [83] independently reported that N-arylglycine derivatives 65 are viable substrates (Scheme 16). They are presumably converted to imines 65a that are intercepted by indoles to give the Mannich-like adducts 67. The conditions used by Li were 10 mol % Ru(bpy)3Cl2 and 1 atm O2 at 40 °C with a 5 W blue LED as the light source. N-arylglycine derivatives 65, including esters and ketones, were successfully converted to the products 67. Rueping used Ir(ppy)2(bpy)PF6 as the photocatalyst, air, and an 11 W fluorescent bulb as the light source. Additionally, Zn(OAc)2 was employed as a Lewis acid cocatalyst. It was postulated that Zn(OAc)2 facilitates the conversion of the initially formed amine radical cation to the imine 65a and subsequently activates the imine for nucelophilic attack. N-arylglycine esters and N-arylglycine derived dipeptides worked quite well under these conditions. However, the ketones failed to provide the desired products.
![[1860-5397-9-234-i16]](/bjoc/content/inline/1860-5397-9-234-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Photoredox-catalyzed α-arylation of glycine derivatives.
Scheme 16: Photoredox-catalyzed α-arylation of glycine derivatives.
Amides 68 are generally much more difficult to be oxidized than amines. Their reduction potentials range from 1.2–1.5 V (vs SCE) for tertiary amides to 2.0 V (vs SCE) for primary amides [84] which makes them less susceptible to oxidation by the photoexcited state of Ru(II) or Ir(III) complexes (Ru(bpy)32+*/Ru1+: 0.77 V vs SCE; Ir(ppy)2(dtbbpy)+*/Ir(ppy)2(dtbbpy), 0.66 V vs SCE) [35]. Stephenson and coworkers devised a strategy by reversing the order of oxidation and C–H abstraction to overcome this issue (Scheme 17) [85]. The first intermediate formed is a strongly reducing α-amino radical 68a that is oxidizable by the photoexcited state of Ru(II) or Ir(III). The α-amino radical 68a is formed via C–H abstraction by the sulfate radical anion (SO4−·), which is generated by exposure of Ru2+* to persulfate (S2O82−), an oxidative quencher. Electron-rich arenes and indoles are then added to the N-acyliminium ions 68b to provide the amidoalkylation products 69. Alternatively, the use of only persulfate at 55 °C afforded the same products. However, higher yields and better selectivities were generally observed with the photocatalytic process.
![[1860-5397-9-234-i17]](/bjoc/content/inline/1860-5397-9-234-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Photoredox-catalyzed α-arylation of amides.
Scheme 17: Photoredox-catalyzed α-arylation of amides.
Intercepted by nitrogen, oxygen, or phosphorus nucleophiles
In addition to carbon nucleophiles, heteronucleophiles including nitrogen, oxygen, and phosphorus are susceptible to interception of the photogenically formed iminium ions. The Xiao group developed a highly diastereroselective route to substituted tetrahydroimidazoles 72 based on intramolecular interception of the iminium ions by a tethered sulfonamide (71, Scheme 18) [86]. Ru(bpy)3Cl2 was employed as the photocatalyst with oxygen as the stoichiometric oxidant. The use of a base in an alcohol solvent, such as MeOH, was also the key to the success of this reaction. The diastereoselectivities were greatly improved by prolonging the reaction time, which would allow for epimerization leading to the thermodynamically more stable products. The starting materials, 1,2-diamines 70, were readily prepared from natural amino acids in enantiomerically pure form.
![[1860-5397-9-234-i18]](/bjoc/content/inline/1860-5397-9-234-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Intramolecular interception of iminium ions by sulfonamides.
Scheme 18: Intramolecular interception of iminium ions by sulfonamides.
Xiao and coworkers then applied the same strategy to prepare two other types of heterocycles, isoquino[2,1-a][3,1]oxazine and isoquino[2,1-a]pyrimidine (75, Scheme 19) [87]. The use of [Ir(ppy)2(dtbbpy)](PF6) with air as the external oxidant was found to be optimal for catalyzing the reaction. Later, the Marvin group reported an identical synthesis of isoquino[2,1-a][3,1]oxazine using Ru(bpy)3Cl2 instead [88]. The tethered nucleophiles, primary alcohols or sulfonamides, are part of the N-aryl group of tetrahydroisoquinolines 73. Similar to the synthesis of tetrahydroimidazoles 72, MeOH was the optimal solvent. No external base was needed when the alcohol was the nucleophile. However, if the sulfonamide was the nucleophile, an external base such as t-BuOK was required presumably to increase the nucleophilicity of the sulfonamide.
![[1860-5397-9-234-i19]](/bjoc/content/inline/1860-5397-9-234-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Intramolecular interception of iminium ions by alcohols and sulfonamides.
Scheme 19: Intramolecular interception of iminium ions by alcohols and sulfonamides.
The Rueping group trapped the iminium ions using phosphites 76 to produce α-amino phosphonates 78 (Scheme 20) [89]. [Ir(ppy)2(bpy)](PF6) was found to be the most effective catalyst. Interestingly, a biphasic mixture of toluene and water turned out to be the optimal solvent. The often-observed byproducts, amides derived from over-oxidation of the iminium ions, were suppressed [65,66]. The reactions were also sensitive to the steric and electronic nature of phosphites. Phosphites are quite acidic; their pKas are similar to alcohols. Less sterically hindered phosphites reacted faster as did more acidic phosphites (e.g., diphenylphosphite).
![[1860-5397-9-234-i20]](/bjoc/content/inline/1860-5397-9-234-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Intermolecular interception of iminium ions by phosphites.
Scheme 20: Intermolecular interception of iminium ions by phosphites.
The König group applied the organic dye Eosin Y as the photocatalyst to catalyze the same reactions (Scheme 21) [67]. The reactions were irradiated in DMF with green LED light, which overlapped with the λmax of Eosin Y. The yields are comparable for the two catalyst systems, but the reactions catalyzed by Eosin Y are much faster (note: the conclusion is based on 2 mol % Eosin Y vs 1 mol % [Ir(ppy)2(bpy)](PF6)).
![[1860-5397-9-234-i21]](/bjoc/content/inline/1860-5397-9-234-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Photoredox-catalyzed oxidative phosphonylation by Eosin Y.
Scheme 21: Photoredox-catalyzed oxidative phosphonylation by Eosin Y.
α-Amino radicals
α-Amino radicals are another class of downstream intermediates produced from amine radical cations. They are also the key intermediate in one of two potential pathways for the conversion of amine radical cations to iminium ions (Scheme 1). In contrast to the electrophilic nature of iminum ions, α-amino radicals are nulceophilic. They tend to add to Michael acceptors in a 1,4 fashion. Since the addition is overall redox neutral, no external oxidant is required. Most of these addition reactions are conducted under degassing conditions. This is in contrast to the chemistries involving iminium ions, which are often performed with exposure to air or oxygen. The reactivity umpolung at the carbon α to the nitrogen atom has expanded the repertoire of amine radical cations’ modes of reactivity. Compared to iminium ions, α-amino radicals have been much less exploited as synthetic intermediates. Their synthetic applications remain limited.
Pandey and Reiser revealed that α-amino radicals derived from N-aryltetrahydroisoquinolines were added intermolecularly to Michael acceptors (Scheme 22) [66]. A blue LED was used as the light source. Both Ru(bpy)3Cl2 and [Ir(ppy)2(dtbbpy)](PF6) were found to catalyze the reactions. However, in some of the examples, the Ir catalyst gave better yields. Mechanistically, reductive quenching of the photoexcited Ru(II) or Ir(III) complex by N-aryltetrahydroisoquinolineamine yields amine radical cation 14, which is converted to α-amino radical 17. Conjugated addition of 17 to methyl vinyl ketone produces radical 79, which is reduced by the Ru(I) or Ir(II) complex with concomitant regeneration of the Ru(II) or Ir(III) complex. Protonation of the resulting enolate furnishes the adduct 80, thus completing the catalytic cycle. The authors performed two control studies to probe the involvement of the α-amino radical 17. The first study was to irradiate N-phenyltetrahydroisoquinoline 13 in the absence of the Michael acceptor under otherwise identical conditions. The dimer, 81, was formed as a mixture of diastereomers. The second study involved irradiation of N-phenyltetrahydroisoquinoline 13 only without degassing the reaction solution. The amide, 82, was produced instead. Both findings lend credence to the intermediacy of the α-amino radical 17.
![[1860-5397-9-234-i22]](/bjoc/content/inline/1860-5397-9-234-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Conjugated addition of α-amino radicals to Michael acceptors.
Scheme 22: Conjugated addition of α-amino radicals to Michael acceptors.
The Yoon group independently discovered that the efficiency of the same Michael reaction was greatly improved in the presence of a Brønsted acid (Scheme 23) [90]. Some of the improvements included shorter reaction time, higher yields, and use of a weaker light source (CFL). The most effective acid catalysts, of which TFA was found to be optimal, lie within a narrow range of pKa values. The authors suggested that TFA protonates the enone 83, thus accelerating the addition of the α-amino radical to the enone (84).
![[1860-5397-9-234-i23]](/bjoc/content/inline/1860-5397-9-234-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Conjugated addition of α-amino radicals to Michael acceptors assisted by a Brønsted acid.
Scheme 23: Conjugated addition of α-amino radicals to Michael acceptors assisted by a Brønsted acid.
The Nishibayashi group reported that α-amino radicals generated from a different class of amines, anilines 87, were also added intermolecularly to Michael acceptors 86 (Scheme 24) [91]. In this reaction, the Michael acceptors 86 were limited to those activated by two electron-withdrawing groups. [Ir(ppy)2(dtbbpy)](BF4) was found to be the most effective photocatalyst. Solvents were also critical to the outcome of the reaction; NMP produced much higher yields of the products 89 than DMF, while no products were formed in MeCN or MeOH. The authors interrogated the intermediacy of the α-amino radical 88 by treatment of diphenylmethylaniline with a Michael acceptor incorporating a cyclopropyl ring 95. The ring-opening product 98 was isolated in 64% yield, which is consistent with the involvement of the α-amino radical 88.
![[1860-5397-9-234-i24]](/bjoc/content/inline/1860-5397-9-234-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Conjugated addition of α-amino radicals derived from anilines to Michael acceptors.
Scheme 24: Conjugated addition of α-amino radicals derived from anilines to Michael acceptors.
Oxygen has been suggested to play multiple roles in the oxidation of amines under photoredox conditions (vide supra). The Rueping group recently reported a new role that oxygen played in the intermolecular addition of α-amino radicals to Michael acceptors (Scheme 25) [92]. Oxygen was found to act as a chemical switch to two competing reaction pathways from the same starting anilines. Irradiation of a degassed solution of aniline 99, 2-benzylidenemalononitrile 100, 5 mol % Ir(ppy)2(bpy)](PF6) in MeCN furnished the typical Michael adduct 102. This result is similar to those reported by Pandey and Reiser [66], and Nishibayashi [91]. However, when the irradiation was conducted in the presence of air, a different reaction pathway occurred, resulting in the formation of N-alkyltetrahydroquinoline 101. The two reaction pathways diverge from the radical intermediate 105 generated from the Michael addition of α-amino radical 104 to 2-benzylidenemalononitrile 100. Without oxygen, the radical undergoes a one-electron reduction by Ir(II) to produce a stabilized anion, which is protonated to afford the Michael adduct 106. Alternatively, the radical is added onto arene to form a cyclohexadienyl radical 107. This step is reversible in the absence of oxygen. However, in the presence of oxygen, superoxide is formed via one-electron reduction of oxygen by Ir(II). The cyclohexadienyl radical 107 is converted to the cyclization product 108 irreversibly by giving one electron and one proton to the superoxide.
![[1860-5397-9-234-i25]](/bjoc/content/inline/1860-5397-9-234-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Oxygen switch between two pathways involving α-amino radicals.
Scheme 25: Oxygen switch between two pathways involving α-amino radicals.
The Nishibayashi group also successfully trapped α-amino radicals derived from N-aryltetrahydroquinolines and N-arylindolines using di-tert-butyl azodicarboxylate 110 to form N,N-acetal products 111 (Scheme 26) [93]. Functionalization of the sp3 C–H bond α to the nitrogen atom in tetrahydroquinolines and indolines via iminium ions is challenging because the corresponding iminium ions are enolizable and thus tend to tautomerize to enamines [94,95] and/or aromatize [96,97]. The authors adopted a strategy to bypass the iminium ions and use α-amino radicals such as 112 instead to construct C–N bonds. Treatment of N,N-acetal product 111 with Grignard reagents (Scheme 26, entry 1) or indoles in the presence of TsOH (Scheme 26, entry 2) provided nucleophilic substitution products at the α carbon. This provides an indirect approach for α-C–H functionalization of N-aryltetrahydroquinolines and N-arylindolines. Based on the feasibility of oxidation of aromatic amines as well as reduction of di-tert-butyl azodicarboxylate (110) by the photoexcited Ir(III) complex [98,99], the authors favored a mechanism that does not involve the direct addition of α-amino radical 112 to di-tert-butyl azodicarboxylate (110). Oxidation of N-phenyltetrahydroquinoline by the photoexcited Ir(III) complex followed by deprotonation provides α-amino radical 112 with the concomitant formation of the Ir(II) complex. Di-tert-butyl azodicarboxylate (110) is reduced by the Ir(II) complex to generate radical anion 113, which couples with α-amino radical 112 to yield nitrogen anion 114. Concurrently, the Ir(III) complex is regenerated. Protonation of 114 furnishes N,N-acetal 111.
![[1860-5397-9-234-i26]](/bjoc/content/inline/1860-5397-9-234-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: Interception of α-amino radicals by azodicarboxylates.
Scheme 26: Interception of α-amino radicals by azodicarboxylates.
α-Amino radicals have been mainly used in conjugate addition reactions. Recently, the MacMillan group has nicely expanded the scope of the reactions to include addition of the radicals to aryl rings (Scheme 27) [100]. Using Ir(ppy)3 as the photocatalyst and a 26 W fluorescent light bulb as the light source, cyclic amines with a variety of ring sizes and acyclic amines underwent the α-arylation reaction to provide benzylic amines. The arylating reagents were benzonitriles substituted with an electron-withdrawing group. The nitrile group functioned as the leaving group. In some classes of five-membered heteroaromatics, a chloride was capable of replacing the nitrile group as the leaving group.
![[1860-5397-9-234-i27]](/bjoc/content/inline/1860-5397-9-234-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
The authors proposed a mechanistic pathway that is initiated by oxidative quenching of the photoexcited state of Ir(ppy)3 by benzonitrile 121 to generate radical anion 123 and Ir4+(ppy)3 (Scheme 28). Amine 122 is then oxidized to amine radical cation 124 by Ir4+(ppy)3 that is reduced to the initial catalyst, Ir(ppy)3. Deprotonation of amine radical cation 124 by NaOAc produces α-amino radical 125, which is coupled with radical anion 123 to form the key C–C bond in 126. Finally, aromatization via expulsion of the nitrile group provides benzylic amine 127.
![[1860-5397-9-234-i28]](/bjoc/content/inline/1860-5397-9-234-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: Plausible mechanism for α-arylation of amines.
Scheme 28: Plausible mechanism for α-arylation of amines.
Cleavage of C–C and N–N bonds
The dominant reaction pathway involving the photogenically formed amine radical cations is deprotonation at the carbon α to the nitrogen atom to produce the strongly reducing α-amino radicals (e.g., 128 to 129). α-Amino radicals can be then intercepted by Michael acceptors or undergo one-electron oxidation to yield iminium ions (vide supra). An alternative yet much less exploited reaction pathway concerning amine radical cations 129 is the cleavage of the C–C bond α to the nitrogen atom to generate a neutral carbon radical (e.g., 130) and an iminium ion (e.g., 131). The iminum ion is subsequently reduced to α-amino radical 132 by Ru(I). Back in 1986, the Whitten group established this pathway by irradiation of three substituted tertiary amines with Ru[4,4’-CO2Et(bpy)]3(PF6)2 respectively using visible light (Scheme 29) [101]. The identity of carbon radicals 130a and 130b was established by trapping them with a spin trap and then analyzing using EPR. Additionally, detection of benzaldehyde by HPLC and VPC provided further evidence for their formation. In contrast, no products from the amine half (e.g., 131 and 132) were detected.
![[1860-5397-9-234-i29]](/bjoc/content/inline/1860-5397-9-234-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 29: Photoinduced C–C bond cleavage of tertiary amines.
Scheme 29: Photoinduced C–C bond cleavage of tertiary amines.
Li and Wang recently applied this cleavage reaction to 1,2-diamines, simultaneously generating two classes of synthetically useful intermediates, iminium ions (e.g., 133, Scheme 30) and α-amino radicals (e.g., 134, Scheme 30) [102]. The authors then exploited the synthetic utility of these two classes of intermediates. Irradiation of nitroalkanes, TMEDA, and Ru(bpy)3Cl2 in 1 atm oxygen afforded the aza-Henry products 135, presumably by trapping the Me2N=CH2 iminium ion 133 that is formed by cleaving TMEDA (Scheme 30, entry 1). Separately, irradiation of 2-hydroxyethylacrylate (HEA), TMEDA, and Ru(bpy)3Cl2 in air produced a polymer incorporating a dimethylamino group (136, Scheme 30, entry 2). The dimethylamino radical, the other intermediate generated by cleaving TMEDA, most likely induced the polymerization. The chemistries involving the iminium ions and α-amino radicals, generated under visible light photoredox conditions, are often limited by the substrate scope of the amine precursors, since aromatic amines are typically required (vide supra). The cleavage reaction, as demonstrated by Li and Wang’s work, has the potential to produce different types of iminium ions and α-amino radicals that are not accessible by oxidizing amines directly.
![[1860-5397-9-234-i30]](/bjoc/content/inline/1860-5397-9-234-i30.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 30: Photoredox cleavage of C–C bonds of 1,2-diamines.
Scheme 30: Photoredox cleavage of C–C bonds of 1,2-diamines.
The reaction is proposed to proceed through the initial oxidation of TMEDA to amine radical cation 137 by the photoexcited state of the Ru(II) complex (Scheme 31). Amine radical cation 137 subsequently induces cleavage of the C–C bond α to the nitrogen atom to form iminium ion 133 and α-amino radical 134 concurrently. By carefully selecting reagents/conditions, either reactive intermediate can selectively participate in the designated reaction. As shown in Li and Wang’s work, iminium ion 133 is intercepted by nitroalkane to afford the aza-Henry product 135 while α-amino radical 134 is used to initialize radical polymerization of HEA.
![[1860-5397-9-234-i31]](/bjoc/content/inline/1860-5397-9-234-i31.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 31: Proposed mechanism photoredox cleavage of C–C bonds.
Scheme 31: Proposed mechanism photoredox cleavage of C–C bonds.
Because of ring strain, cyclopropanes are prone to ring opening via cleavage of one of the three C–C bonds. The resulting reactive intermediates have been shown to participate in a number of synthetic/mechanistic applications [103,104]. One of these applications is a radical clock, which is centered on the cyclopropylcarbonyl to homoallyl radical rearrangement [105]. A homologous rearrangement based on the amine radical cation of N-cyclopropylanilines permits cleavage of the C–C bond α to the nitrogen atom but generating a γ-carbon radical iminium ion (distonic ion) [106]. We have applied Ru(bpz)3(PF6)2-catalyzed photooxidation of N-cyclopropylanilines to induce this rearrangement reaction. The resulting distonic ion was then intercepted by alkenes to produce [3 + 2] annulation products (Scheme 32) [107]. An aryl group on the amine was required for the reaction. Both mono- and bicyclic cyclopropylanilines were viable substrates to provide the annulation products in good to excellent yields. The former gave little to poor diastereoselectivity whereas the later produced modest diastereoselectivity. The reaction has 100% atom economy. It is also overall redox-neutral and thus does not require an external oxidant.
![[1860-5397-9-234-i32]](/bjoc/content/inline/1860-5397-9-234-i32.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 32: Intermolecular [3 + 2] annulation of cyclopropylamines with olefins.
Scheme 32: Intermolecular [3 + 2] annulation of cyclopropylamines with olefins.
We believe that the annulation reaction proceeds first via reductive quenching of the photoexcited state of Ru(II) by cyclopropylaniline 147 to generate amine radical cation 148 and Ru(I) (Scheme 33). Amine radical cation 148 then triggers the ring opening to release the ring strain while producing a distonic ion 149 with a primary radical. Distonic ion 149 is added via a Giese-type radical addition to an alkene, yielding a more stable distonic ion 151 with a secondary radical. Intramolecular addition of the secondary radical to the iminium ion furnishes a new amine radical cation 152. Finally, amine radical cation 152 is reduced by Ru(I) to provide the annulation product 153 and regenerate Ru(II), thus completing the catalytic cycle.
![[1860-5397-9-234-i33]](/bjoc/content/inline/1860-5397-9-234-i33.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 33: Proposed mechanism for intermolecular [3 + 2] annulation.
Scheme 33: Proposed mechanism for intermolecular [3 + 2] annulation.
Our group also realized cleavage of N–N bonds by irradiation of aromatic hydrazines or hydrazides in the presence of Ru(bpz)3(PF6)2 and air (Scheme 34) [108]. A 13 W compact fluorescent light was sufficient as the light source. N,N-disubstituted hydrazines and hydrazides were suitable substrates provided that at least one of the two substituents on the nitrogen atom was an aryl group. Electron-richer hydrazines were found to be more reactive than hydrazides. This is consistent with our expectation that, similar to amines, hydrazines and hydrazides act as an electron donor to reductively quench the photoexcited Ru(II) complex. The photoexcited state of Ru(bpz)3 (E1/2*II/I = 1.45 V vs SCE) is more oxidizing than that of Ru(bpy)3 (E1/2*II/I = 0.77 V vs SCE). However, the two catalysts showed a divergent pattern of reactivity in the reaction. Ru(bpy)3 was the more active catalyst for hydrazines, whereas Ru(bpz)3 was more active for hydrazides. The use of MeOH in addition to CH3CN significantly shortened the reaction time for less reactive hydrazides, but showed little effect for hydrazines. We believe that the cleavage reaction is initialized via the oxidation of hydrazines or hydrazides to a amine radical cation 156 by the photoexcited Ru(II) complex. Deprotonation of the amine radical cation 156 produces a neutral nitrogen radical 157 that reacts with oxygen to furnish the radical 158. The radical 158 then rearranges to a new oxygen-based radical 159, which undergoes a cleavage reaction to yield nitrous acid and a secondary amine radical 160. Finally, one-electron reduction of the amine radical by Ru(I), followed by protonation provides a secondary amine 155.
![[1860-5397-9-234-i34]](/bjoc/content/inline/1860-5397-9-234-i34.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 34: Photoinduced clevage of N–N bonds of aromatic hydrazines and hydrazides.
Scheme 34: Photoinduced clevage of N–N bonds of aromatic hydrazines and hydrazides.
Conclusion
Visible light photoredox catalysis provides a unique way to activate small molecules such as amines. The dual nature of the photocatalyst’s photoexcited state as both oxidant and reductant allows accepting or donating one electron strictly dependent upon the small molecules encountered. Amines typically act as an electron donor to reductively quench the photoexcited state while they are oxidized to the corresponding amine radical cations. The resulting nitrogen radical cations are highly useful reactive intermediates that are capable of initializing multiple downstream pathways leading to diverse synthetic intermediates such as electrophilic iminium ions, nucleophilic α-amino radicals, and distonic ions possessing both an iminium ion and a carbon radical. Interception of these intermediates allows a variety of synthetic transformations to produce a diverse array of amines. Moreover, visible light photoredox catalysis has been merged with other types of catalysis, including enamine catalysis, N-heterocyclic carbene (NHC) catalysis, or copper acetylide formation. This dual catalysis approach has significantly expanded the type of bonds that can be formed, particularly bonds formed asymmetrically. In summary, the utility of amine radical cations formed via photooxidation of the amines has been amply demonstrated in a number of synthetic methods. With the organic community’s increasing interest in visible light photoredox catalysis, new and innovative applications of this reactive intermediate will continue to develop.
References
-
Chow, Y. L.; Danen, W. C.; Nelson, S. F.; Rosenblatt, D. H. Chem. Rev. 1978, 78, 243–274. doi:10.1021/cr60313a003
Return to citation in text: [1] -
Stella, L. Angew. Chem., Int. Ed. Engl. 1983, 22, 337–350. doi:10.1002/anie.198303373
Return to citation in text: [1] -
Bauld, N. L. Tetrahedron 1989, 45, 5307–5363. doi:10.1016/S0040-4020(01)89486-2
Return to citation in text: [1] -
Schmittel, M.; Burghart, A. Angew. Chem., Int. Ed. Engl. 1997, 36, 2550–2589. doi:10.1002/anie.199725501
Return to citation in text: [1] -
Fallis, A. G.; Brinza, I. M. Tetrahedron 1997, 53, 17543–17594. doi:10.1016/S0040-4020(97)10060-6
Return to citation in text: [1] -
Moeller, K. D. Tetrahedron 2000, 56, 9527–9554. doi:10.1016/S0040-4020(00)00840-1
Return to citation in text: [1] -
Hoffmann, N. Pure Appl. Chem. 2007, 79, 1949–1958. doi:10.1351/pac200779111949
Return to citation in text: [1] [2] [3] -
Stella, L. Nitrogen-centered radicals. In Radicals in Organic Synthesis; Renaud, P.; Sibi, M. P., Eds.; Wiley-VCH: Weinheim, Germany, 2001; Vol. 2, pp 407–426. doi:10.1002/9783527618293.ch45
Return to citation in text: [1] -
Chiba, T.; Takata, Y. J. Org. Chem. 1977, 42, 2973–2977. doi:10.1021/jo00438a005
Return to citation in text: [1] -
Shono, T.; Matsumura, Y.; Tsubata, K. J. Am. Chem. Soc. 1981, 103, 1172–1176. doi:10.1021/ja00395a029
Return to citation in text: [1] -
Baslé, O.; Borduas, N.; Dubois, P.; Chapuzet, J. M.; Chan, T.-H.; Lessard, J.; Li, C.-J. Chem.–Eur. J. 2010, 16, 8162–8166. doi:10.1002/chem.201000240
Return to citation in text: [1] -
Tsang, A. S.-K.; Todd, M. H. Tetrahedron Lett. 2009, 50, 1199–1202. doi:10.1016/j.tetlet.2008.12.101
Return to citation in text: [1] -
Richter, T.; Mancheño, O. G. Eur. J. Org. Chem. 2010, 4460–4467. doi:10.1002/ejoc.201000548
Return to citation in text: [1] -
Shu, X.-Z.; Xia, X.-F.; Yang, Y.-F.; Ji, K.-G.; Liu, X.-Y.; Liang, Y.-M. J. Org. Chem. 2009, 74, 7464–7469. doi:10.1021/jo901583r
Return to citation in text: [1] -
Murahashi, S.-I.; Zhang, D. Chem. Soc. Rev. 2008, 37, 1490–1501. doi:10.1039/b706709g
Return to citation in text: [1] -
Li, C.-J. Acc. Chem. Res. 2009, 42, 335–344. doi:10.1021/ar800164n
Return to citation in text: [1] -
Boess, E.; Schmitz, C.; Klussmann, M. J. Am. Chem. Soc. 2012, 134, 5317–5325. doi:10.1021/ja211697s
Return to citation in text: [1] -
Ratnikov, M. O.; Doyle, M. P. J. Am. Chem. Soc. 2013, 135, 1549–1557. doi:10.1021/ja3113559
Return to citation in text: [1] -
Cho, D. W.; Yoon, U. C.; Mariano, P. S. Acc. Chem. Res. 2011, 44, 204–215. doi:10.1021/ar100125j
Return to citation in text: [1] [2] -
Pandey, G.; Gadre, S. R. ARKIVOC 2003, 45–54. doi:10.3998/ark.5550190.0004.306
Return to citation in text: [1] [2] -
Hoshikawa, T.; Yoshioka, S.; Kamijo, S.; Inoue, M. Synthesis 2013, 45, 874–887. doi:10.1055/s-0032-1318325
Return to citation in text: [1] [2] -
Shi, L.; Xia, W. Chem. Soc. Rev. 2012, 41, 7687–7697. doi:10.1039/c2cs35203f
Return to citation in text: [1] [2] -
Maity, S.; Zheng, N. Synlett 2012, 23, 1851–1856. doi:10.1055/s-0032-1316592
Return to citation in text: [1] [2] -
Campagana, S.; Puntoriero, F.; Nastasi, F.; Bergamini, G.; Balzani, V. Top. Curr. Chem. 2007, 280, 117–214. doi:10.1007/128_2007_133
Return to citation in text: [1] -
Juris, A.; Balzani, V.; Barigelletti, F.; Campagna, S.; Belser, P.; von Zelewsky, A. Coord. Chem. Rev. 1988, 84, 85–277. doi:10.1016/0010-8545(88)80032-8
Return to citation in text: [1] -
Kalyanasundaram, K. Coord. Chem. Rev. 1982, 46, 159–244. doi:10.1016/0010-8545(82)85003-0
Return to citation in text: [1] -
Lowry, M. S.; Bernhard, S. Chem.–Eur. J. 2006, 12, 7970–7977. doi:10.1002/chem.200600618
Return to citation in text: [1] -
Flamigni, L.; Barbieri, A.; Sabatini, C.; Ventura, B.; Barigelletti, F. Top. Curr. Chem. 2007, 281, 143–203. doi:10.1007/128_2007_131
Return to citation in text: [1] -
Hedstrand, D. M.; Kruizinga, W. H.; Kellogg, R. M. Tetrahedron Lett. 1978, 19, 1255–1258. doi:10.1016/S0040-4039(01)94515-0
Return to citation in text: [1] -
Neumann, M.; Füldner, S.; König, B.; Zeitler, K. Angew. Chem., Int. Ed. 2011, 50, 951–954. doi:10.1002/anie.201002992
Return to citation in text: [1] -
Meyer, T. J. Acc. Chem. Res. 1989, 22, 163–170. doi:10.1021/ar00161a001
Return to citation in text: [1] -
Bard, A. J.; Fox, M. A. Acc. Chem. Res. 1995, 28, 141–145. doi:10.1021/ar00051a007
Return to citation in text: [1] -
Lehn, J. M.; Ziessel, R. Proc. Natl. Acad. Sci. U. S. A. 1982, 79, 701–704. doi:10.1073/pnas.79.2.701
Return to citation in text: [1] -
Willner, I.; Maidan, R.; Mandler, D.; Duerr, H.; Doerr, G.; Zengerle, K. J. Am. Chem. Soc. 1987, 109, 6080–6086. doi:10.1021/ja00254a029
Return to citation in text: [1] -
Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r
Return to citation in text: [1] [2] [3] -
Yoon, T. P.; Ischay, M. A.; Du, J. Nat. Chem. 2010, 2, 527–532. doi:10.1038/nchem.687
Return to citation in text: [1] [2] -
Tucker, J. W.; Stephenson, C. R. J. J. Org. Chem. 2012, 77, 1617–1622. doi:10.1021/jo202538x
Return to citation in text: [1] [2] -
Xuan, J.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 6828–6838. doi:10.1002/anie.201200223
Return to citation in text: [1] [2] -
Telpý, F. Collect. Czech. Chem. Commun. 2011, 76, 859–917. doi:10.1135/cccc2011078
Return to citation in text: [1] [2] -
Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/b913880n
Return to citation in text: [1] [2] -
Zeitler, K. Angew. Chem., Int. Ed. 2009, 48, 9785–9789. doi:10.1002/anie.200904056
Return to citation in text: [1] [2] -
Xi, Y.; Yi, H.; Lei, A. Org. Biomol. Chem. 2013, 11, 2387–2403. doi:10.1039/c3ob40137e
Return to citation in text: [1] [2] -
Gould, I. R.; Ege, D.; Moser, J. E.; Farid, S. J. Am. Chem. Soc. 1990, 112, 4290–4301. doi:10.1021/ja00167a027
Return to citation in text: [1] -
Kellett, M. A.; Whitten, D. G.; Gould, I. R.; Bergmark, W. R. J. Am. Chem. Soc. 1991, 113, 358–359. doi:10.1021/ja00001a052
Return to citation in text: [1] -
DeLaive, P. J.; Lee, J. T.; Springtschnik, H. W.; Abruña, H.; Meyer, T. J.; Whitten, D. G. J. Am. Chem. Soc. 1977, 99, 7094–7097. doi:10.1021/ja00463a070
Return to citation in text: [1] [2] -
DeLaive, P. J.; Foreman, T. K.; Giannotti, C.; Whitten, D. G. J. Am. Chem. Soc. 1980, 102, 5627–5631. doi:10.1021/ja00537a037
Return to citation in text: [1] [2] -
Mashraqui, S. H.; Kellog, R. M. Tetrahedron Lett. 1985, 26, 1453–1456. doi:10.1016/S0040-4039(00)99069-5
Return to citation in text: [1] -
Fukuzumi, S.; Mochizuki, S.; Tanaka, T. J. Phys. Chem. 1990, 94, 722–726. doi:10.1021/j100365a039
Return to citation in text: [1] -
Willner, I.; Tsfania, T.; Eichen, Y. J. Org. Chem. 1990, 55, 2656–2662. doi:10.1021/jo00296a023
Return to citation in text: [1] [2] -
Narayanam, J. M. R.; Tucker, J. W.; Stephenson, C. R. J. J. Am. Chem. Soc. 2009, 131, 8756–8757. doi:10.1021/ja9033582
Return to citation in text: [1] -
Nguyen, J. D.; D’Amato, E. M.; Narayanam, J. M. R.; Stephenson, C. R. J. Nat. Chem. 2012, 4, 854–859. doi:10.1038/nchem.1452
Return to citation in text: [1] -
Tucker, J. W.; Nguyen, J. D.; Narayanam, J. M. R.; Krabbe, S. W.; Stephenson, C. R. J. Chem. Commun. 2010, 46, 4985–4987. doi:10.1039/c0cc00981d
Return to citation in text: [1] -
Tucker, J. W.; Stephenson, C. R. J. Org. Lett. 2011, 13, 5468–5471. doi:10.1021/ol202178t
Return to citation in text: [1] -
Kim, H.; Lee, C. Angew. Chem., Int. Ed. 2012, 51, 12303–12306. doi:10.1002/anie.201203599
Return to citation in text: [1] -
Chen, Y.; Kamlet, A. S.; Steinman, J. B.; Liu, D. R. Nat. Chem. 2011, 3, 146–153. doi:10.1038/nchem.932
Return to citation in text: [1] -
Nelson, S. F.; Ippoliti, J. T. J. Am. Chem. Soc. 1986, 108, 4879–4881. doi:10.1021/ja00276a028
Return to citation in text: [1] -
Lewis, F. D. Acc. Chem. Res. 1986, 19, 401–405. doi:10.1021/ar00132a004
Return to citation in text: [1] -
Parker, V. D.; Tilset, M. J. Am. Chem. Soc. 1991, 113, 8778–8781. doi:10.1021/ja00023a026
Return to citation in text: [1] [2] -
Dombrowski, G. W.; Dinnocenzo, J. P.; Zielinski, P. A.; Farid, S.; Wosinska, Z. M.; Gould, I. R. J. Org. Chem. 2005, 70, 3791–3800. doi:10.1021/jo047813g
Return to citation in text: [1] -
Freeman, D. B.; Furst, L.; Condie, A. G.; Stephenson, C. R. J. Org. Lett. 2012, 14, 94–97. doi:10.1021/ol202883v
Return to citation in text: [1] -
Dinnocenzo, J. P.; Banach, T. E. J. Am. Chem. Soc. 1989, 111, 8646–8653. doi:10.1021/ja00205a014
Return to citation in text: [1] -
Zhang, X.; Yeh, S.-R.; Hong, S.; Freccero, M.; Albini, A.; Falvey, D. E.; Mariano, P. S. J. Am. Chem. Soc. 1994, 116, 4211–4220. doi:10.1021/ja00089a010
Return to citation in text: [1] -
Wayner, D. D. M.; Dannenberg, J. J.; Griller, D. Chem. Phys. Lett. 1986, 131, 189–191. doi:10.1016/0009-2614(86)80542-5
Return to citation in text: [1] -
Condie, A. G.; González-Gómez, J. C.; Stephenson, C. R. J. J. Am. Chem. Soc. 2010, 132, 1464–1465. doi:10.1021/ja909145y
Return to citation in text: [1] -
Rueping, M.; Vila, C.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C. Chem. Commun. 2011, 47, 2360–2362. doi:10.1039/c0cc04539j
Return to citation in text: [1] [2] [3] [4] -
Kohls, P.; Jadhav, D.; Pandey, G.; Reiser, O. Org. Lett. 2012, 14, 672–675. doi:10.1021/ol202857t
Return to citation in text: [1] [2] [3] [4] [5] -
Hari, D. P.; König, B. Org. Lett. 2011, 13, 3852–3855. doi:10.1021/ol201376v
Return to citation in text: [1] [2] -
Pan, Y.; Kee, C. W.; Chen, L.; Tan, C.-H. Green Chem. 2011, 13, 2682–2685. doi:10.1039/c1gc15489c
Return to citation in text: [1] -
Liu, Q.; Li, Y.-N.; Zhang, H.-H.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Chem.–Eur. J. 2012, 18, 620–627. doi:10.1002/chem.201102299
Return to citation in text: [1] -
Pan, Y.; Wang, S.; Kee, C. W.; Dubuisson, E.; Yang, Y.; Loh, K. P.; Tan, C.-H. Green Chem. 2011, 13, 3341–3344. doi:10.1039/c1gc15865a
Return to citation in text: [1] -
Dreyer, D. R.; Jia, H.-P.; Bielawski, C. W. Angew. Chem., Int. Ed. 2010, 49, 6813–6816. doi:10.1002/anie.201002160
Return to citation in text: [1] -
Jia, H.-P.; Dreyer, D. R.; Bielawski, C. W. Adv. Synth. Catal. 2011, 353, 528–532. doi:10.1002/adsc.201000748
Return to citation in text: [1] -
Dreyer, D. R.; Jia, H.-P.; Todd, A. D.; Geng, J.; Bielawski, C. W. Org. Biomol. Chem. 2011, 9, 7292–7295. doi:10.1039/c1ob06102j
Return to citation in text: [1] -
Todd, A. D.; Bielawski, C. W. Catal. Sci. Technol. 2013, 3, 135–139. doi:10.1039/c2cy20474f
Return to citation in text: [1] -
To, W.-P.; Tong, G. S.-M.; Lu, W.; Ma, C.; Liu, J.; Chow, A. L.-F.; Che, C.-M. Angew. Chem., Int. Ed. 2012, 51, 2654–2657. doi:10.1002/anie.201108080
Return to citation in text: [1] -
Xue, Q.; Xie, J.; Jin, H.; Cheng, Y.; Zhu, C. Org. Biomol. Chem. 2013, 11, 1606–1609. doi:10.1039/c3ob27400d
Return to citation in text: [1] -
Rueping, M.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C.; Leonori, D.; Vila, C. Chem.–Eur. J. 2012, 18, 5170–5174. doi:10.1002/chem.201200050
Return to citation in text: [1] -
DiRocco, D. A.; Rovis, T. J. Am. Chem. Soc. 2012, 134, 8094–8097. doi:10.1021/ja3030164
Return to citation in text: [1] -
Zou, Y.-Q.; Lu, L.-Q.; Fu, L.; Chang, N.-J.; Rong, J.; Chen, J.-R.; Xiao, W.-J. Angew. Chem., Int. Ed. 2011, 50, 7171–7175. doi:10.1002/anie.201102306
Return to citation in text: [1] -
Rueping, M.; Leonori, D.; Poisson, T. Chem. Commun. 2011, 47, 9615–9617. doi:10.1039/c1cc13660g
Return to citation in text: [1] -
Xie, J.; Xue, Q.; Jin, H.; Li, H.; Cheng, Y.; Zhu, C. Chem. Sci. 2013, 4, 1281–1286. doi:10.1039/c2sc22131d
Return to citation in text: [1] -
Wang, Z.-Q.; Hu, M.; Huang, X.-C.; Gong, L.-B.; Xie, Y.-X.; Li, J.-H. J. Org. Chem. 2012, 77, 8705–8711. doi:10.1021/jo301691h
Return to citation in text: [1] -
Zhu, S.; Rueping, M. Chem. Commun. 2012, 48, 11960–11962. doi:10.1039/c2cc36995h
Return to citation in text: [1] -
O’Donnell, J. F.; Mann, C. K. J. Electroanal. Chem. 1967, 13, 157–162. doi:10.1016/0022-0728(67)80108-6
Return to citation in text: [1] -
Dai, C.; Meschini, F.; Narayanam, J. M. R.; Stephenson, C. R. J. J. Org. Chem. 2012, 77, 4425–4431. doi:10.1021/jo300162c
Return to citation in text: [1] -
Xuan, J.; Cheng, Y.; An, J.; Lu, L.-Q.; Zhang, X.-X.; Xiao, W.-J. Chem. Commun. 2011, 47, 8337–8339. doi:10.1039/c1cc12203g
Return to citation in text: [1] -
Xuan, J.; Feng, Z.-J.; Duan, S.-W.; Xiao, W.-J. RSC Adv. 2012, 2, 4065–4068. doi:10.1039/c2ra20403g
Return to citation in text: [1] -
Mathis, C. L.; Gist, B. M.; Frederickson, C. K.; Midkiff, K. M.; Marvin, C. C. Tetrahedron Lett. 2013, 54, 2101–2104. doi:10.1016/j.tetlet.2013.02.031
Return to citation in text: [1] -
Rueping, M.; Zhu, S.; Koenigs, R. M. Chem. Commun. 2011, 47, 8679–8681. doi:10.1039/c1cc12907d
Return to citation in text: [1] -
Espelt, L. R.; Wiensch, E. M.; Yoon, T. P. J. Org. Chem. 2013, 78, 4107–4114. doi:10.1021/jo400428m
Return to citation in text: [1] -
Miyake, Y.; Nakajima, K.; Nishibayashi, Y. J. Am. Chem. Soc. 2012, 134, 3338–3341. doi:10.1021/ja211770y
Return to citation in text: [1] [2] -
Zhu, S.; Das, A.; Bui, L.; Zhou, H.; Curran, D. P.; Rueping, M. J. Am. Chem. Soc. 2013, 135, 1823–1829. doi:10.1021/ja309580a
Return to citation in text: [1] -
Miyake, Y.; Nakajima, K.; Nishibayashi, Y. Chem.–Eur. J. 2012, 18, 16473–16477. doi:10.1002/chem.201203066
Return to citation in text: [1] -
Minakata, S.; Ohshima, Y.; Takemiya, A.; Ryu, I.; Komatsu, M.; Ohshiro, Y. Chem. Lett. 1997, 26, 311–312. doi:10.1246/cl.1997.311
Return to citation in text: [1] -
Kerr, G. H.; Meth-Cohn, O.; Mullock, E. B.; Suschitzky, H. J. Chem. Soc., Perkin Trans. 1 1974, 1614–1619. doi:10.1039/P19740001614
Return to citation in text: [1] -
Murahashi, S.; Oda, T.; Sugahara, T.; Masui, Y. J. Org. Chem. 1990, 55, 1744–1749. doi:10.1021/jo00293a014
Return to citation in text: [1] -
Izumi, T.; Kohei, K.; Murakami, S. J. Heterocycl. Chem. 1993, 30, 1133–1136. doi:10.1002/jhet.5570300453
Return to citation in text: [1] -
Eberson, L.; Persson, O.; Svensson, J. O. Acta Chem. Scand. 1998, 52, 1293–1300. doi:10.3891/acta.chem.scand.52-1293
Return to citation in text: [1] -
Lowry, M. S.; Goldsmith, J. I.; Slinker, J. D.; Rohl, R.; Pascal, R. A., Jr.; Malliaras, G. G.; Bernhard, S. Chem. Mater. 2005, 17, 5712–5719. doi:10.1021/cm051312+
Return to citation in text: [1] -
McNally, A.; Prier, C. K.; MacMillan, D. W. C. Science 2011, 334, 1114–1117. doi:10.1126/science.1213920
Return to citation in text: [1] -
Lee, L. Y. C.; Ci, X.; Giannotti, C.; Whitten, D. G. J. Am. Chem. Soc. 1986, 108, 175–177. doi:10.1021/ja00261a029
Return to citation in text: [1] -
Cai, S.; Zhao, X.; Wang, X.; Liu, Q.; Li, Z.; Wang, D. Z. Angew. Chem., Int. Ed. 2012, 51, 8050–8053. doi:10.1002/anie.201202880
Return to citation in text: [1] -
Carson, C. A.; Kerr, M. A. Chem. Soc. Rev. 2009, 38, 3051–3060. doi:10.1039/b901245c
Return to citation in text: [1] -
Yu, M.; Pagenkopf, B. L. Tetrahedron 2005, 61, 321–347. doi:10.1016/j.tet.2004.10.077
Return to citation in text: [1] -
Griller, D.; Ingold, K. U. Acc. Chem. Res. 1980, 13, 317–323. doi:10.1021/ar50153a004
Return to citation in text: [1] -
Li, X.; Grimm, M. L.; Igarashi, K.; Castagnoli, N., Jr.; Tanko, J. M. Chem. Commun. 2007, 2648–2650. doi:10.1039/b702157g
Return to citation in text: [1] -
Maity, S.; Zheng, N. Angew. Chem., Int. Ed. 2012, 51, 9562–9566. doi:10.1002/anie.201205137
Return to citation in text: [1] -
Zhu, M.; Zheng, N. Synthesis 2011, 2223–2236. doi:10.1055/s-0030-1260082
Return to citation in text: [1]
| 68. | Pan, Y.; Kee, C. W.; Chen, L.; Tan, C.-H. Green Chem. 2011, 13, 2682–2685. doi:10.1039/c1gc15489c |
| 69. | Liu, Q.; Li, Y.-N.; Zhang, H.-H.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Chem.–Eur. J. 2012, 18, 620–627. doi:10.1002/chem.201102299 |
| 65. | Rueping, M.; Vila, C.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C. Chem. Commun. 2011, 47, 2360–2362. doi:10.1039/c0cc04539j |
| 66. | Kohls, P.; Jadhav, D.; Pandey, G.; Reiser, O. Org. Lett. 2012, 14, 672–675. doi:10.1021/ol202857t |
| 78. | DiRocco, D. A.; Rovis, T. J. Am. Chem. Soc. 2012, 134, 8094–8097. doi:10.1021/ja3030164 |
| 76. | Xue, Q.; Xie, J.; Jin, H.; Cheng, Y.; Zhu, C. Org. Biomol. Chem. 2013, 11, 1606–1609. doi:10.1039/c3ob27400d |
| 77. | Rueping, M.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C.; Leonori, D.; Vila, C. Chem.–Eur. J. 2012, 18, 5170–5174. doi:10.1002/chem.201200050 |
| 65. | Rueping, M.; Vila, C.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C. Chem. Commun. 2011, 47, 2360–2362. doi:10.1039/c0cc04539j |
| 75. | To, W.-P.; Tong, G. S.-M.; Lu, W.; Ma, C.; Liu, J.; Chow, A. L.-F.; Che, C.-M. Angew. Chem., Int. Ed. 2012, 51, 2654–2657. doi:10.1002/anie.201108080 |
| 70. | Pan, Y.; Wang, S.; Kee, C. W.; Dubuisson, E.; Yang, Y.; Loh, K. P.; Tan, C.-H. Green Chem. 2011, 13, 3341–3344. doi:10.1039/c1gc15865a |
| 71. | Dreyer, D. R.; Jia, H.-P.; Bielawski, C. W. Angew. Chem., Int. Ed. 2010, 49, 6813–6816. doi:10.1002/anie.201002160 |
| 72. | Jia, H.-P.; Dreyer, D. R.; Bielawski, C. W. Adv. Synth. Catal. 2011, 353, 528–532. doi:10.1002/adsc.201000748 |
| 73. | Dreyer, D. R.; Jia, H.-P.; Todd, A. D.; Geng, J.; Bielawski, C. W. Org. Biomol. Chem. 2011, 9, 7292–7295. doi:10.1039/c1ob06102j |
| 74. | Todd, A. D.; Bielawski, C. W. Catal. Sci. Technol. 2013, 3, 135–139. doi:10.1039/c2cy20474f |
| 79. | Zou, Y.-Q.; Lu, L.-Q.; Fu, L.; Chang, N.-J.; Rong, J.; Chen, J.-R.; Xiao, W.-J. Angew. Chem., Int. Ed. 2011, 50, 7171–7175. doi:10.1002/anie.201102306 |
| 80. | Rueping, M.; Leonori, D.; Poisson, T. Chem. Commun. 2011, 47, 9615–9617. doi:10.1039/c1cc13660g |
| 81. | Xie, J.; Xue, Q.; Jin, H.; Li, H.; Cheng, Y.; Zhu, C. Chem. Sci. 2013, 4, 1281–1286. doi:10.1039/c2sc22131d |
| 87. | Xuan, J.; Feng, Z.-J.; Duan, S.-W.; Xiao, W.-J. RSC Adv. 2012, 2, 4065–4068. doi:10.1039/c2ra20403g |
| 88. | Mathis, C. L.; Gist, B. M.; Frederickson, C. K.; Midkiff, K. M.; Marvin, C. C. Tetrahedron Lett. 2013, 54, 2101–2104. doi:10.1016/j.tetlet.2013.02.031 |
| 85. | Dai, C.; Meschini, F.; Narayanam, J. M. R.; Stephenson, C. R. J. J. Org. Chem. 2012, 77, 4425–4431. doi:10.1021/jo300162c |
| 86. | Xuan, J.; Cheng, Y.; An, J.; Lu, L.-Q.; Zhang, X.-X.; Xiao, W.-J. Chem. Commun. 2011, 47, 8337–8339. doi:10.1039/c1cc12203g |
| 84. | O’Donnell, J. F.; Mann, C. K. J. Electroanal. Chem. 1967, 13, 157–162. doi:10.1016/0022-0728(67)80108-6 |
| 35. | Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r |
| 82. | Wang, Z.-Q.; Hu, M.; Huang, X.-C.; Gong, L.-B.; Xie, Y.-X.; Li, J.-H. J. Org. Chem. 2012, 77, 8705–8711. doi:10.1021/jo301691h |
| 83. | Zhu, S.; Rueping, M. Chem. Commun. 2012, 48, 11960–11962. doi:10.1039/c2cc36995h |
| 65. | Rueping, M.; Vila, C.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C. Chem. Commun. 2011, 47, 2360–2362. doi:10.1039/c0cc04539j |
| 66. | Kohls, P.; Jadhav, D.; Pandey, G.; Reiser, O. Org. Lett. 2012, 14, 672–675. doi:10.1021/ol202857t |
| 89. | Rueping, M.; Zhu, S.; Koenigs, R. M. Chem. Commun. 2011, 47, 8679–8681. doi:10.1039/c1cc12907d |
| 1. | Chow, Y. L.; Danen, W. C.; Nelson, S. F.; Rosenblatt, D. H. Chem. Rev. 1978, 78, 243–274. doi:10.1021/cr60313a003 |
| 2. | Stella, L. Angew. Chem., Int. Ed. Engl. 1983, 22, 337–350. doi:10.1002/anie.198303373 |
| 3. | Bauld, N. L. Tetrahedron 1989, 45, 5307–5363. doi:10.1016/S0040-4020(01)89486-2 |
| 4. | Schmittel, M.; Burghart, A. Angew. Chem., Int. Ed. Engl. 1997, 36, 2550–2589. doi:10.1002/anie.199725501 |
| 5. | Fallis, A. G.; Brinza, I. M. Tetrahedron 1997, 53, 17543–17594. doi:10.1016/S0040-4020(97)10060-6 |
| 6. | Moeller, K. D. Tetrahedron 2000, 56, 9527–9554. doi:10.1016/S0040-4020(00)00840-1 |
| 7. | Hoffmann, N. Pure Appl. Chem. 2007, 79, 1949–1958. doi:10.1351/pac200779111949 |
| 8. | Stella, L. Nitrogen-centered radicals. In Radicals in Organic Synthesis; Renaud, P.; Sibi, M. P., Eds.; Wiley-VCH: Weinheim, Germany, 2001; Vol. 2, pp 407–426. doi:10.1002/9783527618293.ch45 |
| 7. | Hoffmann, N. Pure Appl. Chem. 2007, 79, 1949–1958. doi:10.1351/pac200779111949 |
| 19. | Cho, D. W.; Yoon, U. C.; Mariano, P. S. Acc. Chem. Res. 2011, 44, 204–215. doi:10.1021/ar100125j |
| 20. | Pandey, G.; Gadre, S. R. ARKIVOC 2003, 45–54. doi:10.3998/ark.5550190.0004.306 |
| 21. | Hoshikawa, T.; Yoshioka, S.; Kamijo, S.; Inoue, M. Synthesis 2013, 45, 874–887. doi:10.1055/s-0032-1318325 |
| 47. | Mashraqui, S. H.; Kellog, R. M. Tetrahedron Lett. 1985, 26, 1453–1456. doi:10.1016/S0040-4039(00)99069-5 |
| 48. | Fukuzumi, S.; Mochizuki, S.; Tanaka, T. J. Phys. Chem. 1990, 94, 722–726. doi:10.1021/j100365a039 |
| 49. | Willner, I.; Tsfania, T.; Eichen, Y. J. Org. Chem. 1990, 55, 2656–2662. doi:10.1021/jo00296a023 |
| 50. | Narayanam, J. M. R.; Tucker, J. W.; Stephenson, C. R. J. J. Am. Chem. Soc. 2009, 131, 8756–8757. doi:10.1021/ja9033582 |
| 51. | Nguyen, J. D.; D’Amato, E. M.; Narayanam, J. M. R.; Stephenson, C. R. J. Nat. Chem. 2012, 4, 854–859. doi:10.1038/nchem.1452 |
| 93. | Miyake, Y.; Nakajima, K.; Nishibayashi, Y. Chem.–Eur. J. 2012, 18, 16473–16477. doi:10.1002/chem.201203066 |
| 15. | Murahashi, S.-I.; Zhang, D. Chem. Soc. Rev. 2008, 37, 1490–1501. doi:10.1039/b706709g |
| 16. | Li, C.-J. Acc. Chem. Res. 2009, 42, 335–344. doi:10.1021/ar800164n |
| 17. | Boess, E.; Schmitz, C.; Klussmann, M. J. Am. Chem. Soc. 2012, 134, 5317–5325. doi:10.1021/ja211697s |
| 18. | Ratnikov, M. O.; Doyle, M. P. J. Am. Chem. Soc. 2013, 135, 1549–1557. doi:10.1021/ja3113559 |
| 52. | Tucker, J. W.; Nguyen, J. D.; Narayanam, J. M. R.; Krabbe, S. W.; Stephenson, C. R. J. Chem. Commun. 2010, 46, 4985–4987. doi:10.1039/c0cc00981d |
| 53. | Tucker, J. W.; Stephenson, C. R. J. Org. Lett. 2011, 13, 5468–5471. doi:10.1021/ol202178t |
| 54. | Kim, H.; Lee, C. Angew. Chem., Int. Ed. 2012, 51, 12303–12306. doi:10.1002/anie.201203599 |
| 12. | Tsang, A. S.-K.; Todd, M. H. Tetrahedron Lett. 2009, 50, 1199–1202. doi:10.1016/j.tetlet.2008.12.101 |
| 13. | Richter, T.; Mancheño, O. G. Eur. J. Org. Chem. 2010, 4460–4467. doi:10.1002/ejoc.201000548 |
| 14. | Shu, X.-Z.; Xia, X.-F.; Yang, Y.-F.; Ji, K.-G.; Liu, X.-Y.; Liang, Y.-M. J. Org. Chem. 2009, 74, 7464–7469. doi:10.1021/jo901583r |
| 43. | Gould, I. R.; Ege, D.; Moser, J. E.; Farid, S. J. Am. Chem. Soc. 1990, 112, 4290–4301. doi:10.1021/ja00167a027 |
| 44. | Kellett, M. A.; Whitten, D. G.; Gould, I. R.; Bergmark, W. R. J. Am. Chem. Soc. 1991, 113, 358–359. doi:10.1021/ja00001a052 |
| 66. | Kohls, P.; Jadhav, D.; Pandey, G.; Reiser, O. Org. Lett. 2012, 14, 672–675. doi:10.1021/ol202857t |
| 9. | Chiba, T.; Takata, Y. J. Org. Chem. 1977, 42, 2973–2977. doi:10.1021/jo00438a005 |
| 10. | Shono, T.; Matsumura, Y.; Tsubata, K. J. Am. Chem. Soc. 1981, 103, 1172–1176. doi:10.1021/ja00395a029 |
| 11. | Baslé, O.; Borduas, N.; Dubois, P.; Chapuzet, J. M.; Chan, T.-H.; Lessard, J.; Li, C.-J. Chem.–Eur. J. 2010, 16, 8162–8166. doi:10.1002/chem.201000240 |
| 45. | DeLaive, P. J.; Lee, J. T.; Springtschnik, H. W.; Abruña, H.; Meyer, T. J.; Whitten, D. G. J. Am. Chem. Soc. 1977, 99, 7094–7097. doi:10.1021/ja00463a070 |
| 46. | DeLaive, P. J.; Foreman, T. K.; Giannotti, C.; Whitten, D. G. J. Am. Chem. Soc. 1980, 102, 5627–5631. doi:10.1021/ja00537a037 |
| 91. | Miyake, Y.; Nakajima, K.; Nishibayashi, Y. J. Am. Chem. Soc. 2012, 134, 3338–3341. doi:10.1021/ja211770y |
| 29. | Hedstrand, D. M.; Kruizinga, W. H.; Kellogg, R. M. Tetrahedron Lett. 1978, 19, 1255–1258. doi:10.1016/S0040-4039(01)94515-0 |
| 30. | Neumann, M.; Füldner, S.; König, B.; Zeitler, K. Angew. Chem., Int. Ed. 2011, 50, 951–954. doi:10.1002/anie.201002992 |
| 33. | Lehn, J. M.; Ziessel, R. Proc. Natl. Acad. Sci. U. S. A. 1982, 79, 701–704. doi:10.1073/pnas.79.2.701 |
| 34. | Willner, I.; Maidan, R.; Mandler, D.; Duerr, H.; Doerr, G.; Zengerle, K. J. Am. Chem. Soc. 1987, 109, 6080–6086. doi:10.1021/ja00254a029 |
| 91. | Miyake, Y.; Nakajima, K.; Nishibayashi, Y. J. Am. Chem. Soc. 2012, 134, 3338–3341. doi:10.1021/ja211770y |
| 27. | Lowry, M. S.; Bernhard, S. Chem.–Eur. J. 2006, 12, 7970–7977. doi:10.1002/chem.200600618 |
| 28. | Flamigni, L.; Barbieri, A.; Sabatini, C.; Ventura, B.; Barigelletti, F. Top. Curr. Chem. 2007, 281, 143–203. doi:10.1007/128_2007_131 |
| 35. | Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r |
| 36. | Yoon, T. P.; Ischay, M. A.; Du, J. Nat. Chem. 2010, 2, 527–532. doi:10.1038/nchem.687 |
| 37. | Tucker, J. W.; Stephenson, C. R. J. J. Org. Chem. 2012, 77, 1617–1622. doi:10.1021/jo202538x |
| 38. | Xuan, J.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 6828–6838. doi:10.1002/anie.201200223 |
| 39. | Telpý, F. Collect. Czech. Chem. Commun. 2011, 76, 859–917. doi:10.1135/cccc2011078 |
| 40. | Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/b913880n |
| 41. | Zeitler, K. Angew. Chem., Int. Ed. 2009, 48, 9785–9789. doi:10.1002/anie.200904056 |
| 42. | Xi, Y.; Yi, H.; Lei, A. Org. Biomol. Chem. 2013, 11, 2387–2403. doi:10.1039/c3ob40137e |
| 92. | Zhu, S.; Das, A.; Bui, L.; Zhou, H.; Curran, D. P.; Rueping, M. J. Am. Chem. Soc. 2013, 135, 1823–1829. doi:10.1021/ja309580a |
| 24. | Campagana, S.; Puntoriero, F.; Nastasi, F.; Bergamini, G.; Balzani, V. Top. Curr. Chem. 2007, 280, 117–214. doi:10.1007/128_2007_133 |
| 25. | Juris, A.; Balzani, V.; Barigelletti, F.; Campagna, S.; Belser, P.; von Zelewsky, A. Coord. Chem. Rev. 1988, 84, 85–277. doi:10.1016/0010-8545(88)80032-8 |
| 26. | Kalyanasundaram, K. Coord. Chem. Rev. 1982, 46, 159–244. doi:10.1016/0010-8545(82)85003-0 |
| 66. | Kohls, P.; Jadhav, D.; Pandey, G.; Reiser, O. Org. Lett. 2012, 14, 672–675. doi:10.1021/ol202857t |
| 22. | Shi, L.; Xia, W. Chem. Soc. Rev. 2012, 41, 7687–7697. doi:10.1039/c2cs35203f |
| 23. | Maity, S.; Zheng, N. Synlett 2012, 23, 1851–1856. doi:10.1055/s-0032-1316592 |
| 31. | Meyer, T. J. Acc. Chem. Res. 1989, 22, 163–170. doi:10.1021/ar00161a001 |
| 32. | Bard, A. J.; Fox, M. A. Acc. Chem. Res. 1995, 28, 141–145. doi:10.1021/ar00051a007 |
| 90. | Espelt, L. R.; Wiensch, E. M.; Yoon, T. P. J. Org. Chem. 2013, 78, 4107–4114. doi:10.1021/jo400428m |
| 56. | Nelson, S. F.; Ippoliti, J. T. J. Am. Chem. Soc. 1986, 108, 4879–4881. doi:10.1021/ja00276a028 |
| 57. | Lewis, F. D. Acc. Chem. Res. 1986, 19, 401–405. doi:10.1021/ar00132a004 |
| 58. | Parker, V. D.; Tilset, M. J. Am. Chem. Soc. 1991, 113, 8778–8781. doi:10.1021/ja00023a026 |
| 59. | Dombrowski, G. W.; Dinnocenzo, J. P.; Zielinski, P. A.; Farid, S.; Wosinska, Z. M.; Gould, I. R. J. Org. Chem. 2005, 70, 3791–3800. doi:10.1021/jo047813g |
| 60. | Freeman, D. B.; Furst, L.; Condie, A. G.; Stephenson, C. R. J. Org. Lett. 2012, 14, 94–97. doi:10.1021/ol202883v |
| 49. | Willner, I.; Tsfania, T.; Eichen, Y. J. Org. Chem. 1990, 55, 2656–2662. doi:10.1021/jo00296a023 |
| 55. | Chen, Y.; Kamlet, A. S.; Steinman, J. B.; Liu, D. R. Nat. Chem. 2011, 3, 146–153. doi:10.1038/nchem.932 |
| 98. | Eberson, L.; Persson, O.; Svensson, J. O. Acta Chem. Scand. 1998, 52, 1293–1300. doi:10.3891/acta.chem.scand.52-1293 |
| 99. | Lowry, M. S.; Goldsmith, J. I.; Slinker, J. D.; Rohl, R.; Pascal, R. A., Jr.; Malliaras, G. G.; Bernhard, S. Chem. Mater. 2005, 17, 5712–5719. doi:10.1021/cm051312+ |
| 100. | McNally, A.; Prier, C. K.; MacMillan, D. W. C. Science 2011, 334, 1114–1117. doi:10.1126/science.1213920 |
| 94. | Minakata, S.; Ohshima, Y.; Takemiya, A.; Ryu, I.; Komatsu, M.; Ohshiro, Y. Chem. Lett. 1997, 26, 311–312. doi:10.1246/cl.1997.311 |
| 95. | Kerr, G. H.; Meth-Cohn, O.; Mullock, E. B.; Suschitzky, H. J. Chem. Soc., Perkin Trans. 1 1974, 1614–1619. doi:10.1039/P19740001614 |
| 96. | Murahashi, S.; Oda, T.; Sugahara, T.; Masui, Y. J. Org. Chem. 1990, 55, 1744–1749. doi:10.1021/jo00293a014 |
| 97. | Izumi, T.; Kohei, K.; Murakami, S. J. Heterocycl. Chem. 1993, 30, 1133–1136. doi:10.1002/jhet.5570300453 |
| 65. | Rueping, M.; Vila, C.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C. Chem. Commun. 2011, 47, 2360–2362. doi:10.1039/c0cc04539j |
| 66. | Kohls, P.; Jadhav, D.; Pandey, G.; Reiser, O. Org. Lett. 2012, 14, 672–675. doi:10.1021/ol202857t |
| 58. | Parker, V. D.; Tilset, M. J. Am. Chem. Soc. 1991, 113, 8778–8781. doi:10.1021/ja00023a026 |
| 46. | DeLaive, P. J.; Foreman, T. K.; Giannotti, C.; Whitten, D. G. J. Am. Chem. Soc. 1980, 102, 5627–5631. doi:10.1021/ja00537a037 |
| 106. | Li, X.; Grimm, M. L.; Igarashi, K.; Castagnoli, N., Jr.; Tanko, J. M. Chem. Commun. 2007, 2648–2650. doi:10.1039/b702157g |
| 64. | Condie, A. G.; González-Gómez, J. C.; Stephenson, C. R. J. J. Am. Chem. Soc. 2010, 132, 1464–1465. doi:10.1021/ja909145y |
| 107. | Maity, S.; Zheng, N. Angew. Chem., Int. Ed. 2012, 51, 9562–9566. doi:10.1002/anie.201205137 |
| 22. | Shi, L.; Xia, W. Chem. Soc. Rev. 2012, 41, 7687–7697. doi:10.1039/c2cs35203f |
| 23. | Maity, S.; Zheng, N. Synlett 2012, 23, 1851–1856. doi:10.1055/s-0032-1316592 |
| 35. | Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r |
| 36. | Yoon, T. P.; Ischay, M. A.; Du, J. Nat. Chem. 2010, 2, 527–532. doi:10.1038/nchem.687 |
| 37. | Tucker, J. W.; Stephenson, C. R. J. J. Org. Chem. 2012, 77, 1617–1622. doi:10.1021/jo202538x |
| 38. | Xuan, J.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 6828–6838. doi:10.1002/anie.201200223 |
| 39. | Telpý, F. Collect. Czech. Chem. Commun. 2011, 76, 859–917. doi:10.1135/cccc2011078 |
| 40. | Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/b913880n |
| 41. | Zeitler, K. Angew. Chem., Int. Ed. 2009, 48, 9785–9789. doi:10.1002/anie.200904056 |
| 42. | Xi, Y.; Yi, H.; Lei, A. Org. Biomol. Chem. 2013, 11, 2387–2403. doi:10.1039/c3ob40137e |
| 103. | Carson, C. A.; Kerr, M. A. Chem. Soc. Rev. 2009, 38, 3051–3060. doi:10.1039/b901245c |
| 104. | Yu, M.; Pagenkopf, B. L. Tetrahedron 2005, 61, 321–347. doi:10.1016/j.tet.2004.10.077 |
| 7. | Hoffmann, N. Pure Appl. Chem. 2007, 79, 1949–1958. doi:10.1351/pac200779111949 |
| 19. | Cho, D. W.; Yoon, U. C.; Mariano, P. S. Acc. Chem. Res. 2011, 44, 204–215. doi:10.1021/ar100125j |
| 20. | Pandey, G.; Gadre, S. R. ARKIVOC 2003, 45–54. doi:10.3998/ark.5550190.0004.306 |
| 21. | Hoshikawa, T.; Yoshioka, S.; Kamijo, S.; Inoue, M. Synthesis 2013, 45, 874–887. doi:10.1055/s-0032-1318325 |
| 105. | Griller, D.; Ingold, K. U. Acc. Chem. Res. 1980, 13, 317–323. doi:10.1021/ar50153a004 |
| 61. | Dinnocenzo, J. P.; Banach, T. E. J. Am. Chem. Soc. 1989, 111, 8646–8653. doi:10.1021/ja00205a014 |
| 62. | Zhang, X.; Yeh, S.-R.; Hong, S.; Freccero, M.; Albini, A.; Falvey, D. E.; Mariano, P. S. J. Am. Chem. Soc. 1994, 116, 4211–4220. doi:10.1021/ja00089a010 |
| 101. | Lee, L. Y. C.; Ci, X.; Giannotti, C.; Whitten, D. G. J. Am. Chem. Soc. 1986, 108, 175–177. doi:10.1021/ja00261a029 |
| 45. | DeLaive, P. J.; Lee, J. T.; Springtschnik, H. W.; Abruña, H.; Meyer, T. J.; Whitten, D. G. J. Am. Chem. Soc. 1977, 99, 7094–7097. doi:10.1021/ja00463a070 |
| 63. | Wayner, D. D. M.; Dannenberg, J. J.; Griller, D. Chem. Phys. Lett. 1986, 131, 189–191. doi:10.1016/0009-2614(86)80542-5 |
| 102. | Cai, S.; Zhao, X.; Wang, X.; Liu, Q.; Li, Z.; Wang, D. Z. Angew. Chem., Int. Ed. 2012, 51, 8050–8053. doi:10.1002/anie.201202880 |
© 2013 Hu et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)