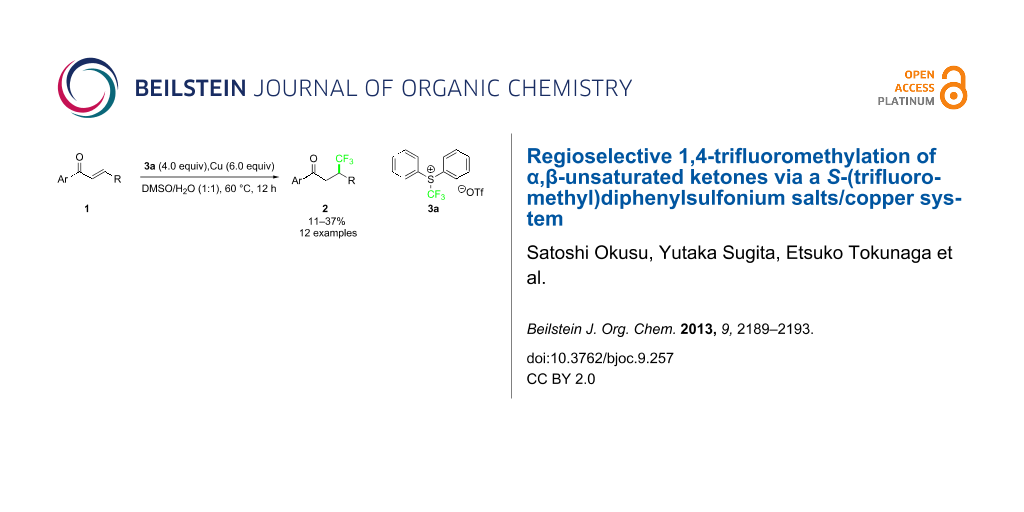
Abstract
Regioselective conjugate 1,4-trifluoromethylation of α,β-unsaturated ketones by the use of shelf-stable electrophilic trifluoromethylating reagents, S-(trifluoromethyl)diphenylsulfonium salts and copper under mild conditions is described. A wide range of acyclic aryl–aryl–enones and aryl–alkyl–enones were converted into β-trifluoromethylated ketones in low to moderate yields.

Graphical Abstract
Introduction
One of the challenges in synthetic organic chemistry is the nucleophilic 1,4-addition of the trifluoromethyl (CF3) group into electron-deficient internal alkenes as represented by the Michael addition reaction, even in a racemic, non-stereoselective fashion [1-5]. The nucleophilic trifluoromethylation to conjugated alkenes essentially occurs solely via a 1,2-addition [1-11], not a 1,4-addition (Scheme 1), with the exception of non-general examples of 1,4-additive trifluoromethylation of (trifluoromethyl)trimethylsilane (Me3SiCF3, Ruppert–Prakash reagent) to very specific substrates such as trans-1-benzoyl-2-(dimethylamino)ethylene [12], 2-polyfluoroalkylchromones [13,14], isoxazoles with a nitro group at the 4-position [15], and Morita–Baylis–Hillman adducts (via SN2’ [16] or successive SN2’/SN2’ mode [17]).
![[1860-5397-9-257-i1]](/bjoc/content/inline/1860-5397-9-257-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Trifluoromethylation of α,β-unsaturated ketones.
Scheme 1: Trifluoromethylation of α,β-unsaturated ketones.
Sevenard and co-workers reported the nucleophilic 1,4-trifluoromethylation to chromones, coumarins and cyclohex-2-enone using the Ruppert–Prakash reagent, which was achieved by blocking the carbonyl moiety of the substrates with a bulky aluminium-centered Lewis acid with low to moderate yields [18]. Dilman and co-workers partially overcame this problem by using highly electrophilic alkenes bearing either Meldrum’ acids [19], or two geminal nitrile groups [20]. However, direct 1,4-trifluoromethylation to conventional α,β-unsaturated ketones such as chalcone is very tough, presumably due to the hardness of the CF3 anion. Recently, we reported the copper-mediated trifluoromethylation at the benzylic position by using shelf-stable electrophilic trifluoromethylating reagents, S-(trifluoromethyl)diphenylsulfonium salts, in good to high yields under mild conditions [21]. In this reaction, a bromide at the benzylic position would be replaced by a CF3 anion mediated by a copper via SET process, although the reaction mechanism is not clear. We envisaged that the system could be applicable to the conjugated 1,4-trifluoromethylation to simple chalcones. During the preparation of this article, the Nicewicz group showed a single example of conjugate trifluoromethylation of chalcone with sodium trifluoromethanesulfinate salt in the presence of N-methyl-9-mesitylacridinium as a photoredox catalyst resulting in a low product yield of 31% as a mixture of regioisomers (C2/C3 1.1:1) [22]. We disclose herein the regioselective 1,4-addition of the CF3 group into simple conjugated acyclic enones including chalcones using S-(trifluoromethyl)diphenylsulfonium salt 3 and a copper system in 11–37% yields (12 examples).
Results and Discussion
We initiated our investigation with the reaction of chalcone (1a) using a series of electrophilic trifluoromethylating reagents 3 [23-26] in the presence of copper in DMF at 60 °C (Table 1), based on previously reported conditions [21]. First, the trifluoromethylation of 1a with S-(trifluoromethyl)diphenylsulfonium salt 3a was attempted, and a desired product 2a was obtained in only 4% yield (Table 1, entry 1). Next the solvent was screened for yield improvement. We attempted the same reaction using NMP and DMSO, and the desired product 2a was obtained in 6% and 11% yields (Table 1, entries 2 and 3, respectively). Interestingly, adding water (DMF/H2O 1:1) effectively improved the yield to 23% (Table 1, entry 4). Reactions mediated by other metals, such as Ni and Zn, either gave poor yields (Table 1, entries 5 and 6). The use of larger excesses of S-(trifluoromethyl)diphenylsulfonium salt 3a (4.0 equiv) and Cu (6.0 equiv) in DMF/H2O (1:1) led to an increase in the yield of 2a (Table 1, entry 9). The best result was obtained by treating 1a at 60 °C in DMSO/H2O (1:1) in the presence of S-(trifluoromethyl)diphenylsulfonium salt 3a (4.0 equiv) and Cu (6.0 equiv), leading to the isolation of 2a in 37% yield (Table 1, entry 10). Using 4.0 equiv of Umemoto’s reagent 3b instead of 3a gave the product 2a in 27% yield (Table 1, entry 12). S-(Trifluoromethyl)benzothiophenium salt 3c [24], trifluoromethylsulfoxinium salt 3d [25], and hypervalent iodine(III) CF3 reagent 3e [26] did not proceed or provided only a trace amount of the desired product 2a under the same reaction conditions (Table 1, entries 13–15). No reaction was observed using Ruppert–Prakash reagent in the presence of Cu under the same conditions (Table 1, entry 16). In all the cases, the reaction was regioselective and a trace amount of regioisomers and/or byproducts was detected in a crude mixture analyzed by 19F NMR. Under the best conditions shown in entry 10, we re-examined the reaction, but in the presence of TEMPO. The product formation was inhibited by TEMPO and O-trifluoromethylated TEMPO was detected in 2% by 19F NMR analysis (Table 1, entry 17).
Table 1: Optimization of CF3 reagents, metal, and solvent for copper-mediated conjugate trifluoromethylation of chalcone (1a).a
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-257-i3.svg?max-width=637&scale=1.0)
|
||||
| Entry | CF3 reagent | metal | solvent | Yield (%)b |
|---|---|---|---|---|
| 1 | 3a | Cu | DMF | 4 |
| 2 | 3a | Cu | NMP | 6 |
| 3 | 3a | Cu | DMSO | 11 |
| 4 | 3a | Cu | DMF/H2O (1:1) | 23 |
| 5 | 3a | Ni | DMF/H2O (1:1) | 9 |
| 6 | 3a | Zn | DMF/H2O (1:1) | trace |
| 7 | 3a | Cu | NMP/H2O (1:1) | trace |
| 8 | 3a | Cu | DMSO/H2O (1:1) | 5 |
| 9c | 3a | Cu | DMF/H2O (1:1) | 25 |
| 10c | 3a | Cu | DMSO/H2O (1:1) | 37 |
| 11 | 3b | Cu | DMSO/H2O (1:1) | 24 |
| 12c | 3b | Cu | DMSO/H2O (1:1) | 27 |
| 13 | 3c | Cu | DMSO/H2O (1:1) | trace |
| 14 | 3d | Cu | DMSO/H2O (1:1) | 0 |
| 15 | 3e | Cu | DMSO/H2O (1:1) | trace |
| 16 | Me3SiCF3 | Cu | DMSO/H2O (1:1) | 0 |
| 17c,d | 3a | Cu | DMSO/H2O (1:1) | tracee |
aThe reaction of 1a with 3 (2.0 equiv) was carried out in the presence of metal (3.0 equiv) at 60 °C. bIsolated yield. c3 (4.0 equiv) and metal (6.0 equiv) were used. dThe reaction was performed in the presence of TEMPO (4.0 equiv). eO-Trifluoromethylated TEMPO was detected in 2% by 19F NMR.
With suitable conditions in hand, the scope of copper-mediated conjugate trifluoromethylation of α,β-unsaturated ketones 1 with 3a was explored with a variety of substrates selected in order to establish the generality of the process (Table 2). With respect to the aryl ketone group, aromatic rings substituted with either electron-donating or -withdrawing substituents, such as methyl, methoxy, fluoro and chloro were tolerated (Table 2, entries 2–4). A heteroaromatic, furanyl-substituted enone was compatible with the same reaction conditions (Table 2, entry 6). We next examined substrates differing in the nature of the β-aryl substituents under the same reaction conditions. A series of compounds with aromatic rings substituted with either electron-donating or -withdrawing substituents, such as methyl, methoxy, fluoro and chloro were also acceptable. Furthermore, the β-alkyl-substituted enone also produced the desired product 2l (Table 2, entry 12).
Table 2: Copper-mediated conjugate trifluoromethylation of α,β-unsaturated ketones 1 with 3a.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-257-i4.svg?max-width=637&scale=1.0)
|
|||||
| Entry | 1 | Ar | R | 2 | Yield (%)b |
|---|---|---|---|---|---|
| 1 | 1a | Ph | Ph | 2a | 37 |
| 2 | 1b | 4-MeC6H4 | Ph | 2b | 20 |
| 3 | 1c | 4-MeOC6H4 | Ph | 2c | 11 |
| 4 | 1d | 4-FC6H4 | Ph | 2d | 13 |
| 5 | 1e | 4-ClC6H4 | Ph | 2e | 22 |
| 6 | 1f | 2-furanyl | Ph | 2f | 13 |
| 7 | 1g | Ph | 4-MeC6H4 | 2g | 12 |
| 8 | 1h | Ph | 4-MeOC6H4 | 2h | 12 |
| 9 | 1i | Ph | 4-FC6H4 | 2i | 17 |
| 10 | 1j | Ph | 3-ClC6H4 | 2j | 18 |
| 11 | 1k | Ph | 4-ClC6H4 | 2k | 13 |
| 12 | 1l | Ph | Me | 2l | 36 |
aThe reaction of 1a with 3a (4.0 equiv) was carried out in the presence of Cu (6.0 equiv) at 60 °C. bIsolated yield.
Based on these results, we hypothesized the reaction mechanism as shown in Scheme 2. First, the conjugate trifluoromethylation of α,β-unsaturated ketones would be initiated by a single-electron transfer between S-(trifluoromethyl)diphenylsulfonium salt 3a and copper. The intermediate 4 decomposes to give the CF3 radical whose generation is supported by the TEMPO inhibition experiment. Ph2S was formed and checked by the 1H NMR spectroscopy. The resulting CF3 radical reacted directly with α,β-unsaturated ketones 1 and/or through the formation of CuCF3 species to provide the 1,4-adduct 2 in low to moderate yield. Although the true reactive species including CF3 radical and/or CuCF3 are not clear, the naked CF3 radical should be ruled out since high regioselectivity was observed, otherwise, a 1:1 mixture of regioisomers (C2/C3) should be detected like in the photoredox trifluoromethylation reaction [22].
![[1860-5397-9-257-i2]](/bjoc/content/inline/1860-5397-9-257-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed mechanism for the conjugate trifluoromethylation of α,β-unsaturated ketones by S-(trifluoromethyl)diphenylsulfonium salt and copper.
Scheme 2: Proposed mechanism for the conjugate trifluoromethylation of α,β-unsaturated ketones by S-(trifluor...
Conclusion
We developed for the first time the copper-mediated conjugate trifluoromethylation of simple α,β-unsaturated ketones through the use of shelf-stable electrophilic trifluoromethylating reagent 3a under mild conditions. Although the yields are low, wide substrate generality was observed. Getting higher yields for this chemistry [27-29] and extension to asymmetric conjugate trifluoromethylation to simple α,β-unsaturated ketones are both our subsequent challenges and we are currently working in these directions.
Supporting Information
| Supporting Information File 1: Experimental section. | ||
| Format: PDF | Size: 3.9 MB | Download |
Acknowledgements
This study was financially supported in part by the Platform for Drug Discovery, Informatics, and Structural Life Science from the Ministry of Education, Culture, Sports, Science and Technology, Japan, by Grants-in-Aid for Scientific Research from MEXT (Ministry of Education, Culture, Sports, Science and Technology) (24105513, Project No. 2304: Advanced Molecular Transformation by Organocatalysts) and JST (ACT-C: Creation of Advanced Catalytic Transformation for the Sustainable Manufacturing at Low Energy, Low Environmental Load).
References
-
Ma, J.-A.; Cahard, D. Chem. Rev. 2008, 108, PR1–PR43. doi:10.1021/cr800221v
Return to citation in text: [1] [2] -
Shibata, N.; Mizuta, S.; Kawai, H. Tetrahedron: Asymmetry 2008, 19, 2633–2644. doi:10.1016/j.tetasy.2008.11.011
Return to citation in text: [1] [2] -
Prakash, G. K. S.; Yudin, A. K. Chem. Rev. 1997, 97, 757–786. doi:10.1021/cr9408991
Return to citation in text: [1] [2] -
Prakash, G. K. S.; Mandal, M. J. Fluorine Chem. 2001, 112, 123–131. doi:10.1016/S0022-1139(01)00477-8
Return to citation in text: [1] [2] -
Billard, T.; Langlois, B. R. Eur. J. Org. Chem. 2007, 891–897. doi:10.1002/ejoc.200600643
Return to citation in text: [1] [2] -
Kawai, H.; Yuan, Z.; Tokunaga, E.; Shibata, N. Org. Biomol. Chem. 2013, 11, 1446–1450. doi:10.1039/c3ob27368g
Return to citation in text: [1] -
Prakash, G. K. S.; Jog, P. V.; Batamack, P. T. D.; Olah, G. A. Science 2012, 338, 1324–1327. doi:10.1126/science.1227859
Return to citation in text: [1] -
Billard, T.; Bruns, S.; Langlois, B. R. Org. Lett. 2000, 2, 2101–2103. doi:10.1021/ol005987o
Return to citation in text: [1] -
Singh, R. P.; Kirchmeier, R. L.; Shreeve, J. M. Org. Lett. 1999, 1, 1047–1049. doi:10.1021/ol990844r
Return to citation in text: [1] -
Matsukawa, S.; Saijo, M. Tetrahedron Lett. 2008, 49, 4655–4657. doi:10.1016/j.tetlet.2008.05.053
Return to citation in text: [1] -
Kawano, Y.; Kaneko, N.; Mukaiyama, T. Bull. Chem. Soc. Jpn. 2006, 79, 1133–1145. doi:10.1246/bcsj.79.1133
Return to citation in text: [1] -
Large, S.; Roques, N.; Langlois, B. R. J. Org. Chem. 2000, 65, 8848–8856. doi:10.1021/jo000150s
Return to citation in text: [1] -
Sosnovskikh, V. Y.; Sevenard, D. V.; Usachev, B. I.; Röschenthaler, G.-V. Tetrahedron Lett. 2003, 44, 2097–2099. doi:10.1016/S0040-4039(03)00184-9
Return to citation in text: [1] -
Sosnovskikh, V. Y.; Usachev, B. I.; Sevenard, D. V.; Röschenthaler, G.-V. J. Org. Chem. 2003, 68, 7747–7754. doi:10.1021/jo034591y
Return to citation in text: [1] -
Kawai, H.; Tachi, K.; Tokunaga, E.; Shiro, M.; Shibata, N. Angew. Chem., Int. Ed. 2011, 50, 7803–7806. doi:10.1002/anie.201102442
Return to citation in text: [1] -
Zemtsov, A. A.; Levin, V. V.; Dilman, A. D.; Struchkova, M. I.; Belyakov, P. A.; Tartakovsky, V. A.; Hu, J. Eur. J. Org. Chem. 2010, 6779–6785. doi:10.1002/ejoc.201001051
Return to citation in text: [1] -
Furukawa, T.; Nishimine, T.; Tokunaga, E.; Hasegawa, K.; Shiro, M.; Shibata, N. Org. Lett. 2011, 13, 3972–3975. doi:10.1021/ol201490e
Return to citation in text: [1] -
Sevenard, D. V.; Sosnovskikh, V. Y.; Kolomeitsev, A. A.; Königsmann, M. H.; Röschenthaler, G.-V. Tetrahedron Lett. 2003, 44, 7623–7627. doi:10.1016/j.tetlet.2003.08.050
Return to citation in text: [1] -
Zemtsov, A. A.; Levin, V. V.; Dilman, A. D.; Struchkova, M. I.; Belyakov, P. A.; Tartakovsky, V. A. Tetrahedron Lett. 2009, 50, 2998–3000. doi:10.1016/j.tetlet.2009.03.188
Return to citation in text: [1] -
Dilman, A. D.; Levin, V. V.; Belyakov, P. A.; Struchkova, M. I.; Tartakovsky, V. A. Tetrahedron Lett. 2008, 49, 4352–4354. doi:10.1016/j.tetlet.2008.05.039
Return to citation in text: [1] -
Kawai, H.; Tachi, K.; Tokunaga, E.; Shiro, M.; Shibata, N. Org. Lett. 2010, 12, 5104–5107. doi:10.1021/ol102189c
Return to citation in text: [1] [2] -
Wilger, D. J.; Gesmundo, N. J.; Nicewicz, D. A. Chem. Sci. 2013, 4, 3160–3165. doi:10.1039/c3sc51209f
Return to citation in text: [1] [2] -
Shibata, N.; Matsnev, A.; Cahard, D. Beilstein J. Org. Chem. 2010, 6, No. 65. doi:10.3762/bjoc.6.65
Return to citation in text: [1] -
Matsnev, A.; Noritake, S.; Nomura, Y.; Tokunaga, E.; Nakamura, S.; Shibata, N. Angew. Chem., Int. Ed. 2010, 49, 572–576. doi:10.1002/anie.200905225
Return to citation in text: [1] [2] -
Noritake, S.; Shibata, N.; Nakamura, S.; Toru, T.; Shiro, M. Eur. J. Org. Chem. 2008, 3465–3468. doi:10.1002/ejoc.200800419
Return to citation in text: [1] [2] -
Eisenberger, P.; Gischig, S.; Togni, A. Chem.–Eur. J. 2006, 12, 2579–2586. doi:10.1002/chem.200501052
Return to citation in text: [1] [2] -
This chemistry is complement the previous one based in the isomerization of trifluoromethylated allylic alcohols (see reference [28]) or the carbanion addition to trifluormethyl enones (see reference [29]).
Return to citation in text: [1] -
Bizet, V.; Pannecoucke, X.; Renaud, J.-L.; Cahard, D. J. Fluorine Chem. 2013, 152, 56–61. doi:10.1016/j.jfluchem.2013.01.004
Return to citation in text: [1] [2] -
Maruoka, K.; Shimada, I.; Imoto, H.; Yamamoto, H. Synlett 1994, 519–520. doi:10.1055/s-1994-22912
Return to citation in text: [1] [2]
| 27. | This chemistry is complement the previous one based in the isomerization of trifluoromethylated allylic alcohols (see reference [28]) or the carbanion addition to trifluormethyl enones (see reference [29]). |
| 28. | Bizet, V.; Pannecoucke, X.; Renaud, J.-L.; Cahard, D. J. Fluorine Chem. 2013, 152, 56–61. doi:10.1016/j.jfluchem.2013.01.004 |
| 29. | Maruoka, K.; Shimada, I.; Imoto, H.; Yamamoto, H. Synlett 1994, 519–520. doi:10.1055/s-1994-22912 |
| 26. | Eisenberger, P.; Gischig, S.; Togni, A. Chem.–Eur. J. 2006, 12, 2579–2586. doi:10.1002/chem.200501052 |
| 22. | Wilger, D. J.; Gesmundo, N. J.; Nicewicz, D. A. Chem. Sci. 2013, 4, 3160–3165. doi:10.1039/c3sc51209f |
| 1. | Ma, J.-A.; Cahard, D. Chem. Rev. 2008, 108, PR1–PR43. doi:10.1021/cr800221v |
| 2. | Shibata, N.; Mizuta, S.; Kawai, H. Tetrahedron: Asymmetry 2008, 19, 2633–2644. doi:10.1016/j.tetasy.2008.11.011 |
| 3. | Prakash, G. K. S.; Yudin, A. K. Chem. Rev. 1997, 97, 757–786. doi:10.1021/cr9408991 |
| 4. | Prakash, G. K. S.; Mandal, M. J. Fluorine Chem. 2001, 112, 123–131. doi:10.1016/S0022-1139(01)00477-8 |
| 5. | Billard, T.; Langlois, B. R. Eur. J. Org. Chem. 2007, 891–897. doi:10.1002/ejoc.200600643 |
| 15. | Kawai, H.; Tachi, K.; Tokunaga, E.; Shiro, M.; Shibata, N. Angew. Chem., Int. Ed. 2011, 50, 7803–7806. doi:10.1002/anie.201102442 |
| 24. | Matsnev, A.; Noritake, S.; Nomura, Y.; Tokunaga, E.; Nakamura, S.; Shibata, N. Angew. Chem., Int. Ed. 2010, 49, 572–576. doi:10.1002/anie.200905225 |
| 13. | Sosnovskikh, V. Y.; Sevenard, D. V.; Usachev, B. I.; Röschenthaler, G.-V. Tetrahedron Lett. 2003, 44, 2097–2099. doi:10.1016/S0040-4039(03)00184-9 |
| 14. | Sosnovskikh, V. Y.; Usachev, B. I.; Sevenard, D. V.; Röschenthaler, G.-V. J. Org. Chem. 2003, 68, 7747–7754. doi:10.1021/jo034591y |
| 25. | Noritake, S.; Shibata, N.; Nakamura, S.; Toru, T.; Shiro, M. Eur. J. Org. Chem. 2008, 3465–3468. doi:10.1002/ejoc.200800419 |
| 12. | Large, S.; Roques, N.; Langlois, B. R. J. Org. Chem. 2000, 65, 8848–8856. doi:10.1021/jo000150s |
| 23. | Shibata, N.; Matsnev, A.; Cahard, D. Beilstein J. Org. Chem. 2010, 6, No. 65. doi:10.3762/bjoc.6.65 |
| 24. | Matsnev, A.; Noritake, S.; Nomura, Y.; Tokunaga, E.; Nakamura, S.; Shibata, N. Angew. Chem., Int. Ed. 2010, 49, 572–576. doi:10.1002/anie.200905225 |
| 25. | Noritake, S.; Shibata, N.; Nakamura, S.; Toru, T.; Shiro, M. Eur. J. Org. Chem. 2008, 3465–3468. doi:10.1002/ejoc.200800419 |
| 26. | Eisenberger, P.; Gischig, S.; Togni, A. Chem.–Eur. J. 2006, 12, 2579–2586. doi:10.1002/chem.200501052 |
| 1. | Ma, J.-A.; Cahard, D. Chem. Rev. 2008, 108, PR1–PR43. doi:10.1021/cr800221v |
| 2. | Shibata, N.; Mizuta, S.; Kawai, H. Tetrahedron: Asymmetry 2008, 19, 2633–2644. doi:10.1016/j.tetasy.2008.11.011 |
| 3. | Prakash, G. K. S.; Yudin, A. K. Chem. Rev. 1997, 97, 757–786. doi:10.1021/cr9408991 |
| 4. | Prakash, G. K. S.; Mandal, M. J. Fluorine Chem. 2001, 112, 123–131. doi:10.1016/S0022-1139(01)00477-8 |
| 5. | Billard, T.; Langlois, B. R. Eur. J. Org. Chem. 2007, 891–897. doi:10.1002/ejoc.200600643 |
| 6. | Kawai, H.; Yuan, Z.; Tokunaga, E.; Shibata, N. Org. Biomol. Chem. 2013, 11, 1446–1450. doi:10.1039/c3ob27368g |
| 7. | Prakash, G. K. S.; Jog, P. V.; Batamack, P. T. D.; Olah, G. A. Science 2012, 338, 1324–1327. doi:10.1126/science.1227859 |
| 8. | Billard, T.; Bruns, S.; Langlois, B. R. Org. Lett. 2000, 2, 2101–2103. doi:10.1021/ol005987o |
| 9. | Singh, R. P.; Kirchmeier, R. L.; Shreeve, J. M. Org. Lett. 1999, 1, 1047–1049. doi:10.1021/ol990844r |
| 10. | Matsukawa, S.; Saijo, M. Tetrahedron Lett. 2008, 49, 4655–4657. doi:10.1016/j.tetlet.2008.05.053 |
| 11. | Kawano, Y.; Kaneko, N.; Mukaiyama, T. Bull. Chem. Soc. Jpn. 2006, 79, 1133–1145. doi:10.1246/bcsj.79.1133 |
| 21. | Kawai, H.; Tachi, K.; Tokunaga, E.; Shiro, M.; Shibata, N. Org. Lett. 2010, 12, 5104–5107. doi:10.1021/ol102189c |
| 19. | Zemtsov, A. A.; Levin, V. V.; Dilman, A. D.; Struchkova, M. I.; Belyakov, P. A.; Tartakovsky, V. A. Tetrahedron Lett. 2009, 50, 2998–3000. doi:10.1016/j.tetlet.2009.03.188 |
| 21. | Kawai, H.; Tachi, K.; Tokunaga, E.; Shiro, M.; Shibata, N. Org. Lett. 2010, 12, 5104–5107. doi:10.1021/ol102189c |
| 18. | Sevenard, D. V.; Sosnovskikh, V. Y.; Kolomeitsev, A. A.; Königsmann, M. H.; Röschenthaler, G.-V. Tetrahedron Lett. 2003, 44, 7623–7627. doi:10.1016/j.tetlet.2003.08.050 |
| 22. | Wilger, D. J.; Gesmundo, N. J.; Nicewicz, D. A. Chem. Sci. 2013, 4, 3160–3165. doi:10.1039/c3sc51209f |
| 17. | Furukawa, T.; Nishimine, T.; Tokunaga, E.; Hasegawa, K.; Shiro, M.; Shibata, N. Org. Lett. 2011, 13, 3972–3975. doi:10.1021/ol201490e |
| 28. | Bizet, V.; Pannecoucke, X.; Renaud, J.-L.; Cahard, D. J. Fluorine Chem. 2013, 152, 56–61. doi:10.1016/j.jfluchem.2013.01.004 |
| 16. | Zemtsov, A. A.; Levin, V. V.; Dilman, A. D.; Struchkova, M. I.; Belyakov, P. A.; Tartakovsky, V. A.; Hu, J. Eur. J. Org. Chem. 2010, 6779–6785. doi:10.1002/ejoc.201001051 |
| 20. | Dilman, A. D.; Levin, V. V.; Belyakov, P. A.; Struchkova, M. I.; Tartakovsky, V. A. Tetrahedron Lett. 2008, 49, 4352–4354. doi:10.1016/j.tetlet.2008.05.039 |
| 29. | Maruoka, K.; Shimada, I.; Imoto, H.; Yamamoto, H. Synlett 1994, 519–520. doi:10.1055/s-1994-22912 |
© 2013 Okusu et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)