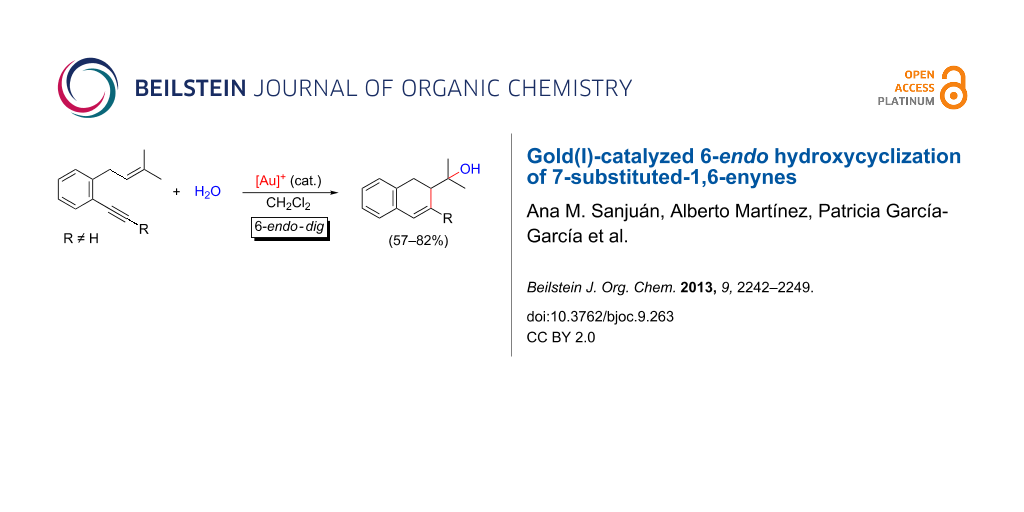
Abstract
The cyclization of o-(alkynyl)-3-(methylbut-2-enyl)benzenes, 1,6-enynes having a condensed aromatic ring at C3–C4 positions, has been studied under the catalysis of cationic gold(I) complexes. The selective 6-endo-dig mode of cyclization observed for the 7-substituted substrates in the presence of water or methanol giving rise to hydroxy(methoxy)-functionalized dihydronaphthalene derivatives is highly remarkable in the context of the observed reaction pathways for the cycloisomerizations of 1,6-enynes bearing a trisubstituted olefin.

Graphical Abstract
Introduction
The cycloisomerization reactions of enynes catalyzed by gold complexes are a powerful tool for accessing complex products from rather simple starting materials under soft and straightforward conditions [1-4]. In this context, 1,6-enynes have been extensively studied, mainly by Echavarren and co-workers, as substrates in the identification of new reactivities catalyzed by gold and other transition metal complexes [5-13]. Cyclopropyl metal carbenes II are usually formed by exo-dig processes from enynes I bearing a terminal alkyne, which in the absence of external nucleophiles undergo skeletal rearrangements to afford products such as III (single cleavage) [14]. However, reactions of II with alcohols or water give the corresponding products of alkoxy(hydroxy)cyclization IV [15-17] (Scheme 1). The less common 6-endo cyclization via metal carbenes V was also observed in particular cases affording methylenecyclohexene derivatives like VI [14]. On the other hand, 1,6-enynes VII, bearing an aryl substituent at the alkyne, undergo a formal intramolecular [4 + 2] cycloaddition through an initial 5-exo cyclization followed by a Friedel–Crafts-type reaction to cyclopenta[b]naphthalenes VIII or, alternatively, a 6-endo cyclization to bicyclo[4.1.0]hept-4-enes like IX [18,19] (Scheme 1). In the case that MeOH is present a 5-exo methoxycyclization is observed, e.g., in the formation of X resembling the behaviour of I [16,20]. In addition, the gold-catalyzed reaction of 7-phenyl-1,6-enynes with a terminal double bond gives rise to bicyclo[3.2.0]heptene derivatives [19,21].
![[1860-5397-9-263-i1]](/bjoc/content/inline/1860-5397-9-263-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Gold(I)-catalyzed reactions of 1,6-enynes.
Scheme 1: Gold(I)-catalyzed reactions of 1,6-enynes.
Despite the numerous studies about the metal-catalyzed transformations of 1,6-enynes, o-(alkynyl)-(3-methylbut-2-enyl)benzenes 1 that are also 1,6-enynes bearing an attached aryl ring at the C3–C4 positions, have been scarcely studied. Only Liu and co-workers have reported the behaviour of terminal substrates 1 (R = H) under ruthenium catalysis, which afford the corresponding metathesis-type product XI [22] (Scheme 2). More recently, the same authors have described the gold-catalyzed [2 + 2 + 3] cycloaddition reaction of these compounds with nitrones giving rise to functionalized 1,2-oxazepane derivatives XIII. This cascade process takes place through the interception of the 1,4-dipole equivalent XII generated by an initial 5-exo cyclization, although with some gold catalysts minor amounts of XI were also obtained [23] (Scheme 2). Following our interest in the development of new gold-catalyzed reactions [24-31], in this context we thought that it could be interesting to study if the cyclization of easily available compounds 1 bearing an internal acetylene moiety would take place through an initial 5-exo cyclization that in the case of aryl-substituted enynes (R = Ar) would give rise to a formal [4 + 2] cycloaddition product XIV [18,19], or alternatively, through a relatively less common 6-endo-dig pathway via gold species XV, which could be represented as two resonance structures highlighting both the carbocation or carbenoid nature of this intermediate (Scheme 2).
![[1860-5397-9-263-i2]](/bjoc/content/inline/1860-5397-9-263-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Cyclization of o-(alkynyl)-(3-methylbut-2-enyl)benzenes 1. Previous work and proposed pathways.
Scheme 2: Cyclization of o-(alkynyl)-(3-methylbut-2-enyl)benzenes 1. Previous work and proposed pathways.
Results and Discussion
As established in Scheme 2, we were intrigued by the possibility that o-(alkynyl)-(3-methylbut-2-enyl)benzenes 1 could undergo a 6-endo-dig cyclization in the presence of cationic gold(I) complexes instead of the usually more favoured 5-exo-dig pathway. So, we initially prepared a variety of these o-disubstituted benzene derivatives 1 by two approaches (see Supporting Information File 1) (Scheme 3). First, o-(bromo)-3-(methylbut-2-enyl)benzene was prepared by the reaction of commercially available 2-methyl-1-propenylmagnesium bromide with 2-bromobenzyl bromide in the presence of CuI and 2,2’-bipyridyl [32]. This aryl bromide could be coupled with selected terminal alkynes by using cesium carbonate as a base and PdCl2(MeCN)2/XPhos as a catalytic system [33]. Alternatively, several o-(alkynyl)bromobenzenes [34] could be transformed into the corresponding derivatives 1 by bromine–lithium exchange and further treatment with 3,3-dimethylallyl bromide in the presence of TMEDA [23].
![[1860-5397-9-263-i3]](/bjoc/content/inline/1860-5397-9-263-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of o-(alkynyl)-(3-methylbut-2-enyl)benzenes 1.
Scheme 3: Synthesis of o-(alkynyl)-(3-methylbut-2-enyl)benzenes 1.
We selected 1-(2-(2-(3-methylbut-2-enyl)phenyl)ethynyl)benzene (1a) as model substrate for the initial experiments (Scheme 4). Its reaction with (Ph3P)AuNTf2, reported by Gagosz and co-workers as a very active catalyst for the cycloisomerization of closely related 7-aryl-1,6-enynes [35], gave rise to a ca. 3:1 mixture of dihydronaphthalene derivative 2a and tetracyclic compound 3a along with some other unidentified minor products. The two major products resulted to be inseparable by column chromatography and were isolated in 68% overall yield. It is remarkable that compound 2a, derived from a 6-endo cyclization and further proton elimination from intermediate resonance structures 4a and 4a’, is generated in preference to 3a which would be the expected product derived from a formal [4 + 2] cycloaddition initiated by a 5-exo cyclization followed by a Friedel–Crafts-type process in intermediate 5a or 5a’, as described by Echavarren and co-workers [18,19].
![[1860-5397-9-263-i4]](/bjoc/content/inline/1860-5397-9-263-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Gold(I)-catalyzed cycloisomerization of 1a.
Scheme 4: Gold(I)-catalyzed cycloisomerization of 1a.
Prompted by this result and taking into account the reported results about the 5-endo hydroxy- and alkoxycyclization of 1,5-enynes [36], as well as our recent report about the alkoxycyclization of 1,3-dien-5-ynes [31], we wondered if the presence of an external protic nucleophile, such as methanol or water, could have an important influence on controlling the selectivity of the reaction. Encouragingly, when we treated model substrate 1a with (Ph3P)AuNTf2 in a 10:1 mixture of CH2Cl2 and MeOH as the solvent, the methoxyalkyl-substituted derivative 6a was obtained as the major product along with minor amounts of 3a (ca. 6:1 ratio) (Scheme 5) [37]. Moreover, the use of H2O (20 equiv) also led to a high yield of the hydroxyalkyl-substituted dihydronaphthalene derivative 7a, whose structure was further confirmed by X-ray analysis [38]. In both cases the high selectivity (>5:1) of these reactions for the 6-endo-type cyclization should be noted and only minor amounts (10–15%) of 3a were also formed.
![[1860-5397-9-263-i5]](/bjoc/content/inline/1860-5397-9-263-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Initial experiments and proof of concept.
Scheme 5: Initial experiments and proof of concept.
Due to the unexpected 6-endo-favored pathway found for substrate 1a [39], we attempted to further improve this selectivity in the hydroxycyclization process (Table 1). Switching the ligand from Ph3P to XPhos or N-heterocyclic carbene (IPr) slightly decreases the selectivity for the 6-endo cyclization (Table 1, entry 1 vs entries 2 and 3). However, when the cationic gold complex (JohnPhos)(NCMe)AuSbF6, developed by Echavarren and co-workers [40], was employed as a catalyst a moderate increase in the ratio of 7a vs 3a was observed (Table 1, entry 4). Both cationic gold complexes (Ph3P)AuNTf2 and (JohnPhos)(NCMe)AuSbF6 gave rise to a similar yield of isolated alcohol 7a. Changing the solvent from CH2Cl2 to a mixture containing other more polar solvent such as acetone or dioxane (Table 1, entries 5 and 6) did not have a significant influence on the selectivity but led to the formation of minor amounts of alcohol 8a, derived from a 5-exo hydroxycyclization reaction. With a 1:1 mixture of CH2Cl2/dioxane the effect of the selected catalytic systems was checked (Table 1, entries 7–10). We found that the use of JohnPhos as a ligand and SbF6 as a counter ion (Table 1, entries 9 and 10) resulted in a slightly better selectivity, although trace amounts of alcohol 8a were also generated, which make the isolation of 7a more difficult. Overall, we concluded that both commercially available gold complexes (Ph3P)AuNTf2 and (JohnPhos)(NCMe)AuSbF6 lead to comparable good results in the 6-endo hydroxycyclization of 1a. The type of products derived from the 5-exo pathway (3a and 8a) depends on the solvent: in CH2Cl2 3a is mainly obtained, whereas the alcohol 8a appears when a more polar mixture of solvents was used.
Table 1: Effect of the catalyst and reaction conditions on the hydroxycyclization of 1a. 6-Endo vs 5-exo cyclization.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-263-i9.svg?max-width=637&scale=1.0)
|
||||
| Entry | Catalyst | Solvent |
Ratiob
6-endo/5-exo |
Yield (%)c |
|---|---|---|---|---|
| 1 | (Ph3P)AuNTf2 | CH2Cl2 | 5:1d | 76 |
| 2 | XPhosAuNTf2e | CH2Cl2 | 3:1d | — |
| 3 | IPrAuCl/AgSbF6f | CH2Cl2 | 4:1d | — |
| 4 | (JohnPhos)(NCMe)AuSbF6g | CH2Cl2 | 6:1d | 77 |
| 5 | (Ph3P)AuNTf2 | CH2Cl2/Me2CO (1:1) | 4.5:1h | — |
| 6 | (Ph3P)AuNTf2 | CH2Cl2/dioxane (1:1) | 5:1h | 75i |
| 7 | XPhosAuNTf2e | CH2Cl2/dioxane (1:1) | 3:1h | — |
| 8 | JohnPhosAuNTf2g | CH2Cl2/dioxane (1:1) | 3:1h | — |
| 9 | (JohnPhos)(NCMe)AuSbF6g | CH2Cl2/dioxane (1:1) | 7:1h | 77i |
| 10 | JohnPhosAuCl/AgSbF6g | CH2Cl2/dioxane (1:1) | 7:1h | — |
aReactions were carried out by treatment of 1a (0.1 mmol) with H2O (2.2 mmol, 0.04 mL) in 0.4 mL of solvent until complete consumption of the starting material, as judged by GC–MS and/or TLC analysis (overnight). bDetermined by 1H NMR analysis of the crude reaction mixture. cIsolated yield of 7a. dThe 5-exo pathway gives rise to 3a. eXPhos = 2-dicyclohexylphosphino-2’,4’,6’-tri-isopropylbiphenyl. fIPr = 1,3-bis-(2,6-di-isopropylphenyl)imidazol-2-ylidene. gJohnPhos = 2-(di-tert-butylphosphino)biphenyl. hA mixture of 3a and 8a was obtained through the 5-exo pathway. iApproximately 5% of 8a was also isolated.
Once we have selected the best conditions to favor the 6-endo hydroxycyclization reaction, a selection of substrates 1a–k, bearing different groups at the triple bond, were reacted under the established conditions (Table 2). When aromatic or alkenyl groups are present as the substituents of the alkyne (Table 2, entries 1–7) the 6-endo cyclization takes place in selective or almost exclusively fashion allowing the isolation of 2-(1,2-dihydro-3-substituted naphthalen-2-yl)propan-2-ol derivatives 7 in usually high yields. Interestingly, we have also observed that when starting with enynes possessing an electron-rich aromatic ring or an alkenyl group at the C7-position of the 1,6-enyne the cyclization results almost completely selective via the 6-endo mode (Table 2, entries 2,3 and entries 6,7). However, in the case of halogen-containing aromatic substituents at C7 the formation of the corresponding products 3 or 8, derived from an initial 5-exo cyclization, becomes more competitive (Table 2, entries 4 and 5). Then, we turned our attention to alkyl-substituted alkynes (Table 2, entries 8 and 9), which could not undergo the formal [4 + 2] cycloaddition leading to 3. In these cases, and after some optimization studies, we surprisingly found that the solvent has an important role on the selectivity of the cyclization. When a 1:1 mixture of CH2Cl2/dioxane was used the 5-exo hydroxycyclization that gives rise to alcohols 8 was competitive with the 6-endo process (3:1 for 1h and 1.7:1 for 1i), allowing the isolation of the corresponding methyleneindene derivatives 8h and 8i in 21% and 30% yield, respectively [41]. Gratifyingly, we found that when the same reactions were performed in CH2Cl2 the 6-endo cyclization was completely selective leading to the corresponding alcohols 7 in high yields (Table 2, entries 8 and 9). On the other hand, the reaction of trimethylsilyl-substituted enyne 1j did not proceed at all (Table 2, entry 10), whereas the presence of a phenylthio group as an R substituent mainly afforded the corresponding 6-endo product 7k although the reaction was significantly slower (Table 2, entry 11). As expected [10-12] the terminal enyne 1l (R = H) underwent exclusively the 5-exo cyclization leading to the corresponding alcohol 8l in 55% yield (Table 2, entry 12).
Table 2: Synthesis of 2-(1,2-dihydro-3-substituted-naphthalen-2-yl)propan-2-ol derivatives 7 by gold-catalyzed 6-endo hydroxycyclization of enynes 1.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-263-i10.svg?max-width=637&scale=1.0)
|
||||
| Entry | Starting material | R | Product | Yield (%)b |
|---|---|---|---|---|
| 1 | 1a | Ph | 7a | 77 (12)c |
| 2 | 1b | 4-MeOC6H4 | 7b | 80 |
| 3 | 1c | 2,4,5-(Me)3C6H2 | 7c | 71 |
| 4 | 1d | 3-ClC6H4 | 7d | 63 (22)d |
| 5 | 1e | 2,4-(F)2C6H3 | 7e | 75e |
| 6 | 1f | thiophen-3-yl | 7f | 82 |
| 7 | 1g | c-C6H9 | 7g | 79 |
| 8f | 1h | c-C3H5 | 7h | 77 |
| 9 | 1i | n-Bu | 7i | 82 |
| 10 | 1j | SiMe3 | — | —g |
| 11h | 1k | SPh | 7k | 60 |
| 12 | 1l | H | 8l | 55 |
aReactions were carried out by treatment of 1 (0.3 mmol) with H2O (22 equiv, 0.12 mL) in 1.2 mL of solvent until complete consumption of the starting material, as judged by GC–MS and/or TLC analysis (overnight). bIsolated yield of compounds 7 after column chromatography. cYield of 3a which could not be isolated in pure form. dIsolated yield of 3d which was obtained as a mixture of regioisomers with respect to the chlorine atom position. eIsolated along with ≈10% of 8e. ≈10% of 2e is also observed. fCarried out with (Ph3P)AuNTf2. Slightly lower yield (ca. 5%) was obtained with (JohnPhos)(NCMe)AuSbF6. gStarting material was recovered. hReaction time: 48 h.
At this point we wondered if the cyclization would be diastereoselective, so we prepared enynes 1m by reacting 2-(phenylethynyl)phenyllithium with geranyl bromide and 1n by two Wittig reactions from 2’-(phenylethynyl)acetophenone (see Supporting Information File 1). First, the hydroxycyclization of 1m, as a pure E isomer, under the previously established conditions afforded the dihydronaphthalene derivative 7m as a single isomer (Scheme 6), whose relative configuration was assigned by analogy with previously related results reported by Gagosz and co-workers [37]. On the other hand, the reaction of 1n afforded a ca. 2.5:1 mixture of alcohol 7n [42] and the tetracyclic product 3n, derived from an initial 5-exo cyclization and subsequent Friedel–Crafts reaction (Scheme 6). Both compounds were isolated as single stereoisomers with a high overall yield [43]. In this case, the 5-exo pathway was more competitive compared to the result of model substrate 1a, probably due to the Thorpe–Ingold-type effect caused by the methyl group at the allylic position. To account for the stereoselectivity of these reactions we proposed the generation of a stabilized gold–carbenoid intermediate such as A that undergoes stereoselective attack by water (Scheme 6).
![[1860-5397-9-263-i6]](/bjoc/content/inline/1860-5397-9-263-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Gold(I)-catalyzed hydroxycyclization of enynes 1m,n.
Scheme 6: Gold(I)-catalyzed hydroxycyclization of enynes 1m,n.
Furthermore, we have also carried out the methoxycyclization of selected 1,6-enynes 1 by their treatment with catalytic amounts of (JohnPhos)(NCMe)AuSbF6 in a 30:1 mixture of CH2Cl2 and MeOH as the solvent (Scheme 7) [44]. The corresponding methoxy-functionalized dihydronaphthalene derivatives 6 were obtained in high yields although the corresponding minor isomer derived from a 5-exo cyclization could not be separated in the case of 6a and 6h.
![[1860-5397-9-263-i7]](/bjoc/content/inline/1860-5397-9-263-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Gold(I)-catalyzed methoxycyclization of selected 1,6-enynes 1 [45].
Scheme 7: Gold(I)-catalyzed methoxycyclization of selected 1,6-enynes 1 [45].
Finally, to support the proposed intermediacy of gold–carbenoid intermediate 4 or 4’ (Scheme 4), we treated enyne 1b with D2O instead of water and under the same catalytic conditions we observed the exclusive formation of the deuterated compound [D]-7b in 75% yield (>90% deuterium incorporation at C4). The generation of that compound could be explained by deuterodemetallation of the vinylgold species B generated by an attack of the nucleophile on intermediate 4b or 4b’ (Scheme 8).
![[1860-5397-9-263-i8]](/bjoc/content/inline/1860-5397-9-263-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Labelling experiment and proposed mechanism.
Scheme 8: Labelling experiment and proposed mechanism.
Conclusion
We described an efficient gold(I)-catalyzed 6-endo hydroxycyclization of 7-substituted 1,6-enynes bearing a condensed aromatic ring at the C3–C4 position of the enyne. This type of cyclization has not been previously observed for 1,6-enynes bearing trisubstituted olefins and represents a new addition to the observed reaction topologies in the gold-catalyzed cycloisomerization of these substrates. The new oxygen-functionalized dihydronaphthalene derivatives have been synthesized in high yields.
Supporting Information
Experimental procedures and spectroscopic data for all new compounds. Copies of 1H NMR and 13C NMR spectra for new compounds.
| Supporting Information File 1: Experimental and analytical data. | ||
| Format: PDF | Size: 262.7 KB | Download |
| Supporting Information File 2: NMR spectra. | ||
| Format: PDF | Size: 1.6 MB | Download |
Acknowledgements
We gratefully acknowledge the Ministerio de Ciencia e Innovación (MICINN) and FEDER (CTQ2010-15358) for financial support. A. M. S. thanks the Junta de Castilla y León (Consejería de Educación) and the Fondo Social Europeo for a PIRTU contract. P. G.-G. and M. A. F.-R. thank MICINN for “Juan de la Cierva” and “Ramón y Cajal” contracts.
References
-
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319
Return to citation in text: [1] -
Zhang, L.; Sun, J.; Kozmin, S. A. Adv. Synth. Catal. 2006, 348, 2271–2296. doi:10.1002/adsc.200600368
Return to citation in text: [1] -
Michelet, V.; Toullec, P. Y.; Genét, J.-P. Angew. Chem., Int. Ed. 2008, 47, 4268–4315. doi:10.1002/anie.200701589
Return to citation in text: [1] -
Sohel, S. M. A.; Liu, R.-S. Chem. Soc. Rev. 2009, 38, 2269–2281. doi:10.1039/b807499m
Return to citation in text: [1] -
Nieto-Oberhuber, C.; López, S.; Muñoz, M. P.; Cárdenas, D. J.; Buñuel, E.; Nevado, C.; Echavarren, A. M. Angew. Chem., Int. Ed. 2005, 44, 6146–6148. doi:10.1002/anie.200501937
Return to citation in text: [1] -
Fürstner, A.; Davies, P. W.; Gress, T. J. Am. Chem. Soc. 2005, 127, 8244–8245. doi:10.1021/ja050845g
Return to citation in text: [1] -
Jiménez-Núñez, E.; Claverie, C. K.; Bour, C.; Cárdenas, D. J.; Echavarren, A. M. Angew. Chem., Int. Ed. 2008, 47, 7892–7895. doi:10.1002/anie.200803269
Return to citation in text: [1] -
Jiménez-Núñez, E.; Raducan, M.; Lauterbach, T.; Molawi, K.; Solorio, C. R.; Echavarren, A. M. Angew. Chem., Int. Ed. 2009, 48, 6152–6155. doi:10.1002/anie.200902248
Return to citation in text: [1] -
Escribano-Cuesta, A.; López-Carrillo, V.; Janssen, D.; Echavarren, A. M. Chem.–Eur. J. 2009, 15, 5646–5650. doi:10.1002/chem.200900668
Return to citation in text: [1] -
Pérez-Galán, P.; Martin, N. J. A.; Campaña, A. G.; Cárdenas, D. J.; Echavarren, A. M. Chem.–Asian J. 2011, 6, 482–486. doi:10.1002/asia.201000557
Return to citation in text: [1] [2] -
Solorio-Alvarado, C. R.; Echavarren, A. M. J. Am. Chem. Soc. 2010, 132, 11881–11883. doi:10.1021/ja104743k
Return to citation in text: [1] [2] -
Pérez-Galán, P.; Herrero-Gómez, E.; Hog, D. T.; Martin, N. J. A.; Maseras, F.; Echavarren, A. M. Chem. Sci. 2011, 2, 141–149. doi:10.1039/c0sc00335b
Return to citation in text: [1] [2] -
Sakiyama, N.; Noguchi, K.; Tanaka, K. Angew. Chem., Int. Ed. 2012, 51, 5976–5980. doi:10.1002/anie.201201186
Return to citation in text: [1] -
Nieto-Oberhuber, C.; Muñoz, M. P.; Buñuel, E.; Nevado, C.; Cárdenas, D. J.; Echavarren, A. M. Angew. Chem., Int. Ed. 2004, 43, 2402–2406. doi:10.1002/anie.200353207
Return to citation in text: [1] [2] -
Muñoz, M. P.; Adrio, J.; Carretero, J. C.; Echavarren, A. M. Organometallics 2005, 24, 1293–1300. doi:10.1021/om0491645
Return to citation in text: [1] -
Nieto-Oberhuber, C.; Muñoz, M. P.; López, S.; Jiménez-Núñez, E.; Nevado, C.; Herrero-Gómez, E.; Raducan, M.; Echavarren, A. M. Chem.–Eur. J. 2006, 12, 1677–1693. doi:10.1002/chem.200501088
Return to citation in text: [1] [2] -
Chao, C.-M.; Genin, E.; Toullec, P. Y.; Genêt, J.-P.; Michelet, V. J. Organomet. Chem. 2009, 694, 538–545. doi:10.1016/j.jorganchem.2008.08.008
Return to citation in text: [1] -
Nieto-Oberhuber, C.; López, S.; Echavarren, A. M. J. Am. Chem. Soc. 2005, 127, 6178–6179. doi:10.1021/ja042257t
Return to citation in text: [1] [2] [3] -
Nieto-Oberhuber, C.; Pérez-Galán, P.; Herrero-Gómez, E.; Lauterbach, T.; Rodríguez, C.; López, S.; Bour, C.; Rosellón, A.; Cárdenas, D. J.; Echavarren, A. M. J. Am. Chem. Soc. 2008, 130, 269–279. doi:10.1021/ja075794x
Return to citation in text: [1] [2] [3] [4] -
However, the addition of carbon nucleophiles to 1,6-enynes bearing internal alkynes and disubstituted olefins usually takes place through a 6-endo cyclization affording six-membered cycles.
Amijs, C. H. M.; López-Carrillo, V.; Raducan, M.; Pérez-Galán, P.; Ferrer, C.; Echavarren, A. M. J. Org. Chem. 2008, 73, 7721–7730. doi:10.1021/jo8014769
Return to citation in text: [1] -
Brooner, R. E. M.; Brown, T. J.; Widenhoefer, R. A. Angew. Chem., Int. Ed. 2013, 52, 6259–6261. doi:10.1002/anie.201301640
Return to citation in text: [1] -
Madhushaw, R. J.; Lo, C.-Y.; Hwang, C.-W.; Su, M.-D.; Shen, H.-C.; Pal, S.; Shaikh, I. R.; Liu, R.-S. J. Am. Chem. Soc. 2004, 126, 15560–15565. doi:10.1021/ja045516n
Return to citation in text: [1] -
Gawade, S. A.; Bhunia, S.; Liu, R.-S. Angew. Chem., Int. Ed. 2012, 51, 7835–7838. doi:10.1002/anie.201203507
Return to citation in text: [1] [2] -
Sanz, R.; Miguel, D.; Rodríguez, F. Angew. Chem., Int. Ed. 2008, 47, 7354–7357. doi:10.1002/anie.200802660
Return to citation in text: [1] -
Sanz, R.; Miguel, D.; Gohain, M.; García-García, P.; Fernández-Rodríguez, M. A.; González-Pérez, A.; Nieto-Faza, O.; de Lera, A. R.; Rodríguez, F. Chem.–Eur. J. 2010, 16, 9818–9828. doi:10.1002/chem.201001162
Return to citation in text: [1] -
Álvarez, E.; Miguel, D.; García-García, P.; Fernández-Rodríguez, M. A.; Rodríguez, F.; Sanz, R. Beilstein J. Org. Chem. 2011, 7, 786–793. doi:10.3762/bjoc.7.89
Return to citation in text: [1] -
Álvarez, E.; Miguel, D.; García-García, P.; Fernández-Rodríguez, M. A.; Rodríguez, F.; Sanz, R. Synthesis 2012, 1874–1884. doi:10.1055/s-0031-1290950
Return to citation in text: [1] -
Martínez, A.; García-García, P.; Fernández-Rodríguez, M. A.; Rodríguez, F.; Sanz, R. Angew. Chem., Int. Ed. 2010, 49, 4633–4637. doi:10.1002/anie.201001089
Return to citation in text: [1] -
García-García, P.; Martínez, A.; Sanjuán, A. M.; Fernández-Rodríguez, M. A.; Sanz, R. Org. Lett. 2011, 13, 4970–4973. doi:10.1021/ol202129n
Return to citation in text: [1] -
García-García, P.; Rashid, M. A.; Sanjuán, A. M.; Fernández-Rodríguez, M. A.; Sanz, R. Org. Lett. 2012, 14, 4778–4781. doi:10.1021/ol3020682
Return to citation in text: [1] -
Sanjuán, A. M.; García-García, P.; Fernández-Rodríguez, M. A.; Sanz, R. Adv. Synth. Catal. 2013, 355, 1955–1962. doi:10.1002/adsc.201300448
Return to citation in text: [1] [2] -
Knight, J.; Parsons, P. J. J. Chem. Soc., Perkin Trans. 1 1989, 979–984. doi:10.1039/P19890000979
Return to citation in text: [1] -
Gelman, D.; Buchwald, S. L. Angew. Chem., Int. Ed. 2003, 42, 5993–5996. doi:10.1002/anie.200353015
Return to citation in text: [1] -
Prepared by Sonogashira coupling of o-bromoiodobenzene with terminal acetylenes. See, for instance.
Guilarte, V.; Fernández-Rodríguez, M. A.; García-García, P.; Hernando, E.; Sanz, R. Org. Lett. 2011, 13, 5100–5103. doi:10.1021/ol201970m
Return to citation in text: [1] -
Mézailles, N.; Ricard, L.; Gagosz, F. Org. Lett. 2005, 7, 4133–4136. doi:10.1021/ol0515917
Return to citation in text: [1] -
Buzas, A. K.; Istrate, F. M.; Gagosz, F. Angew. Chem., Int. Ed. 2007, 46, 1141–1144. doi:10.1002/anie.200604140
Return to citation in text: [1] -
However, 6a was isolated with trace amounts of a by-product that could be the corresponding methoxyalkyl-substituted product derived from a 5-exo cyclization.
Return to citation in text: [1] [2] -
CCDC-945504 contains the supplementary crystallographic data for compound 7a. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre at http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Jeganmohan, M.; Bhuwaneswari, S.; Cheng, C.-H. Angew. Chem., Int. Ed. 2009, 48, 391–394. doi:10.1002/anie.200804873
Return to citation in text: [1] -
Nieto-Oberhuber, C.; López, S.; Jiménez-Núñez, E.; Echavarren, A. M. Chem.–Eur. J. 2006, 12, 5916–5923. doi:10.1002/chem.200600174
Return to citation in text: [1] -
Under these conditions with an CH2Cl2/dioxane as solvent, 7h and 7i were isolated in 61% and 52% yield, respectively.
Return to citation in text: [1] -
Isolated with trace amounts of 8n.
Return to citation in text: [1] -
Their structures were established by NMR experiments.
Return to citation in text: [1] -
A brief screening of gold catalysts showed that, in analogy with the hydroxycyclization reaction, (JohnPhos)(NCMe)AuSbF6 afforded the best results in terms of selectivity and chemical yield.
Return to citation in text: [1] -
In the methoxycyclization of 1h the 6-endo methoxy ether 6h and its 5-membered isomer derived from the 5-exo cyclization were obtained approximately in a 4:1 ratio (80% overall yield). For 1a only trace amounts of the 5-membered ring were observed.
Return to citation in text: [1]
| 44. | A brief screening of gold catalysts showed that, in analogy with the hydroxycyclization reaction, (JohnPhos)(NCMe)AuSbF6 afforded the best results in terms of selectivity and chemical yield. |
| 1. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319 |
| 2. | Zhang, L.; Sun, J.; Kozmin, S. A. Adv. Synth. Catal. 2006, 348, 2271–2296. doi:10.1002/adsc.200600368 |
| 3. | Michelet, V.; Toullec, P. Y.; Genét, J.-P. Angew. Chem., Int. Ed. 2008, 47, 4268–4315. doi:10.1002/anie.200701589 |
| 4. | Sohel, S. M. A.; Liu, R.-S. Chem. Soc. Rev. 2009, 38, 2269–2281. doi:10.1039/b807499m |
| 14. | Nieto-Oberhuber, C.; Muñoz, M. P.; Buñuel, E.; Nevado, C.; Cárdenas, D. J.; Echavarren, A. M. Angew. Chem., Int. Ed. 2004, 43, 2402–2406. doi:10.1002/anie.200353207 |
| 34. |
Prepared by Sonogashira coupling of o-bromoiodobenzene with terminal acetylenes. See, for instance.
Guilarte, V.; Fernández-Rodríguez, M. A.; García-García, P.; Hernando, E.; Sanz, R. Org. Lett. 2011, 13, 5100–5103. doi:10.1021/ol201970m |
| 15. | Muñoz, M. P.; Adrio, J.; Carretero, J. C.; Echavarren, A. M. Organometallics 2005, 24, 1293–1300. doi:10.1021/om0491645 |
| 16. | Nieto-Oberhuber, C.; Muñoz, M. P.; López, S.; Jiménez-Núñez, E.; Nevado, C.; Herrero-Gómez, E.; Raducan, M.; Echavarren, A. M. Chem.–Eur. J. 2006, 12, 1677–1693. doi:10.1002/chem.200501088 |
| 17. | Chao, C.-M.; Genin, E.; Toullec, P. Y.; Genêt, J.-P.; Michelet, V. J. Organomet. Chem. 2009, 694, 538–545. doi:10.1016/j.jorganchem.2008.08.008 |
| 23. | Gawade, S. A.; Bhunia, S.; Liu, R.-S. Angew. Chem., Int. Ed. 2012, 51, 7835–7838. doi:10.1002/anie.201203507 |
| 14. | Nieto-Oberhuber, C.; Muñoz, M. P.; Buñuel, E.; Nevado, C.; Cárdenas, D. J.; Echavarren, A. M. Angew. Chem., Int. Ed. 2004, 43, 2402–2406. doi:10.1002/anie.200353207 |
| 32. | Knight, J.; Parsons, P. J. J. Chem. Soc., Perkin Trans. 1 1989, 979–984. doi:10.1039/P19890000979 |
| 5. | Nieto-Oberhuber, C.; López, S.; Muñoz, M. P.; Cárdenas, D. J.; Buñuel, E.; Nevado, C.; Echavarren, A. M. Angew. Chem., Int. Ed. 2005, 44, 6146–6148. doi:10.1002/anie.200501937 |
| 6. | Fürstner, A.; Davies, P. W.; Gress, T. J. Am. Chem. Soc. 2005, 127, 8244–8245. doi:10.1021/ja050845g |
| 7. | Jiménez-Núñez, E.; Claverie, C. K.; Bour, C.; Cárdenas, D. J.; Echavarren, A. M. Angew. Chem., Int. Ed. 2008, 47, 7892–7895. doi:10.1002/anie.200803269 |
| 8. | Jiménez-Núñez, E.; Raducan, M.; Lauterbach, T.; Molawi, K.; Solorio, C. R.; Echavarren, A. M. Angew. Chem., Int. Ed. 2009, 48, 6152–6155. doi:10.1002/anie.200902248 |
| 9. | Escribano-Cuesta, A.; López-Carrillo, V.; Janssen, D.; Echavarren, A. M. Chem.–Eur. J. 2009, 15, 5646–5650. doi:10.1002/chem.200900668 |
| 10. | Pérez-Galán, P.; Martin, N. J. A.; Campaña, A. G.; Cárdenas, D. J.; Echavarren, A. M. Chem.–Asian J. 2011, 6, 482–486. doi:10.1002/asia.201000557 |
| 11. | Solorio-Alvarado, C. R.; Echavarren, A. M. J. Am. Chem. Soc. 2010, 132, 11881–11883. doi:10.1021/ja104743k |
| 12. | Pérez-Galán, P.; Herrero-Gómez, E.; Hog, D. T.; Martin, N. J. A.; Maseras, F.; Echavarren, A. M. Chem. Sci. 2011, 2, 141–149. doi:10.1039/c0sc00335b |
| 13. | Sakiyama, N.; Noguchi, K.; Tanaka, K. Angew. Chem., Int. Ed. 2012, 51, 5976–5980. doi:10.1002/anie.201201186 |
| 33. | Gelman, D.; Buchwald, S. L. Angew. Chem., Int. Ed. 2003, 42, 5993–5996. doi:10.1002/anie.200353015 |
| 22. | Madhushaw, R. J.; Lo, C.-Y.; Hwang, C.-W.; Su, M.-D.; Shen, H.-C.; Pal, S.; Shaikh, I. R.; Liu, R.-S. J. Am. Chem. Soc. 2004, 126, 15560–15565. doi:10.1021/ja045516n |
| 24. | Sanz, R.; Miguel, D.; Rodríguez, F. Angew. Chem., Int. Ed. 2008, 47, 7354–7357. doi:10.1002/anie.200802660 |
| 25. | Sanz, R.; Miguel, D.; Gohain, M.; García-García, P.; Fernández-Rodríguez, M. A.; González-Pérez, A.; Nieto-Faza, O.; de Lera, A. R.; Rodríguez, F. Chem.–Eur. J. 2010, 16, 9818–9828. doi:10.1002/chem.201001162 |
| 26. | Álvarez, E.; Miguel, D.; García-García, P.; Fernández-Rodríguez, M. A.; Rodríguez, F.; Sanz, R. Beilstein J. Org. Chem. 2011, 7, 786–793. doi:10.3762/bjoc.7.89 |
| 27. | Álvarez, E.; Miguel, D.; García-García, P.; Fernández-Rodríguez, M. A.; Rodríguez, F.; Sanz, R. Synthesis 2012, 1874–1884. doi:10.1055/s-0031-1290950 |
| 28. | Martínez, A.; García-García, P.; Fernández-Rodríguez, M. A.; Rodríguez, F.; Sanz, R. Angew. Chem., Int. Ed. 2010, 49, 4633–4637. doi:10.1002/anie.201001089 |
| 29. | García-García, P.; Martínez, A.; Sanjuán, A. M.; Fernández-Rodríguez, M. A.; Sanz, R. Org. Lett. 2011, 13, 4970–4973. doi:10.1021/ol202129n |
| 30. | García-García, P.; Rashid, M. A.; Sanjuán, A. M.; Fernández-Rodríguez, M. A.; Sanz, R. Org. Lett. 2012, 14, 4778–4781. doi:10.1021/ol3020682 |
| 31. | Sanjuán, A. M.; García-García, P.; Fernández-Rodríguez, M. A.; Sanz, R. Adv. Synth. Catal. 2013, 355, 1955–1962. doi:10.1002/adsc.201300448 |
| 19. | Nieto-Oberhuber, C.; Pérez-Galán, P.; Herrero-Gómez, E.; Lauterbach, T.; Rodríguez, C.; López, S.; Bour, C.; Rosellón, A.; Cárdenas, D. J.; Echavarren, A. M. J. Am. Chem. Soc. 2008, 130, 269–279. doi:10.1021/ja075794x |
| 21. | Brooner, R. E. M.; Brown, T. J.; Widenhoefer, R. A. Angew. Chem., Int. Ed. 2013, 52, 6259–6261. doi:10.1002/anie.201301640 |
| 18. | Nieto-Oberhuber, C.; López, S.; Echavarren, A. M. J. Am. Chem. Soc. 2005, 127, 6178–6179. doi:10.1021/ja042257t |
| 19. | Nieto-Oberhuber, C.; Pérez-Galán, P.; Herrero-Gómez, E.; Lauterbach, T.; Rodríguez, C.; López, S.; Bour, C.; Rosellón, A.; Cárdenas, D. J.; Echavarren, A. M. J. Am. Chem. Soc. 2008, 130, 269–279. doi:10.1021/ja075794x |
| 16. | Nieto-Oberhuber, C.; Muñoz, M. P.; López, S.; Jiménez-Núñez, E.; Nevado, C.; Herrero-Gómez, E.; Raducan, M.; Echavarren, A. M. Chem.–Eur. J. 2006, 12, 1677–1693. doi:10.1002/chem.200501088 |
| 20. |
However, the addition of carbon nucleophiles to 1,6-enynes bearing internal alkynes and disubstituted olefins usually takes place through a 6-endo cyclization affording six-membered cycles.
Amijs, C. H. M.; López-Carrillo, V.; Raducan, M.; Pérez-Galán, P.; Ferrer, C.; Echavarren, A. M. J. Org. Chem. 2008, 73, 7721–7730. doi:10.1021/jo8014769 |
| 45. | In the methoxycyclization of 1h the 6-endo methoxy ether 6h and its 5-membered isomer derived from the 5-exo cyclization were obtained approximately in a 4:1 ratio (80% overall yield). For 1a only trace amounts of the 5-membered ring were observed. |
| 18. | Nieto-Oberhuber, C.; López, S.; Echavarren, A. M. J. Am. Chem. Soc. 2005, 127, 6178–6179. doi:10.1021/ja042257t |
| 19. | Nieto-Oberhuber, C.; Pérez-Galán, P.; Herrero-Gómez, E.; Lauterbach, T.; Rodríguez, C.; López, S.; Bour, C.; Rosellón, A.; Cárdenas, D. J.; Echavarren, A. M. J. Am. Chem. Soc. 2008, 130, 269–279. doi:10.1021/ja075794x |
| 23. | Gawade, S. A.; Bhunia, S.; Liu, R.-S. Angew. Chem., Int. Ed. 2012, 51, 7835–7838. doi:10.1002/anie.201203507 |
| 36. | Buzas, A. K.; Istrate, F. M.; Gagosz, F. Angew. Chem., Int. Ed. 2007, 46, 1141–1144. doi:10.1002/anie.200604140 |
| 35. | Mézailles, N.; Ricard, L.; Gagosz, F. Org. Lett. 2005, 7, 4133–4136. doi:10.1021/ol0515917 |
| 18. | Nieto-Oberhuber, C.; López, S.; Echavarren, A. M. J. Am. Chem. Soc. 2005, 127, 6178–6179. doi:10.1021/ja042257t |
| 19. | Nieto-Oberhuber, C.; Pérez-Galán, P.; Herrero-Gómez, E.; Lauterbach, T.; Rodríguez, C.; López, S.; Bour, C.; Rosellón, A.; Cárdenas, D. J.; Echavarren, A. M. J. Am. Chem. Soc. 2008, 130, 269–279. doi:10.1021/ja075794x |
| 10. | Pérez-Galán, P.; Martin, N. J. A.; Campaña, A. G.; Cárdenas, D. J.; Echavarren, A. M. Chem.–Asian J. 2011, 6, 482–486. doi:10.1002/asia.201000557 |
| 11. | Solorio-Alvarado, C. R.; Echavarren, A. M. J. Am. Chem. Soc. 2010, 132, 11881–11883. doi:10.1021/ja104743k |
| 12. | Pérez-Galán, P.; Herrero-Gómez, E.; Hog, D. T.; Martin, N. J. A.; Maseras, F.; Echavarren, A. M. Chem. Sci. 2011, 2, 141–149. doi:10.1039/c0sc00335b |
| 37. | However, 6a was isolated with trace amounts of a by-product that could be the corresponding methoxyalkyl-substituted product derived from a 5-exo cyclization. |
| 40. | Nieto-Oberhuber, C.; López, S.; Jiménez-Núñez, E.; Echavarren, A. M. Chem.–Eur. J. 2006, 12, 5916–5923. doi:10.1002/chem.200600174 |
| 41. | Under these conditions with an CH2Cl2/dioxane as solvent, 7h and 7i were isolated in 61% and 52% yield, respectively. |
| 38. | CCDC-945504 contains the supplementary crystallographic data for compound 7a. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre at http://www.ccdc.cam.ac.uk/data_request/cif. |
| 39. | Jeganmohan, M.; Bhuwaneswari, S.; Cheng, C.-H. Angew. Chem., Int. Ed. 2009, 48, 391–394. doi:10.1002/anie.200804873 |
| 31. | Sanjuán, A. M.; García-García, P.; Fernández-Rodríguez, M. A.; Sanz, R. Adv. Synth. Catal. 2013, 355, 1955–1962. doi:10.1002/adsc.201300448 |
| 37. | However, 6a was isolated with trace amounts of a by-product that could be the corresponding methoxyalkyl-substituted product derived from a 5-exo cyclization. |
© 2013 Sanjuán et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)