Abstract
A convenient and efficient methodology for the synthesis of densely substituted pyrrole-fused isocoumarins, which employs solid-supported silica sulfuric acid (SSA) as catalyst, has been developed. When the mixture of ninhydrin adducts of acetylacetone/ethyl acetoacetate and primary amines was heated on the solid surface of SSA under solvent-free conditions, the pyrrole-fused isocoumarins were formed in good yields. This synthetic method has several advantages such as the employment of solvent-free reaction conditions without the use of any toxic reagents and metal catalysts, the ease of product isolation, the use of a recyclable catalyst, the low cost, the easy availability of the starting materials, and the excellent yields of products.

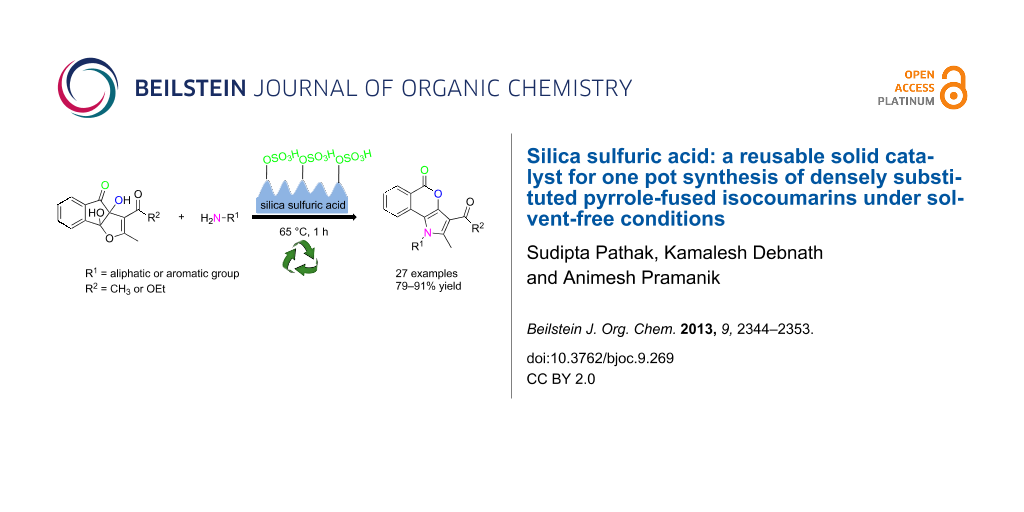
Graphical Abstract
Introduction
Isocoumarins are an important class of naturally occurring lactones [1-3], which has attracted the attention of chemists because of their various biological activities such as antioxidative [4], anticancer [5] and antifungal activities [6]. The development of a new and efficient methodology for the synthesis of biologically potent isocoumarins and their carbo/hetero annulated analogues has drawn great attention of synthetic as well as medicinal chemists [7-9]. Various methodologies for the synthesis of isocoumarins have been reported such as the reaction of o-halobenzoic acids and 1,3-diketones through a copper-catalyzed tandem sequential cyclization/addition/deacylation process [10,11], an iridium-catalyzed oxidative lactonization or an intramolecular cyclization reaction of δ-ketoaldehydes [12], a ruthenium-catalyzed aerobic oxidative cyclization of aromatic acids with alkynes [13], an FeCl3-promoted regioselective annulation of o-(1-alkynyl)benzoates with disulfides [14], a Heck–Matsuda cyclization reaction [15], a 6-endo-dig cyclization of heteroaryl esters to alkynes [16], or a Pd(II)-mediated cyclization of o-allylbenzaldehydes [17]. Salvinorin A, a natural product isolated from the hallucinogenic sage Salvia divinorum, which also contains a saturated isocoumarin ring, has been synthesized [18]. Although these methods are useful for the synthesis of isocoumarin derivatives, the reactions involved in the synthesis still suffer from some serious limitations such as the use of expensive and hazardous reagents [12] and toxic metal catalysts [10,11,15,17]. Some of the reactions need laborious and time consuming procedures [12,13,17], or drastic reaction conditions and with only low to moderate yields [16]. On the other hand, although a number of synthetic methods have been developed for the construction of densely substituted pyrrole rings [19-21], not a single report has been given on the synthesis of pyrrole-fused isocoumarins with the help of green methodology, so far. Therefore, the development of an environmentally friendly and safer reaction methodology following the green chemistry principles is essential for the synthesis of pyrrole-fused isocoumarins.
The employment of a reusable solid supported/heterogeneous catalyst for the efficient synthesis of heterocyclic compounds remains a challenge to chemists in laboratories and in the industry [22,23]. Reactions with reagents that are immobilized on inorganic solid supports show several advantages over the conventional reactions in solution because of simple work-up procedures, improved product yields, greater ease of purification, shorter reaction times, milder reaction conditions, and recyclability of the catalyst [24]. In the recent years, silica sulfuric acid (SSA) has shown immense potentiality as an efficient and easily retrievable solid catalyst in various important organic syntheses under solvent-free conditions [25]. The high catalytic activity, the operational simplicity and the recyclability of SSA can be exploited in the industry for the synthesis of various drugs and pharmaceuticals. SSA, a product that is easily synthesized from silica gel and chlorosulfonic acid [26], was observed to improve the reactivity and selectivity in carbon–carbon bond-formation reactions [27,28], in cycloaddition reactions [29,30], in protection–deprotection reactions of multistep syntheses [31-33], in esterifications [34] and in syntheses of heterocycles [35]. Since we are actively involved in the synthesis of biologically important heterocycles [36-42], we wish to report herein a green methodology for the construction of pyrrole-fused isocoumarins, which uses SSA as a solid-supported acid catalyst under solvent-free conditions (Scheme 1, present work).
![[1860-5397-9-269-i1]](/bjoc/content/inline/1860-5397-9-269-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of pyrrole-fused isocoumarins.
Scheme 1: Synthesis of pyrrole-fused isocoumarins.
Results and Discussion
Recently, we have reported that the enamines 3 generated from acetylacetone (1) and amines 2 react with ninhydrin to form the cyclic hemiaminal dihydroxyindenopyrroles 4. Subsequently intermediates 4 produce the pyrrole-fused isocoumarins 5 upon heating in glacial acetic acid with a catalytic amount of conc. H2SO4 (Scheme 1, previous work) [38]. It was observed that in the above synthesis the intermediate dihydroxyindenopyrroles 4 were needed to be isolated for further reaction to get the final products 5 in pure form. Otherwise some acetylated amines were always produced as byproducts. Besides, the formation of 4 from 3 did not proceed significantly when the enamines of ethyl acetoacetate were employed, because under acidic conditions, the enamines of ethyl acetoacetate readily hydrolyze and the free amines react with ninhydrin to form Schiff bases. To overcome the above problems we have designed an operationally simple one-pot reaction for the synthesis of pyrrole-fused isocoumarins (5 or 8) from the ninhydrin adducts of acetylacetone/ethyl acetoacetate (6 or 7) [43] and primary amines 2 under solvent-free conditions (Scheme 1, present work).
In order to explore the role of the different catalysts and solvents in the preparation of pyrrole-fused isocoumarins, an optimisation study was carried out with the model reaction between dihydroxyindenofuran ethyl ester 7 [43] and aniline in a molar ration of 1.00:1.50 (Scheme 2). When the reaction was carried out in aqueous solution under reflux the reaction did not proceed at all (Table 1, entry 1). Previous results showed that an activation by a Brønsted acid was necessary to carry out the reaction successfully [38]. Therefore, we screened various Brønsted acid catalysts, e.g., lactic acid, formic acid, citric acid and acetic acid in aqueous solution under reflux. But the yields were very low even after prolonged reaction time (Table 1, entries 2–5). On the basis of the assumption that more acidic conditions might be necessary to furnish the desired products in high yields, we carried out the reaction in acetic acid with adding a catalytic amount of H2SO4. Intriguingly, the yield of the product increased from less than 10% to 64% (Table 1, entry 6). The structure of the product 8a was confirmed by IR, 1H NMR and 13C NMR spectroscopy and elemental analysis. Surprisingly, when the above-mentioned reaction was carried out with aliphatic amines, only the acetylated amines were obtained instead of the desired products 8. These results influenced us to carry out the reaction under greener and milder reaction conditions, but with satisfying yield of the desired product, both for aromatic and aliphatic amines. We restrained the reaction to using PEG–OSO3H as a Brønsted acid–surfactant combined catalyst in aqeous solution under refluxing conditions as well as under solvent-free conditions (Table 1, entries 7 and 8). Although under solvent-free conditions the required temperature was lower and the yields of the products were higher, the yields were still only moderate. This encouraged us to execute the optimization study in presence of a solid acid catalyst under solvent-free conditions. This is one important facet of green chemistry: the eradication of solvents in chemical processes. Hence, we have carried out the synthesis by dissolving the substrate 7 and aniline in a minimum volume of chloroform, soaked them on the solid surface of solid Brønsted acid catalysts, such as silica gel and melamine sulfonic acid (MSA), dried the mixture under vacuum, and heated the reaction mixture to 100 °C (Table 1, entries 9 and 10). Unfortunately, the reactions on silica gel and MSA failed to give the desired product 8a. In the search of a suitable solid acid catalyst we employed silica sulfuric acid (SSA) at 100 °C. However, the reaction mixture got charred after 0.5 h and a considerable amount of impurities along with the desired product 8a was formed (Table 1, entry 11). When lowering the reaction temperature (65–100 ºC) and varying the amount (300–500 mg) of solid catalyst (Table 1, entries 12–15), the maximum yield (90%) of 8a was obtained at 65 °C using 400 mg of SSA (Table 1, entry 14).
![[1860-5397-9-269-i2]](/bjoc/content/inline/1860-5397-9-269-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction scheme for the synthesis of pyrrole-fused isocoumarins.
Scheme 2: Reaction scheme for the synthesis of pyrrole-fused isocoumarins.
Table 1: Optimization of reaction conditions for the synthesis of 8a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-269-i4.svg?max-width=637&scale=1.0)
|
||||||
| entry | catalyst | solvent | catalyst load | temperature (°C) | time (h) | yield (%)a |
|---|---|---|---|---|---|---|
| 1 | — | H2O | — | 100 | 24 | — |
| 2 | lactic acid | H2O | 20 mol % | 100 | 24 | 5 |
| 3 | formic acid | H2O | 20 mol % | 100 | 24 | 8 |
| 4 | citric acid | H2O | 20 mol % | 100 | 24 | 5 |
| 5 | acetic acid | H2O | 20 mol % | 100 | 24 | 6 |
| 6 | H2SO4 | acetic acid | 20 mol % | 85 | 1 | 64 |
| 7 | PEG–OSO3H | H2O | 500 mg | 100 | 2 | 45 |
| 8 | PEG–OSO3H | — | 500 mg | 80 | 1.5 | 66 |
| 9 | silica gel | — | 500 mg | 100 | 24 | — |
| 10 | melamine sulphonic acid | — | 500 mg | 100 | 24 | — |
| 11 | silica sulfuric acid | — | 500 mg | 100 | 0.5 | 45 |
| 12 | silica sulfuric acid | — | 500 mg | 85 | 0.5 | 58 |
| 13 | silica sulfuric acid | — | 500 mg | 65 | 1 | 90 |
| 14 | silica sulfuric acid | — | 400 mg | 65 | 1 | 90 |
| 15 | silica sulfuric acid | — | 300 mg | 65 | 1.5 | 83 |
aOptimization studies were carried out with 1.0 mmol 7 and 1.5 mmol of aniline.
Having successfully prepared 8a, we decided to explore the scope and generality of this reaction in the synthesis of other analogues. Accordingly, the ninhydrin adducts of acetylacetone/ethyl acetoacetate (6 and 7) [43] were reacted with a variety of commercially available aliphatic and aromatic primary amines under the optimized conditions (Table 1, entry 14). As becomes evident from Table 2, all the primary amines reacted well with adducts 6 and 7 affording the desired products 5 and 8 in good yields. The results show that solvent-free conditions and the SSA catalyst are crucial carrying out the reaction succesfully even with aliphatic amines. The structures of the new products 8a–o were determined by using spectroscopic data and elemental analysis. X-ray crystal data analysis of compound 8c further confirmed the product formation (Figure 1). The formation of products 5a–l was confirmed by comparing the reported spectral data and melting points (Table 2) [38].
Table 2: Formation of isocoumarins 5 and 8 from adducts 6 and 7 respectively on an SSA surface.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-269-i5.svg?max-width=637&scale=1.0)
|
||||||
| entry | R1 | R2 | adduct | product | yield (%)a | mp observed/lit. [38] (°C) |
|---|---|---|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-269-i6.svg?max-width=637&scale=1.0)
|
Me | 6 | 5a | 91 | 248–250/248 |
| 2 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-269-i7.svg?max-width=637&scale=1.0)
|
Me | 6 | 5b | 82 | 205–207/205 |
| 3 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-269-i8.svg?max-width=637&scale=1.0)
|
Me | 6 | 5c | 88 | 262–264/262 |
| 4 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-269-i9.svg?max-width=637&scale=1.0)
|
Me | 6 | 5d | 89 | 258–260/258 |
| 5 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-269-i10.svg?max-width=637&scale=1.0)
|
Me | 6 | 5e | 84 | 220–222/220 |
| 6 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-269-i11.svg?max-width=637&scale=1.0)
|
Me | 6 | 5f | 80 | 172–174/172 |
| 7 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-269-i12.svg?max-width=637&scale=1.0)
|
Me | 6 | 5g | 79 | 236–238/236 |
| 8 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-269-i13.svg?max-width=637&scale=1.0)
|
Me | 6 | 5h | 82 | 260–262/260 |
| 9 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-269-i14.svg?max-width=637&scale=1.0)
|
Me | 6 | 5i | 86 | >320/>320 |
| 10 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-269-i15.svg?max-width=637&scale=1.0)
|
Me | 6 | 5j | 84 | >320/>320 |
| 11 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-269-i16.svg?max-width=637&scale=1.0)
|
Me | 6 | 5k | 90 | 150–152/150 |
| 12 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-269-i17.svg?max-width=637&scale=1.0)
|
Me | 6 | 5l | 88 | 182–184/182 |
| 13 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-269-i18.svg?max-width=637&scale=1.0)
|
OEt | 7 | 8a | 90 | 208–210 |
| 14 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-269-i19.svg?max-width=637&scale=1.0)
|
OEt | 7 | 8b | 89 | 252–254 |
| 15 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-269-i20.svg?max-width=637&scale=1.0)
|
OEt | 7 | 8c | 79 | 233–235 |
| 16 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-269-i21.svg?max-width=637&scale=1.0)
|
OEt | 7 | 8d | 85 | 230–232 |
| 17 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-269-i22.svg?max-width=637&scale=1.0)
|
OEt | 7 | 8e | 87 | 218–220 |
| 18 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-269-i23.svg?max-width=637&scale=1.0)
|
OEt | 7 | 8f | 80 | 194–196 |
| 19 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-9-269-i24.svg?max-width=637&scale=1.0)
|
OEt | 7 | 8g | 83 | 198–200 |
| 20 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-9-269-i25.svg?max-width=637&scale=1.0)
|
OEt | 7 | 8h | 81 | 254–256 |
| 21 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-9-269-i26.svg?max-width=637&scale=1.0)
|
OEt | 7 | 8i | 86 | 202–204 |
| 22 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-9-269-i27.svg?max-width=637&scale=1.0)
|
OEt | 7 | 8j | 81 | 190–192 |
| 23 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-9-269-i28.svg?max-width=637&scale=1.0)
|
OEt | 7 | 8k | 83 | 212–214 |
| 24 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-9-269-i29.svg?max-width=637&scale=1.0)
|
OEt | 7 | 8l | 86 | 260–262 |
| 25 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-9-269-i30.svg?max-width=637&scale=1.0)
|
OEt | 7 | 8m | 91 | 180–182 |
| 26 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-9-269-i31.svg?max-width=637&scale=1.0)
|
OEt | 7 | 8n | 89 | 132–134 |
| 27 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-9-269-i32.svg?max-width=637&scale=1.0)
|
OEt | 7 | 8o | 87 | 125–127 |
aIsolated yield.
![[1860-5397-9-269-1]](/bjoc/content/figures/1860-5397-9-269-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: ORTEP diagram of 8c with atom numbering scheme. Thermal ellipsoids are shown at 50% probability with CCDC number 949317.
Figure 1: ORTEP diagram of 8c with atom numbering scheme. Thermal ellipsoids are shown at 50% probability wit...
The comparative studies in terms of overall reaction times and product yields show that although the present method takes more time than the previous method [38], the overall yields of the products are larger in the present method (Table 3). More importantly the present method enables us to access a new series of pyrrole-fused isocoumarins with ester functionality (Table 3, compounds 8a–o), which were not possible to synthesize with the previous method. Apart from that the present method is more advantageous in terms of product formation and greener characteristics than the previous one in many respects such as (a) a less laborious and more step-economical reaction for the library synthesis of pyrrole-fused isocoumarin derivatives, since the starting materials dihydroxy indenofurans 6 and 7 need only one step for preparation, (b) the employment of milder acidic conditions, (c) a lower reaction temperature (65 °C), (d) solvent-free conditions, and (e) more cost-effective because of the reusability of the solid-supported SSA. Moreover the starting materials 6 and 7 can also be prepared through a green methodology [43].
Table 3: Comparison between the present and the previous method for the synthesis of pyrrole-fused isocoumarins from ninhydrin.
![[Graphic 30]](/bjoc/content/inline/1860-5397-9-269-i33.svg?max-width=637&scale=1.0)
|
|||||
| entry | product | overall reaction time (min) | overall yield (%) | ||
|---|---|---|---|---|---|
| present method | previous method | present method | previous method | ||
| 1 | 5a | 61 | 37 | 87 | 81 |
| 2 | 5b | 61 | 46 | 79 | 64 |
| 3 | 5c | 61 | 45 | 84 | 74 |
| 4 | 5d | 61 | 37 | 85 | 80 |
| 5 | 5e | 61 | 39 | 81 | 80 |
| 6 | 5f | 61 | 51 | 77 | 66 |
| 7 | 5g | 61 | 37 | 76 | 58 |
| 8 | 5h | 61 | 36 | 79 | 77 |
| 9 | 5i | 61 | 37 | 83 | 56 |
| 10 | 5j | 61 | 46 | 81 | 68 |
| 11 | 5k | 61 | 21 | 86 | 78 |
| 12 | 5l | 61 | 25 | 84 | 84 |
| 13 | 8a | 61 | — | 84 | — |
| 14 | 8b | 61 | — | 83 | — |
| 15 | 8c | 61 | — | 73 | — |
| 16 | 8d | 61 | — | 79 | — |
| 17 | 8e | 61 | — | 81 | — |
| 18 | 8f | 61 | — | 74 | — |
| 19 | 8g | 61 | — | 77 | — |
| 20 | 8h | 61 | — | 75 | — |
| 21 | 8i | 61 | — | 80 | — |
| 22 | 8j | 61 | — | 75 | — |
| 23 | 8k | 61 | — | 77 | — |
| 24 | 8l | 61 | — | 80 | — |
| 25 | 8m | 61 | — | 85 | — |
| 26 | 8n | 61 | — | 83 | — |
| 27 | 8o | 61 | — | 81 | — |
Based on the results of Table 2 and the fact that SSA plays the role of transferring protons from its solid surface, a probable mechanism for the formation of isocoumarins 5 or 8 is explicated in Scheme 3. The protonation and activation of the hydroxy group of dihydroxy indenofuran (6 or 7) by the sulfonic group of SSA generates dehydrated cationic intermediate 9. This reactive intermediate 9 provokes a nucleophilic attack of primary amines to form bicyclo[3.3.0]octanamino compound 10. Then the α-hydroxy group of 10 attacks the adjacent carbonyl carbon to generate epoxy intermediate 11. This unstable epoxy intermediate 11 produces a six-membered lactone intermediate 12 through the breaking of a C–C bond. Subsequently, intermediate 12 tautomerizes to 13 under formation of the isocoumarin skeleton. The dihydropyrrole-fused isocoumarin intermediate 14 is formed through the intramolecular nucleophilic attack of the secondary amine group to the carbonyl carbon of 13. Finally, intermediate 14 loses water to furnish pyrrole-fused isocoumarins 5 or 8. It is worth mentioning that in the previous method instead of the formation of epoxy intermediate like 11 a transannular rearrangement was proposed for the product formation [38], because the formation of epoxy intermediate is less probable in the presence of a strong acid and a nucleophilic solvent as well as at higher temperatures.
![[1860-5397-9-269-i3]](/bjoc/content/inline/1860-5397-9-269-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Mechanism of formation of isocoumarins 5 or 8 on the surface of SSA.
Scheme 3: Mechanism of formation of isocoumarins 5 or 8 on the surface of SSA.
Furthermore, a test with respect to recovery and reusability of SSA for the formation of 8a was carried out. After heating the mixture of aniline and adduct 7 on the solid surface of SSA for 1 h, the product 8a was isolated easily with ethylacetate by sonication of the reaction mixture. The recovered solid-supported SSA was reused five times, and the yield of the product 8a varied from 90–83%, which indicates a substantial retention of catalytic activity and efficiency of SSA even after repeated application (Figure 2).
![[1860-5397-9-269-2]](/bjoc/content/figures/1860-5397-9-269-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Reusability of SSA for the synthesis of pyrrole-fused isocoumarins.
Figure 2: Reusability of SSA for the synthesis of pyrrole-fused isocoumarins.
Conclusion
In conclusion, a facile and convenient methodology has been developed for the synthesis of a diverse range of N-substituted pyrrole-fused isocoumarins in presence of the solid-supported Brønsted acid catalyst silica sulfuric acid (SSA). The methodology has a series of intrinsic advantages such as easy preparation of the solid supported SSA from chlorosulfonic acid and silica gel, less energy and manpower usage, easy product isolation/purification and operational simplicity, which lead to the synthetic route ‘‘benign by design’’. This is the first report, in which a rearrangement reaction has been carried out on the solid surface of SSA. Overall this greener and environmentally friendly method may attract the fellow chemists in chemical and pharmaceutical industries for the synthesis of biologically important pyrrole-fused isocoumarins.
Experimental
General information: Starting materials and solvents were purchased from commercial suppliers and used without further purification. Melting points were determined in open capillary tubes and were uncorrected. IR spectra were recorded on a Perkin-Elmer 782 spectrophotometer. 1H (300 MHz) and 13C NMR (75 MHz) spectra were recorded on a Bruker 300 MHz instrument in CDCl3 and d6-DMSO. Elemental analyses (C, H and N) were performed by using a Perkin-Elmer 240C elemental analyzer. The X-ray diffraction data for crystallized compounds were collected with Mo Kα radiation at 296 K using the Bruker APEX-II CCD System. The crystals were positioned at 50 mm from the CCD. Frames were measured with a counting time of 5 s. Data analyses were carried out with the Bruker APEX2 and Bruker SAINT program. The structures were solved using direct methods with the SHELXS97 program.
General experimental procedure for synthesis of pyrrole-fused isocoumarins 5 and 8: A mixture of primary amines 2 (1.5 mmol) and dihydroxy indenofurans 6 or 7 (1.0 mmol) in chloroform (5 mL) was soaked in SSA (400 mg) by stirring for 10 min and then the solvent was removed under reduced pressure to get a solid mass. The solid mass was heated at 65 °C for 1 h under continuous stirring, until the complete disappearance of dihydroxyindenofuran was observed (as monitored by TLC). After cooling the solid mass to room temperature, ethylacetate (15 mL) was added to it, shaken thoroughly, ultra-sonicated and filtered to remove the SSA catalyst. The separated organic phase was evaporated under reduced pressure to get the crude product 5 or 8 which was purified by column chromatography (hexane/EtOAc).
Reusability of the SSA: After completion of the reaction, the product was isolated by ultra-sonication with ethylacetate and decanted. Then the separated solid catalyst SSA was dried under vacuum and reused directly for a new reaction set. The yield of the product 8a varied from 90–83% after five consecutive cycles without any serious loss of efficiency of the solid supported SSA (Figure 2).
Supporting Information
Supporting Information features detailed analytical data of the prepared compounds and a collection of NMR spectra.
| Supporting Information File 1: Detailed analytical data. | ||
| Format: PDF | Size: 1.8 MB | Download |
Acknowledgements
S.P. and K.D. thank CSIR and UGC, New Delhi, India, for offering a Senior Research Fellowship (SRF) and a Junior Research Fellowship (JRF) respectively. The financial assistance of CSIR, New Delhi is gratefully acknowledged [Major Research Project, No. 02(0007)/11/EMR-II]. Crystallography was performed at the DST-FIST, India-funded Single Crystal Diffractometer Facility at the Department of Chemistry, University of Calcutta.
References
-
Zhang, W.; Krohn, K.; Draeger, S.; Schulz, B. J. Nat. Prod. 2008, 71, 1078–1081. doi:10.1021/np800095g
Return to citation in text: [1] -
Lu, X.; Li, D.; Dalley, N. K.; Wood, S. G.; Owen, N. L. Nat. Prod. Res. 2007, 21, 677–685. doi:10.1080/14786410500463395
Return to citation in text: [1] -
Pal, S.; Chatare, V.; Pal, M. Curr. Org. Chem. 2011, 15, 782–800. doi:10.2174/138527211794518970
Return to citation in text: [1] -
De Abreu, H. A.; dos S. Lago, I. A.; Souza, G. P.; Piló-Veloso, D.; Duarte, H. A.; de C. Alcântara, A. F. Org. Biomol. Chem. 2008, 6, 2713–2718. doi:10.1039/B804385J
Return to citation in text: [1] -
Nakhi, A.; Adepu, R.; Rambabu, D.; Kishore, R.; Vanaja, G. R.; Kalle, A. M.; Pal, M. Bioorg. Med. Chem. Lett. 2012, 22, 4418–4427. doi:10.1016/j.bmcl.2012.04.109
Return to citation in text: [1] -
Yadav, P.; Purohit, N. V. Indian J. Pharm. Sci. 2011, 73, 171–178. doi:10.4103/0250-474X.91586
Return to citation in text: [1] -
Uchiyama, M.; Ozawa, H.; Takuma, K.; Matsumoto, Y.; Yonehara, M.; Hiroya, K.; Sakamoto, T. Org. Lett. 2006, 8, 5517–5520. doi:10.1021/ol062190+
Return to citation in text: [1] -
Özcan, S.; Şahin, E.; Balci, M. Tetrahedron Lett. 2007, 48, 2151–2154. doi:10.1016/j.tetlet.2007.01.098
Return to citation in text: [1] -
Soleimani, E.; Zainali, M. J. Org. Chem. 2011, 76, 10306–10311. doi:10.1021/jo201908f
Return to citation in text: [1] -
Kavala, V.; Wang, C.-C.; Barange, D. K.; Kuo, C.-W.; Lei, P.-M.; Yao, C.-F. J. Org. Chem. 2012, 77, 5022–5029. doi:10.1021/jo300501j
Return to citation in text: [1] [2] -
Chary, R. G.; Reddy, G. R.; Ganesh, Y. S. S.; Prasad, K. V.; Chandra, S. K. P.; Mukherjee, S.; Pal, M. RSC Adv. 2013, 3, 9641–9644. doi:10.1039/C3RA40969D
Return to citation in text: [1] [2] -
Suzuki, T.; Yamada, T.; Watanabe, K.; Katoh, T. Bioorg. Med. Chem. Lett. 2005, 15, 2583–2585. doi:10.1016/j.bmcl.2005.03.043
Return to citation in text: [1] [2] [3] -
Deponti, M.; Kozhushkov, S. I.; Yufit, D. S.; Ackermann, L. Org. Biomol. Chem. 2013, 11, 142–148. doi:10.1039/C2OB26250A
Return to citation in text: [1] [2] -
Li, Z.; Hong, J.; Weng, L.; Zhou, X. Tetrahedron 2012, 68, 1552–1559. doi:10.1016/j.tet.2011.12.003
Return to citation in text: [1] -
da Penha, E. T. D.; Forni, J. A.; Biajoli, A. F. P.; Correia, C. R. D. Tetrahedron Lett. 2011, 52, 6342–6345. doi:10.1016/j.tetlet.2011.09.014
Return to citation in text: [1] [2] -
Hellal, M.; Bourguignon, J.-J.; Bihel, F. J.-J. Tetrahedron Lett. 2008, 49, 62–65. doi:10.1016/j.tetlet.2007.11.020
Return to citation in text: [1] [2] -
Chen, P.-Y.; Huang, K.-S.; Tsai, C.-C.; Wang, T.-P.; Wang, E.-C. Org. Lett. 2012, 14, 4930–4933. doi:10.1021/ol302256y
Return to citation in text: [1] [2] [3] -
Munro, T. A.; Duncanl, K. K.; Staples, R. J.; Xu, W.; Liu-Chen, L.-Y.; Béguin, C.; Carlezon, W. A., Jr.; Cohen, B. M. Beilstein J. Org. Chem. 2007, 3, No. 1. doi:10.1186/1860-5397-3-1
Return to citation in text: [1] -
Reddy, G. R.; Reddy, T. R.; Joseph, S. C.; Reddya, K. S.; Pal, M. RSC Adv. 2012, 2, 3387–3395. doi:10.1039/c2ra00982j
Return to citation in text: [1] -
Reddy, G. R.; Reddy, T. R.; Joseph, S. C.; Reddy, K. S.; Reddy, L. S.; Kumar, P. M.; Krishna, G. R.; Reddy, C. M.; Rambabu, D.; Kapavarapu, R.; Lakshmi, C.; Meda, T.; Priya, K. K.; Parsad, K. V. L.; Pal, M. Chem. Commun. 2011, 47, 7779–7781. doi:10.1039/c1cc12321a
Return to citation in text: [1] -
Maiti, S.; Biswas, S.; Jana, U. J. Org. Chem. 2010, 75, 1674–1683. doi:10.1021/jo902661y
Return to citation in text: [1] -
Varma, R. S. Green Chem. 1999, 1, 43–55. doi:10.1039/A808223E
Return to citation in text: [1] -
Sathishkumar, M.; Nagarajan, S.; Shanmugavelan, P.; Dinesh, M.; Ponnuswamy, A. Beilstein J. Org. Chem. 2013, 9, 689–697. doi:10.3762/bjoc.9.78
Return to citation in text: [1] -
Bougrin, K.; Soufiaoui, M. Tetrahedron Lett. 1995, 36, 3683–3686. doi:10.1016/0040-4039(95)00611-F
Return to citation in text: [1] -
Azizian, J.; Karimi, A. R.; Kazemizadeh, Z.; Mohammadi, A. A.; Mohammadizadeh, M. R. Synthesis 2005, 1095–1098. doi:10.1055/s-2005-861870
Return to citation in text: [1] -
Zolfigol, M. A. Tetrahedron 2001, 57, 9509–9511. doi:10.1016/S0040-4020(01)00960-7
Return to citation in text: [1] -
Chen, W.-Y.; Lu, J. Synlett 2005, 2293–2296. doi:10.1055/s-2005-872654
Return to citation in text: [1] -
Khodaei, M. M.; Khosropour, A. R.; Fattahpour, P. Tetrahedron Lett. 2005, 46, 2105–2108. doi:10.1016/j.tetlet.2005.01.149
Return to citation in text: [1] -
Saheli, P.; Dabiri, M.; Zolfigol, M. A.; Baghbanzadeh, M. Synlett 2005, 1155–1157. doi:10.1055/s-2005-865200
Return to citation in text: [1] -
Saheli, P.; Dabiri, M.; Zolfigol, M. A.; Baghbanzadeh, M. Tetrahedron Lett. 2005, 46, 7051–7053. doi:10.1016/j.tetlet.2005.08.043
Return to citation in text: [1] -
Shirini, F.; Zolfigol, M. A.; Mohammadi, K. Bull. Korean Chem. Soc. 2004, 25, 325–327. doi:10.5012/bkcs.2004.25.2.325
Return to citation in text: [1] -
Mirjalili, B. F.; Zolfigol, M. A.; Bamoniri, A. Molecules 2002, 7, 751–755. doi:10.3390/71000751
Return to citation in text: [1] -
Hajipour, A. R.; Zarei, A.; Khazdooz, L.; Pourmousavi, S. A.; Ruoho, A. E. Bull. Korean Chem. Soc. 2005, 26, 808–810. doi:10.5012/bkcs.2005.26.5.808
Return to citation in text: [1] -
Chakraborti, A. K.; Singh, B.; Chankeshwara, S. V.; Patel, A. R. J. Org. Chem. 2009, 74, 5967–5974. doi:10.1021/jo900614s
Return to citation in text: [1] -
Maleki, B.; Keshvari-Shirvan, H.; Taimazi, F.; Akbarzadeh, E. Int. J. Org. Chem. 2012, 2, 93–99. doi:10.4236/ijoc.2012.21015
Return to citation in text: [1] -
Das, S.; Fröhlich, R.; Pramanik, A. Org. Lett. 2006, 8, 4263–4266. doi:10.1021/ol061520n
Return to citation in text: [1] -
Das, S.; Koley, P.; Pramanik, A. Tetrahedron Lett. 2011, 52, 3243–3246. doi:10.1016/j.tetlet.2011.04.076
Return to citation in text: [1] -
Pathak, S.; Kundu, A.; Pramanik, A. Tetrahedron Lett. 2011, 52, 5180–5183. doi:10.1016/j.tetlet.2011.07.133
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Das, S.; Fröhlich, R.; Pramanik, A. Synlett 2006, 207–210. doi:10.1055/s-2006-926226
Return to citation in text: [1] -
Das, S.; Pramanik, A.; Fröhlich, R.; Patra, A. Tetrahedron 2004, 60, 10197–10205. doi:10.1016/j.tet.2004.09.004
Return to citation in text: [1] -
Pathak, S.; Debnath, K.; Hossain, S. T.; Mukherjee, S. K.; Pramanik, A. Tetrahedron Lett. 2013, 54, 3137–3143. doi:10.1016/j.tetlet.2013.04.015
Return to citation in text: [1] -
Pathak, S.; Pramanik, A. Eur. J. Org. Chem. 2013, 4410–4417. doi:10.1002/ejoc.201300096
Return to citation in text: [1] -
Chakrabarty, M.; Mukherji, A.; Arima, S.; Harigaya, Y.; Pilet, G. Monatsh. Chem. 2009, 140, 189–197. doi:10.1007/s00706-008-0066-6
Return to citation in text: [1] [2] [3] [4]
| 36. | Das, S.; Fröhlich, R.; Pramanik, A. Org. Lett. 2006, 8, 4263–4266. doi:10.1021/ol061520n |
| 37. | Das, S.; Koley, P.; Pramanik, A. Tetrahedron Lett. 2011, 52, 3243–3246. doi:10.1016/j.tetlet.2011.04.076 |
| 38. | Pathak, S.; Kundu, A.; Pramanik, A. Tetrahedron Lett. 2011, 52, 5180–5183. doi:10.1016/j.tetlet.2011.07.133 |
| 39. | Das, S.; Fröhlich, R.; Pramanik, A. Synlett 2006, 207–210. doi:10.1055/s-2006-926226 |
| 40. | Das, S.; Pramanik, A.; Fröhlich, R.; Patra, A. Tetrahedron 2004, 60, 10197–10205. doi:10.1016/j.tet.2004.09.004 |
| 41. | Pathak, S.; Debnath, K.; Hossain, S. T.; Mukherjee, S. K.; Pramanik, A. Tetrahedron Lett. 2013, 54, 3137–3143. doi:10.1016/j.tetlet.2013.04.015 |
| 42. | Pathak, S.; Pramanik, A. Eur. J. Org. Chem. 2013, 4410–4417. doi:10.1002/ejoc.201300096 |
| 38. | Pathak, S.; Kundu, A.; Pramanik, A. Tetrahedron Lett. 2011, 52, 5180–5183. doi:10.1016/j.tetlet.2011.07.133 |
| 43. | Chakrabarty, M.; Mukherji, A.; Arima, S.; Harigaya, Y.; Pilet, G. Monatsh. Chem. 2009, 140, 189–197. doi:10.1007/s00706-008-0066-6 |
| 1. | Zhang, W.; Krohn, K.; Draeger, S.; Schulz, B. J. Nat. Prod. 2008, 71, 1078–1081. doi:10.1021/np800095g |
| 2. | Lu, X.; Li, D.; Dalley, N. K.; Wood, S. G.; Owen, N. L. Nat. Prod. Res. 2007, 21, 677–685. doi:10.1080/14786410500463395 |
| 3. | Pal, S.; Chatare, V.; Pal, M. Curr. Org. Chem. 2011, 15, 782–800. doi:10.2174/138527211794518970 |
| 7. | Uchiyama, M.; Ozawa, H.; Takuma, K.; Matsumoto, Y.; Yonehara, M.; Hiroya, K.; Sakamoto, T. Org. Lett. 2006, 8, 5517–5520. doi:10.1021/ol062190+ |
| 8. | Özcan, S.; Şahin, E.; Balci, M. Tetrahedron Lett. 2007, 48, 2151–2154. doi:10.1016/j.tetlet.2007.01.098 |
| 9. | Soleimani, E.; Zainali, M. J. Org. Chem. 2011, 76, 10306–10311. doi:10.1021/jo201908f |
| 10. | Kavala, V.; Wang, C.-C.; Barange, D. K.; Kuo, C.-W.; Lei, P.-M.; Yao, C.-F. J. Org. Chem. 2012, 77, 5022–5029. doi:10.1021/jo300501j |
| 11. | Chary, R. G.; Reddy, G. R.; Ganesh, Y. S. S.; Prasad, K. V.; Chandra, S. K. P.; Mukherjee, S.; Pal, M. RSC Adv. 2013, 3, 9641–9644. doi:10.1039/C3RA40969D |
| 15. | da Penha, E. T. D.; Forni, J. A.; Biajoli, A. F. P.; Correia, C. R. D. Tetrahedron Lett. 2011, 52, 6342–6345. doi:10.1016/j.tetlet.2011.09.014 |
| 17. | Chen, P.-Y.; Huang, K.-S.; Tsai, C.-C.; Wang, T.-P.; Wang, E.-C. Org. Lett. 2012, 14, 4930–4933. doi:10.1021/ol302256y |
| 43. | Chakrabarty, M.; Mukherji, A.; Arima, S.; Harigaya, Y.; Pilet, G. Monatsh. Chem. 2009, 140, 189–197. doi:10.1007/s00706-008-0066-6 |
| 6. | Yadav, P.; Purohit, N. V. Indian J. Pharm. Sci. 2011, 73, 171–178. doi:10.4103/0250-474X.91586 |
| 12. | Suzuki, T.; Yamada, T.; Watanabe, K.; Katoh, T. Bioorg. Med. Chem. Lett. 2005, 15, 2583–2585. doi:10.1016/j.bmcl.2005.03.043 |
| 13. | Deponti, M.; Kozhushkov, S. I.; Yufit, D. S.; Ackermann, L. Org. Biomol. Chem. 2013, 11, 142–148. doi:10.1039/C2OB26250A |
| 17. | Chen, P.-Y.; Huang, K.-S.; Tsai, C.-C.; Wang, T.-P.; Wang, E.-C. Org. Lett. 2012, 14, 4930–4933. doi:10.1021/ol302256y |
| 38. | Pathak, S.; Kundu, A.; Pramanik, A. Tetrahedron Lett. 2011, 52, 5180–5183. doi:10.1016/j.tetlet.2011.07.133 |
| 5. | Nakhi, A.; Adepu, R.; Rambabu, D.; Kishore, R.; Vanaja, G. R.; Kalle, A. M.; Pal, M. Bioorg. Med. Chem. Lett. 2012, 22, 4418–4427. doi:10.1016/j.bmcl.2012.04.109 |
| 18. | Munro, T. A.; Duncanl, K. K.; Staples, R. J.; Xu, W.; Liu-Chen, L.-Y.; Béguin, C.; Carlezon, W. A., Jr.; Cohen, B. M. Beilstein J. Org. Chem. 2007, 3, No. 1. doi:10.1186/1860-5397-3-1 |
| 38. | Pathak, S.; Kundu, A.; Pramanik, A. Tetrahedron Lett. 2011, 52, 5180–5183. doi:10.1016/j.tetlet.2011.07.133 |
| 4. | De Abreu, H. A.; dos S. Lago, I. A.; Souza, G. P.; Piló-Veloso, D.; Duarte, H. A.; de C. Alcântara, A. F. Org. Biomol. Chem. 2008, 6, 2713–2718. doi:10.1039/B804385J |
| 12. | Suzuki, T.; Yamada, T.; Watanabe, K.; Katoh, T. Bioorg. Med. Chem. Lett. 2005, 15, 2583–2585. doi:10.1016/j.bmcl.2005.03.043 |
| 38. | Pathak, S.; Kundu, A.; Pramanik, A. Tetrahedron Lett. 2011, 52, 5180–5183. doi:10.1016/j.tetlet.2011.07.133 |
| 14. | Li, Z.; Hong, J.; Weng, L.; Zhou, X. Tetrahedron 2012, 68, 1552–1559. doi:10.1016/j.tet.2011.12.003 |
| 16. | Hellal, M.; Bourguignon, J.-J.; Bihel, F. J.-J. Tetrahedron Lett. 2008, 49, 62–65. doi:10.1016/j.tetlet.2007.11.020 |
| 43. | Chakrabarty, M.; Mukherji, A.; Arima, S.; Harigaya, Y.; Pilet, G. Monatsh. Chem. 2009, 140, 189–197. doi:10.1007/s00706-008-0066-6 |
| 13. | Deponti, M.; Kozhushkov, S. I.; Yufit, D. S.; Ackermann, L. Org. Biomol. Chem. 2013, 11, 142–148. doi:10.1039/C2OB26250A |
| 17. | Chen, P.-Y.; Huang, K.-S.; Tsai, C.-C.; Wang, T.-P.; Wang, E.-C. Org. Lett. 2012, 14, 4930–4933. doi:10.1021/ol302256y |
| 38. | Pathak, S.; Kundu, A.; Pramanik, A. Tetrahedron Lett. 2011, 52, 5180–5183. doi:10.1016/j.tetlet.2011.07.133 |
| 12. | Suzuki, T.; Yamada, T.; Watanabe, K.; Katoh, T. Bioorg. Med. Chem. Lett. 2005, 15, 2583–2585. doi:10.1016/j.bmcl.2005.03.043 |
| 43. | Chakrabarty, M.; Mukherji, A.; Arima, S.; Harigaya, Y.; Pilet, G. Monatsh. Chem. 2009, 140, 189–197. doi:10.1007/s00706-008-0066-6 |
| 10. | Kavala, V.; Wang, C.-C.; Barange, D. K.; Kuo, C.-W.; Lei, P.-M.; Yao, C.-F. J. Org. Chem. 2012, 77, 5022–5029. doi:10.1021/jo300501j |
| 11. | Chary, R. G.; Reddy, G. R.; Ganesh, Y. S. S.; Prasad, K. V.; Chandra, S. K. P.; Mukherjee, S.; Pal, M. RSC Adv. 2013, 3, 9641–9644. doi:10.1039/C3RA40969D |
| 15. | da Penha, E. T. D.; Forni, J. A.; Biajoli, A. F. P.; Correia, C. R. D. Tetrahedron Lett. 2011, 52, 6342–6345. doi:10.1016/j.tetlet.2011.09.014 |
| 38. | Pathak, S.; Kundu, A.; Pramanik, A. Tetrahedron Lett. 2011, 52, 5180–5183. doi:10.1016/j.tetlet.2011.07.133 |
| 22. | Varma, R. S. Green Chem. 1999, 1, 43–55. doi:10.1039/A808223E |
| 23. | Sathishkumar, M.; Nagarajan, S.; Shanmugavelan, P.; Dinesh, M.; Ponnuswamy, A. Beilstein J. Org. Chem. 2013, 9, 689–697. doi:10.3762/bjoc.9.78 |
| 16. | Hellal, M.; Bourguignon, J.-J.; Bihel, F. J.-J. Tetrahedron Lett. 2008, 49, 62–65. doi:10.1016/j.tetlet.2007.11.020 |
| 19. | Reddy, G. R.; Reddy, T. R.; Joseph, S. C.; Reddya, K. S.; Pal, M. RSC Adv. 2012, 2, 3387–3395. doi:10.1039/c2ra00982j |
| 20. | Reddy, G. R.; Reddy, T. R.; Joseph, S. C.; Reddy, K. S.; Reddy, L. S.; Kumar, P. M.; Krishna, G. R.; Reddy, C. M.; Rambabu, D.; Kapavarapu, R.; Lakshmi, C.; Meda, T.; Priya, K. K.; Parsad, K. V. L.; Pal, M. Chem. Commun. 2011, 47, 7779–7781. doi:10.1039/c1cc12321a |
| 21. | Maiti, S.; Biswas, S.; Jana, U. J. Org. Chem. 2010, 75, 1674–1683. doi:10.1021/jo902661y |
| 34. | Chakraborti, A. K.; Singh, B.; Chankeshwara, S. V.; Patel, A. R. J. Org. Chem. 2009, 74, 5967–5974. doi:10.1021/jo900614s |
| 35. | Maleki, B.; Keshvari-Shirvan, H.; Taimazi, F.; Akbarzadeh, E. Int. J. Org. Chem. 2012, 2, 93–99. doi:10.4236/ijoc.2012.21015 |
| 29. | Saheli, P.; Dabiri, M.; Zolfigol, M. A.; Baghbanzadeh, M. Synlett 2005, 1155–1157. doi:10.1055/s-2005-865200 |
| 30. | Saheli, P.; Dabiri, M.; Zolfigol, M. A.; Baghbanzadeh, M. Tetrahedron Lett. 2005, 46, 7051–7053. doi:10.1016/j.tetlet.2005.08.043 |
| 31. | Shirini, F.; Zolfigol, M. A.; Mohammadi, K. Bull. Korean Chem. Soc. 2004, 25, 325–327. doi:10.5012/bkcs.2004.25.2.325 |
| 32. | Mirjalili, B. F.; Zolfigol, M. A.; Bamoniri, A. Molecules 2002, 7, 751–755. doi:10.3390/71000751 |
| 33. | Hajipour, A. R.; Zarei, A.; Khazdooz, L.; Pourmousavi, S. A.; Ruoho, A. E. Bull. Korean Chem. Soc. 2005, 26, 808–810. doi:10.5012/bkcs.2005.26.5.808 |
| 26. | Zolfigol, M. A. Tetrahedron 2001, 57, 9509–9511. doi:10.1016/S0040-4020(01)00960-7 |
| 27. | Chen, W.-Y.; Lu, J. Synlett 2005, 2293–2296. doi:10.1055/s-2005-872654 |
| 28. | Khodaei, M. M.; Khosropour, A. R.; Fattahpour, P. Tetrahedron Lett. 2005, 46, 2105–2108. doi:10.1016/j.tetlet.2005.01.149 |
| 24. | Bougrin, K.; Soufiaoui, M. Tetrahedron Lett. 1995, 36, 3683–3686. doi:10.1016/0040-4039(95)00611-F |
| 25. | Azizian, J.; Karimi, A. R.; Kazemizadeh, Z.; Mohammadi, A. A.; Mohammadizadeh, M. R. Synthesis 2005, 1095–1098. doi:10.1055/s-2005-861870 |
© 2013 Pathak et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)