Abstract
Antimycins (>40 members) were discovered nearly 65 years ago but the discovery of the gene cluster encoding antimycin biosynthesis in 2011 has facilitated rapid progress in understanding the unusual biosynthetic pathway. Antimycin A is widely used as a piscicide in the catfish farming industry and also has potent killing activity against insects, nematodes and fungi. The mode of action of antimycins is to inhibit cytochrome c reductase in the electron transport chain and halt respiration. However, more recently, antimycin A has attracted attention as a potent and selective inhibitor of the mitochondrial anti-apoptotic proteins Bcl-2 and Bcl-xL. Remarkably, this inhibition is independent of the main mode of action of antimycins such that an artificial derivative named 2-methoxyantimycin A inhibits Bcl-xL but does not inhibit respiration. The Bcl-2/Bcl-xL family of proteins are over-produced in cancer cells that are resistant to apoptosis-inducing chemotherapy agents, so antimycins have great potential as anticancer drugs used in combination with existing chemotherapeutics. Here we review what is known about antimycins, the regulation of the ant gene cluster and the unusual biosynthetic pathway.

Graphical Abstract
Review
It is estimated that around 60% of all known antibiotics are derived from secondary metabolites made by filamentous actinomycete bacteria, most notably Streptomyces species [1]. Streptomyces species are predominantly known as saprophytic soil bacteria that have a complex differentiating life cycle. The life cycle begins with spore germination and outgrowth of a substrate mycelium and ends with the production of reproductive aerial hyphae, which undergo cell division to form chains of unigenomic spores [2]. Aerial hyphae production and sporulation is triggered by nutritional stress and is accompanied by the production of secondary metabolites. These specialised metabolites likely function both as chemical weapons against competing organisms in the soil and as signaling molecules to neighbouring microbes [3]. In recent years genome sequencing has revealed that each Streptomyces species encodes many more specialised metabolites than it makes in laboratory culture, leading to new efforts to activate these so-called “silent pathways.” The number of known antibiotics made by Streptomyces species is likely to grow rapidly with the advent of genome mining approaches, which start by identifying promising specialised metabolite gene clusters in whole genome sequences and then inducing their expression through chemical or genetic manipulation of the gene cluster in the native or a heterologous host. This approach has already been used to identify novel chemical scaffolds of antibiotics produced by well-studied Streptomyces species [4-6] and to identify the biosynthetic gene clusters for commercially important antibiotics [7-11]. The latter allows cloning, optimisation and engineering of such pathways to generate new derivatives with improved pharmacological properties.
We recently sequenced the genome of Streptomyces albus S4, which we isolated from the cuticle of the leaf-cutter ant Acromyrmex octospinosus [12,13]. Using genome-mining strategies, we identified the biosynthetic gene cluster for a group of compounds called antimycins that were discovered more than 60 years ago [7,14]. Antimycins, including structurally related uranchimycins, kitamycins and splenocins have unique structures comprising a nine-membered dilactone core conjugated to a rare 3-formamidosalicylic acid moiety and they comprise more than 40 known members (Figure 1) [15-22]. Antimycins can undergo base-catalysed decomposition resulting in the production of volatile blastmycinones and butenolides [23]. The main mode of action of antimycins is to inhibit cytochrome c reductase, an enzyme in the electron transport chain in mitochondria and bacteria and as such they are bioactive against a wide range of oragnisms including fish, fungi, insects and nematodes [24].
![[1860-5397-9-290-1]](/bjoc/content/figures/1860-5397-9-290-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Antimycins: Antimycins A1, A2, A3, and A4 and non-natural antimycins referenced in the text. Antimycin A1, R1 = COCH(CH3)CH2CH3, R2 = (CH2)4CH3; Antimycin A2, R1 = COCH(CH3)2, R2 = (CH2)4CH3; Antimycin A3, R1 = COCH2CH(CH3)2, R2 = (CH2)2CH3; Antimycin A4, R1 = COCH(CH3)2, R2 = (CH2)2CH3. For a recent summary of the chemical diversity in the antimycin family see [25].
Figure 1: Antimycins: Antimycins A1, A2, A3, and A4 and non-natural antimycins referenced in the text. Antimy...
Antimycins are widely used as a piscicide (brandname Fintrol) in the catfish farming industry. Catfish are easy to farm and they provide an inexpensive source of food in large parts of Asia and in the Southern USA. Catfish are relatively insensitive to antimycins and Fintrol is used to selectively kill other unwanted scaled fish species during aquaculture [26]. Antimycins are also used as research tools to study the structure and function of cytochromes [27]. More recently antimycins have been shown to be potent and selective inhibitors of the mitochondrial Bcl-2/Bcl-xL-related anti-apoptotic proteins [28]. Over-production of Bcl-2/Bcl-xL-related proteins in cancer cells confers resistance to multiple chemotherapeutic agents whose mode of action is to trigger apoptosis. A small molecule screen identified antimycins as potent inhibitors of Bcl-2-related proteins where they were shown to bind to the hydrophobic groove [28]. A synthetic derivative of antimycin A3, 2-methoxyantimycin A3 (Figure 1), no longer inhibits the respiratory chain, but still promotes apoptosis in cells over-producing Bcl-2-related proteins [29]. This suggests antimycin derivatives could be used alongside traditional apoptosis-inducing chemotherapeutics to block drug resistance and kill cancer cells [30]. Therefore, there is significant interest in better understanding the biosynthesis and regulation of antimycins, with a view toward bioengineering improved pharmacological properties for the treatment of drug-resistant cancers in the future.
The ant gene cluster
Since our discovery of the ant gene cluster in S. albus S4, we and others have identified ant gene clusters in 14 out of 117 fully or partially sequenced genomes available in Genbank for the genus Streptomyces (Figure 2). The 14 ant gene clusters can be classified as long-form (L-form, 17 genes), intermediate-form (I-form, 16 genes) or short-form (S-form, 15 genes). L-form ant gene clusters contain two genes, antP and antQ, which are not encoded by S-form gene clusters [25], and I-form gene clusters contain either antP or antQ, but not both.
![[1860-5397-9-290-2]](/bjoc/content/figures/1860-5397-9-290-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Schematic representation of ant biosynthetic gene clusters. L-form ant gene clusters are encoded by: S. ambofaciens ATCC 23877 (AM238663), S. blastmyceticus NBRC 12747 (AB727666), S. gancidicus BKS 13-15 (AOHP01000135, AOHP01000134, AOHP01000056, [31]), S. griseoflavus Tü4000 (ACFA01000901, ACFA01000902, ACFA01000903, ACFA01000904, ACFA01000905, ACFA01000906, ACFA01000907); S. hygroscopicus subsp. jinggangensis 5008 (NC_017765); S. hygroscopicus subsp. jinggangensis TL01 (NC_020895). I-form ant gene clusters are encoded by: Streptomyces sp. 303MFCol5.2 (ARTR01000061), Streptomyces sp. TOR3209 (AGNH01000419, AGNH01000420, AGNH01000421, [32]). S-form ant gene clusters are encoded by: S. albus S4 (CADY01000091); S. albus J1074 (NC_020990); Streptomyces sp. SM8 (AMPN01000393, AMPN01000430, AMPN01000050); Streptomyces sp. NRRL2288 [25]; Streptomyces sp. LaPpAH-202 (ARDM01000016); Streptomyces sp. CNY228 (ARIN01000033). The Genbank accession numbers provided correspond to either the complete genome sequence or the contig(s) of draft genome sequences encoding the ant gene cluster. It is worth noting that there is a probable sequencing error in the S. gancidicus BKS 13–15 cluster which causes the AntD orthologue not to have a stop codon. There is also a probable sequencing error in S. griseoflavus Tü4000, which truncates the truncates C1 of AntC into a discrete protein. CCR, crotonyl-CoA reductase.
Figure 2: Schematic representation of ant biosynthetic gene clusters. L-form ant gene clusters are encoded by...
L-form ant gene clusters are encoded by six taxa, S. ambofaciens ATCC 23877 [7,33], S. blastmyceticus NBRC 12747 [25], S. gancidicus BKS 13–15 and S. griseoflavus Tü4000 [23]. Interestingly, S. hygroscopicus subsp. jinggangensis 5008 and S. hygroscopicus subsp. jinggangensis TL01 encode L-form ant gene clusters, but do not encode AntE (Figure 2). I-form ant gene clusters are encoded by two species, Streptomyces sp. 303MFCol5.2 and Streptomyces sp. TOR3209, which lack antQ and antP, respectively (Figure 2). There are six taxa, all highly related to S. albus S4, that encode S-form ant gene clusters: S. albus S4 [7], S. albus J1074 [7], Streptomyces sp. SM8, Streptomyces sp. NRRL2288 [25], Streptomyces sp. LaPpAH-202 and Streptomyces sp. CNY228.
Based on our analysis of S. albus S4, the S-form gene cluster is organised into four transcriptional units; antAB, antCDE, antFG and antHIJKLMNO (Seipke and Hutchings, unpublished results; Figure 2). The antFGHIJKLN genes encode the biosynthesis pathway for the unusual starter unit, 3-aminosalicylate, antCD encode the hybrid NRPS/PKS machinery and antE and antM encode a crotonyl-CoA reductase and a discrete ketoreductase, respectively. The antB and antO genes encode tailoring enzymes and antA encodes an extracytoplasmic function (ECF) RNA polymerase σ factor named σAntA. The additional genes found in the 17 gene L-form and I-form ant gene clusters are antP and antQ, which encode a kynureninase and phosphopantetheinyl transferase, respectively [25] (Figure 2).
The availability of 14 ant gene clusters will facilitate a better understanding of how the different forms of the ant gene cluster evolved. More work in this area is required, however it is tempting to speculate that the L-form ant gene cluster is the ancestral gene cluster and antP and antQ were lost, giving rise to the S-form gene cluster. The I-form ant gene clusters encoded by Streptomyces 303MFCol5.2 and Streptomyces sp. TOR3209, which lack antP and antQ, respectively could be midway points toward evolving into S-form clusters, which presumably use the kynureninase involved in tryptophan catabolism and a phosphopantetheinyl transferase encoded elsewhere in the genome to compensate for the loss of antP and antQ, respectively.
Biosynthesis of the antimycin dilactone core
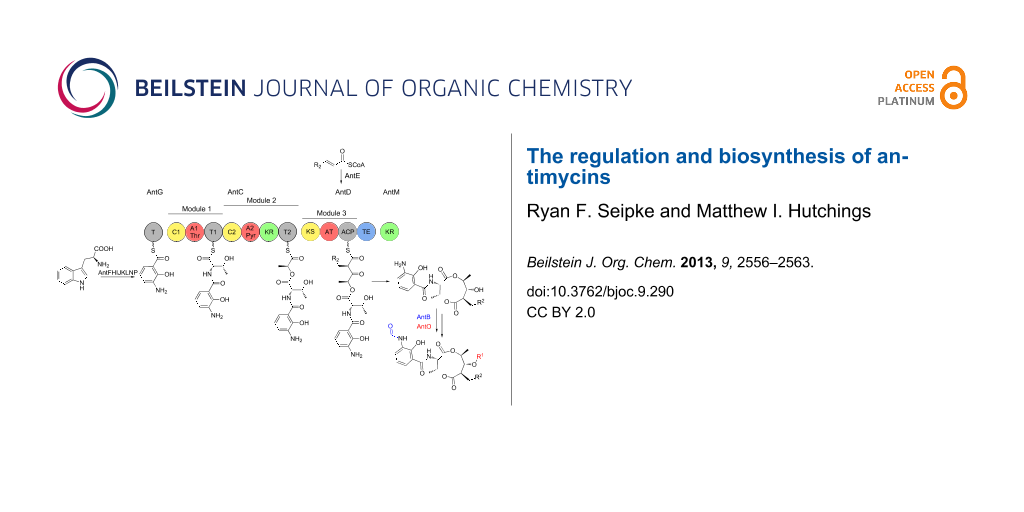
Antimycins are produced by a hybrid non-ribosomal peptide synthetase (NRPS)/polyketide synthase (PKS) assembly line for which the complete biosynthetic pathway has been proposed [25,34] (Figure 3). The biosynthesis of antimycins involves the activities of fourteen proteins, AntBCDEFGHIJKLMNO. The biosynthesis begins with the opening of the indole ring of tryptophan by a pathway-specific tryptophan-2,3-dioxygenase, AntN, to produce N-formyl-L-kynurenine. For L-form ant gene clusters, N-formyl-L-kynurenine is likely converted to anthranilate by the pathway-specific kynureninase, AntP, whereas S-form gene clusters lack AntP and likely use the kynureninase involved in primary tryptophan metabolism. Anthranilate and not N-formylanthranilate is activated by the acyl-CoA ligase protein, AntF and loaded onto its cognate carrier protein, AntG [34]. Once loaded onto AntG, anthranilate is converted to 3-aminosalicylate by a multicomponent oxygenase, AntHIJKL [33,34]. The anthraniloyl-S-AntG carboxylic acid-CoA thioester undergoes a never before seen 1,2-shift. Spiteller and colleagues suggested that AntHIJKL promotes this reaction via an epoxide intermediate similar to a reaction in phenylacetate catabolism [35] resulting in hydryoxylation of C-2 [33]. 3-Aminosalicylate serves as the starter unit and is presented to the NRPS, AntC. The AntC protein possesses two modules organised as follows: C1-A1-T1-C2-A2-KR-T2. The A1 domain activates and loads threonine onto T1, followed by condensation of 3-aminosalicylate and threonine promoted by C1. The A2 domain activates and loads pyruvate onto T2. Pyruvate is subsequently stereospecifically reduced by the KR domain and condensed with threonine by C2. The PKS, AntD posseses one module composed of the domains KS-AT-ACP-TE. The AT domain promotes the transfer of a 2-carboxylated acyl-CoA to ACP. The acyl-CoAs that are utilised by AntDAT are the product of AntE, a crotonyl-CoA reductase homologue, which biosynthesises 'unusual' acylmalonyl-CoAs utilised as PKS extender units [34,36,37]. The AntD AT domain is promiscuous and accepts multiple acylmalonyl-CoAs. In combination with AntE, AntDAT is the source of the large chemical diversity observed at position R2 within the antimycin family. Interestingly, S. hygroscopicus subsp. jinggangensis 5008 and S. hygroscopicus subsp. jinggangensis TL01 do not encode AntE, suggesting that these strains produce antimycins with less chemical diversity at the R2 position (Figure 2 and Figure 3). The KS domain catalyses the decarboxylative condensation between the aminoacyl thioester attached to AntCT2 and the 2-carboxy-acyl moiety attached to AntDACP. Next, a discrete ketoreductase, AntM catalyses the stereoselective reduction of the β-keto group, which precedes AntDTE – promoted regiospecific macrolactonisation and release of the nine-membered dilactone. Sandy et al. heterologously produced and purified AntCDEFGM and used building monomers anthranilate, L-threonine, pyruvate, and 2E-hexenoyl-CoA to conclusively demonstrate the minimum set of enzymes required for biosynthesis of the antimycin dilactone scaffold in vitro [34].
![[1860-5397-9-290-3]](/bjoc/content/figures/1860-5397-9-290-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Proposed biosynthetic pathway for antimycins. The antimycin biosynthetic pathway is described in detail in the text. C = condensation; A, adenylation; T, thiolation; KR, ketoreduction; KS, ketosynthase; AT, acyltransferase; ACP, acyl carrier protein; TE, thioesterase; *AntP is a pathway-specific kynureninase that is encoded by L- and I-form ant gene clusters.
Figure 3: Proposed biosynthetic pathway for antimycins. The antimycin biosynthetic pathway is described in de...
Antimycin tailoring genes
All known antimycin gene clusters contain two tailoring enzymes, AntB and AntO (Figure 2). AntB is a discrete acyltransferase, which catalyses a transesterification reaction resulting in the formation of a C-8 acyloxyl moiety and is responsible for generating the chemical diversity at R1 (Figure 1). AntB likely performs this reaction after assembly of the dilactone core, as wild-type levels of biosynthetic intermediates possessing a C-8 hydroxyl and not a C-8 acyloxyl were detected in a ∆antB mutant [38]. In vitro enzymatic synthesis showed that AntB is incredibly promiscuous and is capable of accepting a wide variety of substrates, including an alkyne-containing moiety, which has not been observed previously in the antimycin family [38]. AntB is also able to accept substrates presented by an alternate acyl carrier, N-acetylcysteamine (SNAC), though the turnover rate of acyl-SNACs is 100-fold lower than acyl-CoAs [38]. Despite the lower turnover rate, acyl-SNAC substrates are cell-permeable and thus AntB's ability to utilise acyl-SNACs provides the possibility to employ feeding studies to introduce new chemistry at C-8. AntO is a lipase homologue and is predicted to install the N-formyl group resulting in the 3-formamidosalicylate moiety. AntO is required for bioactivity against the human pathogenic fungus, Candida albicans (Seipke and Hutchings, unpublished results), but the exact time in which AntO installs the N-formyl group is unclear and requires investigation.
Regulation of the ant gene cluster
Antimycin biosynthesis is linked to development, as is the case for many Streptomyces secondary metabolites. However, antimycin biosynthesis is unusual because all four ant operons are highly expressed in substrate mycelium (after 24 hours growth on solid medium) whereas antimycins are not detected until after the production of aerial mycelium when ant gene expression is switched off (after 48 hours growth) (Seipke and Hutchings, unpublished results). This lag between gene expression and antimycin production may be due to the complex biosynthetic machinery that needs to be assembled to first make the unusual starter unit 3-aminosalicylate and then finally assemble the antimycin scaffold. The down-regulation of all the ant genes in stationary phase also suggests that specific regulatory mechanisms must exist to switch off the expression of all four ant operons. Despite this, the 14 ant gene clusters contain only a single conserved regulatory gene, antA, which encodes an orphan ECF RNA polymerase σ factor named σAntA (Figure 2). This sigma factor appears to be unique to the 14 known ant clusters in the database and they form a new sub-family of ECF sigma factors (Figure 4). Expression of the antFG and antHIJKLMNO genes is completely dependent on σAntA and over-expression of antA results in antFGHIJKLMNO expression in differentiated cultures, suggesting it regulates production of the starter unit, 3-aminosalicylate (Seipke and Hutchings, unpublished results). An unknown regulator, which is not encoded within the ant cluster, controls expression of the antAB and antCDE operons. Despite an earlier report that the S-form ant cluster (from Streptomyces sp. NRRL 2288) can be heterologously expressed in S. lividans and S. coelicolor, we have been unable to replicate this with the cloned S-form ant clusters from S. albus S4 or Streptomyces sp. NRRL 2288 (a kind gift from Professor Wen Liu). This suggests that the regulator of antAB and antCDE is a transcriptional activator that is encoded outside of the ant gene cluster and is absent from the heterologous host strains (Seipke and Hutchings, unpublished results).
![[1860-5397-9-290-4]](/bjoc/content/figures/1860-5397-9-290-4.png?scale=3.5&max-width=1024&background=FFFFFF)
Figure 4: σAntA comprises a new subfamily of ECF RNA polymerase σ factors. σAntA amino acid sequences were aligned to amino acid sequences of random representative of each ECF RNA polymerase σ factor subfamily reported by [39] using ClustalΩ [40]. The phylogenetic tree was created using PhyML 3.0 [41] and visualised using FigTree v1.4. The relevant region of the phylogenetic tree is enhanced for visualisation and reveals that all σAntA protein sequences cluster together and have a distinct phylogenetic lineage and represents a new ECF RNA polymerase σ factor subfamily.
Figure 4: σAntA comprises a new subfamily of ECF RNA polymerase σ factors. σAntA amino acid sequences were al...
Regulation of antibiotic gene clusters by ECF σ factors has only been reported in two rare actinomycete strains and in both cases there is a co-encoded anti-σ factor, which regulates σ factor activity, along with a pathway specific transcriptional activator. In Microbispora corallina production of the lantibiotic microbisporicin is regulated by the pathway-specific transcription factor MibR and the ECF σMibX, whose activity is modulated by its cognate anti-σ factor, MibW [8]. Biosynthesis of the lantibiotic planosporicin by Planomonospora alba is also regulated by a σ and anti-σ factor pair (PspX and PspW) whose closest homologues are MibX and MibW, respectively, and by the pathway-specific LuxR-family regulator, PspR [42]. There are no anti-σ factors encoded in any of the 14 ant gene clusters and no transcriptional regulators other than σAntA (Figure 2). Production of σAntA is regulated at the transcriptional level, by an unknown transcription factor, and it appears that σAntA is rapidly cleared from the cell once transcription of antA is switched off (Seipke and Hutchings, unpublished results). One striking feature of the 14 known σAntA proteins is that they all terminate in Ala–Ala, a well-known signal for the ClpXP protease. Attempts to N-terminally 6xHis-tag σAntA so that we could detect it using monoclonal anti-His antibodies were unsuccessful because His6-σAntA does not complement the antA mutant strain of S. albus S4, suggesting the protein is inactive. Furthermore, polyclonal antibodies raised against purified σAntA reacted with the purified σAntA protein in immunoblotting experiments, but could not detect σAntA in whole cell extracts, even from strains over-expressing antA, perhaps suggesting the protein is very unstable. Intriguingly, a variant of the S. albus S4 σAntA protein in which the C-terminal Ala–Ala was changed to Asp–Asp resulted in higher levels of expression for the antFG and antHIJKLMNO operons suggesting this may increase the stability or activity of σAntA (Seipke and Hutchings, unpublished results). A role for ClpXP in regulating σAntA activity remains to be proven however.
Future perspectives: toward bioengineering antimycins with improved pharmacological properties
There is significant interest in bioengineering new antimycin analogues with improved pharmacological properties for use as antifungal therapeutics and to be used to treat cancers, alongside traditional chemotherapy agents. Substantial progress has been made in a relatively short time and has mostly been driven by studies rooted in better understanding the biosynthesis of these unusual molecules.
Initial success toward bioengineering new antimycins was achieved by feeding modified monomer building blocks to batch bacterial fermentations. Spiteller and colleagues showed that by feeding fluoroanthranilates to S. ambofaciens ATCC 23877 and S. odorifer DSM 40347 cultures they could produce 5-fluoroantimycins and 4-fluoroantimycins [33] (Figure 1). Similarly, feeding 6-fluoro-L-tryptophan to cultures of Streptomyces sp. NRRL 2288 resulted in the production of 5-fluoroantimycins [25]. Liu and coworkers analysed the bioactivity of 5-fluoroantimycins and reported they retained potent antifungal activity against Candida albicans, but were significantly reduced in cytotoxity in a leukemia P388 mouse cell line compared to the parent compounds [25]. Sandy et al. recently showed AntB can accept both acyl-CoAs and acyl-SNACs to form the C-8 acyloxy group and generate diversity at R1 in vitro [38]. Acyl-SNACs are cell permeable, and the ability of AntB to utilise these substrates provides the opportunity to perform feeding studies using synthesised acyl-SNACs to introduce new chemistry at R1 [38].
The gene clusters for JBIR-06 (12-membered ring), neoantimycin (15-membered ring), and 18-membered ringed respirantin were recently identified [43,44]. JBIR-06 and neoantimycin inhibit GRB78 chaperone involved in the unfolded protein response [45,46]. Although the DNA sequence for the gene clusters for these ring-expanded antimycins has not yet been made publically available, they all encode the machinery necessary to assemble the 9-membered antimycin core suggesting a common evolutionary past [44]. The vast chemical diversity in the antimycin family together with the recent characterisations of the promiscuous biosynthetic machinery suggest it is possible to use synthetic biology to bioengineer non-natural analogues in large enough quantity to test their efficacies in the clinic. In line with that view, generating a chemistry-dereplicated culture collection of antimycin-type depsipeptide producers to build a library of swappable biosynthetic gene cassettes to introduce new chemical diversity should result in rapid generation of new analogues in the antimycin family.
Acknowledgements
This work was funded by a Medical Research Council Milstein Award (G0801721) and a Natural Environment Research Council responsive mode grant (NE/J01074X/1) to MIH. We thank all Hutchings' Lab members past and present and Mark Buttner, Mervyn Bibb and Barrie Wilkinson (John Innes Centre, Norwich) for useful discussions as well as Jeroen Dickschat for inviting our contribution to this Thematic Series.
References
-
Challis, G. L.; Hopwood, D. A. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 14555–14561.
Return to citation in text: [1] -
Flärdh, K.; Buttner, M. J. Nat. Rev. Microbiol. 2009, 7, 36–49. doi:10.1038/nrmicro1968
Return to citation in text: [1] -
Davies, J.; Spiegelman, G. B.; Yim, G. Curr. Opin. Microbiol. 2006, 9, 445–453. doi:10.1016/j.mib.2006.08.006
Return to citation in text: [1] -
Gottelt, M.; Kol, S.; Gomez-Escribano, J. P.; Bibb, M.; Takano, E. Microbiology 2010, 156, 2343–2353. doi:10.1099/mic.0.038281-0
Return to citation in text: [1] -
Laureti, L.; Song, L.; Huang, S.; Corre, C.; Leblond, P.; Challis, G. L.; Aigle, B. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6258–6263. doi:10.1073/pnas.1019077108
Return to citation in text: [1] -
Bunet, R.; Song, L.; Mendes, M. V.; Corre, C.; Hotel, L.; Rouhier, N.; Framboisier, X.; Leblond, P.; Challis, G. L.; Aigle, B. J. Bacteriol. 2011, 193, 1142–1153. doi:10.1128/JB.01269-10
Return to citation in text: [1] -
Seipke, R. F.; Barke, J.; Brearley, C.; Hill, L.; Yu, D. W.; Goss, R. J. M.; Hutchings, M. I. PLoS One 2011, 6, e22028. doi:10.1371/journal.pone.0022028
Return to citation in text: [1] [2] [3] [4] [5] -
Foulston, L. C.; Bibb, M. J. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 13461–13466. doi:10.1073/pnas.1008285107
Return to citation in text: [1] [2] -
Sherwood, E. J.; Hesketh, A. R.; Bibb, M. J. J. Bacteriol. 2013, 195, 2309–2321. doi:10.1128/JB.02291-12
Return to citation in text: [1] -
Claesen, J.; Bibb, M. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 16297–16302. doi:10.1073/pnas.1008608107
Return to citation in text: [1] -
Rackham, E. J.; Grüschow, S.; Ragab, A. E.; Dickens, S.; Goss, R. J. M. ChemBioChem 2010, 11, 1700–1709. doi:10.1002/cbic.201000200
Return to citation in text: [1] -
Barke, J.; Seipke, R. F.; Grüschow, S.; Heavens, D.; Drou, N.; Bibb, M. J.; Goss, R. J. M.; Yu, D. W.; Hutchings, M. I. BMC Biol. 2010, 8, No. 109. doi:10.1186/1741-7007-8-109
Return to citation in text: [1] -
Seipke, R. F.; Crossman, L.; Drou, N.; Heavens, D.; Bibb, M. J.; Caccamo, M.; Hutchings, M. I. J. Bacteriol. 2011, 193, 4270–4271. doi:10.1128/JB.05275-11
Return to citation in text: [1] -
Dunshee, B. R.; Leben, C.; Keitt, G. W.; Strong, F. M. J. Am. Chem. Soc. 1949, 71, 2436–2437. doi:10.1021/ja01175a057
Return to citation in text: [1] -
van Tamelen, E. E.; Dickie, J. P.; Loomans, M. E.; Dewey, R. S.; Strong, F. M. J. Am. Chem. Soc. 1961, 83, 1639–1646. doi:10.1021/ja01468a023
Return to citation in text: [1] -
Imamura, N.; Nishijima, M.; Adachi, K.; Sano, H. J. Antibiot. 1993, 46, 241–246. doi:10.7164/antibiotics.46.241
Return to citation in text: [1] -
Hayashi, K.; Nozaki, H. J. Antibiot. 1999, 52, 325–328. doi:10.7164/antibiotics.52.325
Return to citation in text: [1] -
Shiomi, K.; Hatae, K.; Hatano, H.; Matsumoto, A.; Takahashi, Y.; Jiang, C.-L.; Tomoda, H.; Kobayashi, S.; Tanaka, H.; Omura, S. J. Antibiot. 2005, 58, 74–78. doi:10.1038/ja.2005.10
Return to citation in text: [1] -
Hosotani, N.; Kumagai, K.; Nakagawa, H.; Shimatani, T.; Saji, I. J. Antibiot. 2005, 58, 460–467. doi:10.1038/ja.2005.61
Return to citation in text: [1] -
Chen, G.; Lin, B.; Lin, Y.; Xie, F.; Lu, W.; Fong, W.-F. J. Antibiot. 2005, 58, 519–522. doi:10.1038/ja.2005.70
Return to citation in text: [1] -
Yan, L.-L.; Han, N.-N.; Zhang, Y.-Q.; Yu, L.-Y.; Chen, J.; Wei, Y.-Z.; Li, Q.-P.; Tao, L.; Zheng, G.-H.; Yang, S.-E.; Jiang, C.-X.; Zhang, X.-D.; Huang, Q.; Habdin, X.; Hu, Q.-B.; Li, Z.; Liu, S.-W.; Zhang, Z.-Z.; He, Q.-Y.; Si, S.-Y.; Sun, C.-H. J. Antibiot. 2010, 63, 259–261. doi:10.1038/ja.2010.21
Return to citation in text: [1] -
Strangman, W. K.; Kwon, H. C.; Broide, D.; Jensen, P. R.; Fenical, W. J. Med. Chem. 2009, 52, 2317–2327. doi:10.1021/jm801110j
Return to citation in text: [1] -
Riclea, R.; Aigle, B.; Leblond, P.; Schoenian, I.; Spiteller, D.; Dickschat, J. S. ChemBioChem 2012, 13, 1635–1644. doi:10.1002/cbic.201200260
Return to citation in text: [1] [2] -
Tappel, A. L. Biochem. Pharmacol. 1960, 3, 289–296. doi:10.1016/0006-2952(60)90094-0
Return to citation in text: [1] -
Yan, Y.; Zhang, L.; Ito, T.; Qu, X.; Asakawa, Y.; Awakawa, T.; Abe, I.; Liu, W. Org. Lett. 2012, 14, 4142–4145. doi:10.1021/ol301785x
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Finlayson, B. J.; Schnick, R. A.; Cailteux, R. L.; DeMong, L.; Horton, W. D.; McClay, W.; Thompson, C. W. Fisheries 2002, 27, 10–18. doi:10.1577/1548-8446(2002)027<0010:AOAAUI>2.0.CO;2
Return to citation in text: [1] -
Cramer, W. A.; Hasan, S. S.; Yamashita, E. Biochim. Biophys. Acta, Bioenerg. 2011, 1807, 788–802. doi:10.1016/j.bbabio.2011.02.006
Return to citation in text: [1] -
Tzung, S.-P.; Kim, K. M.; Basañez, G.; Giedt, C. D.; Simon, J.; Zimmerberg, J.; Zhang, K. Y.; Hockenbery, D. M. Nat. Cell Biol. 2001, 3, 183–191. doi:10.1038/35055095
Return to citation in text: [1] [2] -
Schwartz, P. S.; Manion, M. K.; Emerson, C. B.; Fry, J. S.; Schulz, C. M.; Sweet, I. R.; Hockenbery, D. M. Mol. Cancer Ther. 2007, 6, 2073–2080. doi:10.1158/1535-7163.MCT-06-0767
Return to citation in text: [1] -
Thomas, S.; Quinn, B. A.; Das, S. K.; Dash, R.; Emdad, L.; Dasgupta, S.; Wang, X.-Y.; Dent, P.; Reed, J. C.; Pellecchia, M.; Sarkar, D.; Fisher, P. B. Expert Opin. Ther. Targets 2013, 17, 61–75. doi:10.1517/14728222.2013.733001
Return to citation in text: [1] -
Kumar, S.; Kaur, N.; Singh, N. K.; Raghava, G. P. S.; Mayilraj, S. Genome Announc. 2013, 1, e00150–13. doi:10.1128/genomeA.00150-13
Return to citation in text: [1] -
Hu, D.; Li, X.; Chang, Y.; He, H.; Zhang, C.; Jia, N.; Li, H.; Wang, Z. J. Bacteriol. 2012, 194, 1627. doi:10.1128/JB.06684-11
Return to citation in text: [1] -
Schoenian, I.; Paetz, C.; Dickschat, J. S.; Aigle, B.; Leblond, P.; Spiteller, D. ChemBioChem 2012, 13, 769–773. doi:10.1002/cbic.201200033
Return to citation in text: [1] [2] [3] [4] -
Sandy, M.; Rui, Z.; Gallagher, J.; Zhang, W. ACS Chem. Biol. 2012, 7, 1956–1961. doi:10.1021/cb300416w
Return to citation in text: [1] [2] [3] [4] [5] -
Teufel, R.; Mascaraque, V.; Ismail, W.; Voss, M.; Perera, J.; Eisenreich, W.; Haehnel, W.; Fuchs, G. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 14390–14395. doi:10.1073/pnas.1005399107
Return to citation in text: [1] -
Quade, N.; Huo, L.; Rachid, S.; Heinz, D. W.; Müller, R. Nat. Chem. Biol. 2012, 8, 117–124. doi:10.1038/nchembio.734
Return to citation in text: [1] -
Wilson, M. C.; Moore, B. S. Nat. Prod. Rep. 2012, 29, 72–86. doi:10.1039/c1np00082a
Return to citation in text: [1] -
Sandy, M.; Zhu, X.; Rui, Z.; Zhang, W. Org. Lett. 2013, 15, 3396–3399. doi:10.1021/ol4014365
Return to citation in text: [1] [2] [3] [4] [5] -
Staro, A.; Sofia, H. J.; Dietrich, S.; Ulrich, L. E.; Liesegang, H.; Mascher, T. Mol. Microbiol. 2009, 74, 557–581. doi:10.1111/j.1365-2958.2009.06870.x
Return to citation in text: [1] -
Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T. J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; Thompson, J. D.; Higgins, D. G. Mol. Syst. Biol. 2011, 7, No. 539. doi:10.1038/msb.2011.75
Return to citation in text: [1] -
Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. Syst. Biol. 2010, 59, 307–321. doi:10.1093/sysbio/syq010
Return to citation in text: [1] -
Sherwood, E. J.; Bibb, M. J. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E2500–E2509. doi:10.1073/pnas.1305392110
Return to citation in text: [1] -
Li, X.; Zvanych, R.; Vanner, S. A.; Wang, W.; Magarvey, N. A. Bioorg. Med. Chem. Lett. 2013, 23, 5123–5127. doi:10.1016/j.bmcl.2013.07.031
Return to citation in text: [1] -
Vanner, S. A.; Li, X.; Zvanych, R.; Torchia, J.; Sang, J.; Andrews, D. W.; Magarvey, N. A. Mol. BioSyst. 2013, 9, 2712–2719. doi:10.1039/c3mb70219g
Return to citation in text: [1] [2] -
Ueda, J.; Nagai, A.; Izumikawa, M.; Chijiwa, S.; Takagi, M.; Shin-ya, K. J. Antibiot. 2008, 61, 241–244. doi:10.1038/ja.2008.35
Return to citation in text: [1] -
Umeda, Y.; Chijiwa, S.; Furihata, K.; Furihata, K.; Sakuda, S.; Nagasawa, H.; Watanabe, H.; Shin-ya, K. J. Antibiot. 2005, 58, 206–209. doi:10.1038/ja.2005.25
Return to citation in text: [1]
| 25. | Yan, Y.; Zhang, L.; Ito, T.; Qu, X.; Asakawa, Y.; Awakawa, T.; Abe, I.; Liu, W. Org. Lett. 2012, 14, 4142–4145. doi:10.1021/ol301785x |
| 25. | Yan, Y.; Zhang, L.; Ito, T.; Qu, X.; Asakawa, Y.; Awakawa, T.; Abe, I.; Liu, W. Org. Lett. 2012, 14, 4142–4145. doi:10.1021/ol301785x |
| 34. | Sandy, M.; Rui, Z.; Gallagher, J.; Zhang, W. ACS Chem. Biol. 2012, 7, 1956–1961. doi:10.1021/cb300416w |
| 34. | Sandy, M.; Rui, Z.; Gallagher, J.; Zhang, W. ACS Chem. Biol. 2012, 7, 1956–1961. doi:10.1021/cb300416w |
| 38. | Sandy, M.; Zhu, X.; Rui, Z.; Zhang, W. Org. Lett. 2013, 15, 3396–3399. doi:10.1021/ol4014365 |
| 38. | Sandy, M.; Zhu, X.; Rui, Z.; Zhang, W. Org. Lett. 2013, 15, 3396–3399. doi:10.1021/ol4014365 |
| 34. | Sandy, M.; Rui, Z.; Gallagher, J.; Zhang, W. ACS Chem. Biol. 2012, 7, 1956–1961. doi:10.1021/cb300416w |
| 38. | Sandy, M.; Zhu, X.; Rui, Z.; Zhang, W. Org. Lett. 2013, 15, 3396–3399. doi:10.1021/ol4014365 |
| 33. | Schoenian, I.; Paetz, C.; Dickschat, J. S.; Aigle, B.; Leblond, P.; Spiteller, D. ChemBioChem 2012, 13, 769–773. doi:10.1002/cbic.201200033 |
| 34. | Sandy, M.; Rui, Z.; Gallagher, J.; Zhang, W. ACS Chem. Biol. 2012, 7, 1956–1961. doi:10.1021/cb300416w |
| 36. | Quade, N.; Huo, L.; Rachid, S.; Heinz, D. W.; Müller, R. Nat. Chem. Biol. 2012, 8, 117–124. doi:10.1038/nchembio.734 |
| 37. | Wilson, M. C.; Moore, B. S. Nat. Prod. Rep. 2012, 29, 72–86. doi:10.1039/c1np00082a |
| 33. | Schoenian, I.; Paetz, C.; Dickschat, J. S.; Aigle, B.; Leblond, P.; Spiteller, D. ChemBioChem 2012, 13, 769–773. doi:10.1002/cbic.201200033 |
| 34. | Sandy, M.; Rui, Z.; Gallagher, J.; Zhang, W. ACS Chem. Biol. 2012, 7, 1956–1961. doi:10.1021/cb300416w |
| 35. | Teufel, R.; Mascaraque, V.; Ismail, W.; Voss, M.; Perera, J.; Eisenreich, W.; Haehnel, W.; Fuchs, G. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 14390–14395. doi:10.1073/pnas.1005399107 |
| 39. | Staro, A.; Sofia, H. J.; Dietrich, S.; Ulrich, L. E.; Liesegang, H.; Mascher, T. Mol. Microbiol. 2009, 74, 557–581. doi:10.1111/j.1365-2958.2009.06870.x |
| 40. | Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T. J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; Thompson, J. D.; Higgins, D. G. Mol. Syst. Biol. 2011, 7, No. 539. doi:10.1038/msb.2011.75 |
| 41. | Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. Syst. Biol. 2010, 59, 307–321. doi:10.1093/sysbio/syq010 |
| 38. | Sandy, M.; Zhu, X.; Rui, Z.; Zhang, W. Org. Lett. 2013, 15, 3396–3399. doi:10.1021/ol4014365 |
| 43. | Li, X.; Zvanych, R.; Vanner, S. A.; Wang, W.; Magarvey, N. A. Bioorg. Med. Chem. Lett. 2013, 23, 5123–5127. doi:10.1016/j.bmcl.2013.07.031 |
| 44. | Vanner, S. A.; Li, X.; Zvanych, R.; Torchia, J.; Sang, J.; Andrews, D. W.; Magarvey, N. A. Mol. BioSyst. 2013, 9, 2712–2719. doi:10.1039/c3mb70219g |
| 25. | Yan, Y.; Zhang, L.; Ito, T.; Qu, X.; Asakawa, Y.; Awakawa, T.; Abe, I.; Liu, W. Org. Lett. 2012, 14, 4142–4145. doi:10.1021/ol301785x |
| 38. | Sandy, M.; Zhu, X.; Rui, Z.; Zhang, W. Org. Lett. 2013, 15, 3396–3399. doi:10.1021/ol4014365 |
| 33. | Schoenian, I.; Paetz, C.; Dickschat, J. S.; Aigle, B.; Leblond, P.; Spiteller, D. ChemBioChem 2012, 13, 769–773. doi:10.1002/cbic.201200033 |
| 25. | Yan, Y.; Zhang, L.; Ito, T.; Qu, X.; Asakawa, Y.; Awakawa, T.; Abe, I.; Liu, W. Org. Lett. 2012, 14, 4142–4145. doi:10.1021/ol301785x |
| 8. | Foulston, L. C.; Bibb, M. J. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 13461–13466. doi:10.1073/pnas.1008285107 |
| 42. | Sherwood, E. J.; Bibb, M. J. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E2500–E2509. doi:10.1073/pnas.1305392110 |
| 44. | Vanner, S. A.; Li, X.; Zvanych, R.; Torchia, J.; Sang, J.; Andrews, D. W.; Magarvey, N. A. Mol. BioSyst. 2013, 9, 2712–2719. doi:10.1039/c3mb70219g |
| 45. | Ueda, J.; Nagai, A.; Izumikawa, M.; Chijiwa, S.; Takagi, M.; Shin-ya, K. J. Antibiot. 2008, 61, 241–244. doi:10.1038/ja.2008.35 |
| 46. | Umeda, Y.; Chijiwa, S.; Furihata, K.; Furihata, K.; Sakuda, S.; Nagasawa, H.; Watanabe, H.; Shin-ya, K. J. Antibiot. 2005, 58, 206–209. doi:10.1038/ja.2005.25 |
| 1. | Challis, G. L.; Hopwood, D. A. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 14555–14561. |
| 7. | Seipke, R. F.; Barke, J.; Brearley, C.; Hill, L.; Yu, D. W.; Goss, R. J. M.; Hutchings, M. I. PLoS One 2011, 6, e22028. doi:10.1371/journal.pone.0022028 |
| 8. | Foulston, L. C.; Bibb, M. J. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 13461–13466. doi:10.1073/pnas.1008285107 |
| 9. | Sherwood, E. J.; Hesketh, A. R.; Bibb, M. J. J. Bacteriol. 2013, 195, 2309–2321. doi:10.1128/JB.02291-12 |
| 10. | Claesen, J.; Bibb, M. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 16297–16302. doi:10.1073/pnas.1008608107 |
| 11. | Rackham, E. J.; Grüschow, S.; Ragab, A. E.; Dickens, S.; Goss, R. J. M. ChemBioChem 2010, 11, 1700–1709. doi:10.1002/cbic.201000200 |
| 28. | Tzung, S.-P.; Kim, K. M.; Basañez, G.; Giedt, C. D.; Simon, J.; Zimmerberg, J.; Zhang, K. Y.; Hockenbery, D. M. Nat. Cell Biol. 2001, 3, 183–191. doi:10.1038/35055095 |
| 4. | Gottelt, M.; Kol, S.; Gomez-Escribano, J. P.; Bibb, M.; Takano, E. Microbiology 2010, 156, 2343–2353. doi:10.1099/mic.0.038281-0 |
| 5. | Laureti, L.; Song, L.; Huang, S.; Corre, C.; Leblond, P.; Challis, G. L.; Aigle, B. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6258–6263. doi:10.1073/pnas.1019077108 |
| 6. | Bunet, R.; Song, L.; Mendes, M. V.; Corre, C.; Hotel, L.; Rouhier, N.; Framboisier, X.; Leblond, P.; Challis, G. L.; Aigle, B. J. Bacteriol. 2011, 193, 1142–1153. doi:10.1128/JB.01269-10 |
| 29. | Schwartz, P. S.; Manion, M. K.; Emerson, C. B.; Fry, J. S.; Schulz, C. M.; Sweet, I. R.; Hockenbery, D. M. Mol. Cancer Ther. 2007, 6, 2073–2080. doi:10.1158/1535-7163.MCT-06-0767 |
| 3. | Davies, J.; Spiegelman, G. B.; Yim, G. Curr. Opin. Microbiol. 2006, 9, 445–453. doi:10.1016/j.mib.2006.08.006 |
| 27. | Cramer, W. A.; Hasan, S. S.; Yamashita, E. Biochim. Biophys. Acta, Bioenerg. 2011, 1807, 788–802. doi:10.1016/j.bbabio.2011.02.006 |
| 2. | Flärdh, K.; Buttner, M. J. Nat. Rev. Microbiol. 2009, 7, 36–49. doi:10.1038/nrmicro1968 |
| 28. | Tzung, S.-P.; Kim, K. M.; Basañez, G.; Giedt, C. D.; Simon, J.; Zimmerberg, J.; Zhang, K. Y.; Hockenbery, D. M. Nat. Cell Biol. 2001, 3, 183–191. doi:10.1038/35055095 |
| 23. | Riclea, R.; Aigle, B.; Leblond, P.; Schoenian, I.; Spiteller, D.; Dickschat, J. S. ChemBioChem 2012, 13, 1635–1644. doi:10.1002/cbic.201200260 |
| 25. | Yan, Y.; Zhang, L.; Ito, T.; Qu, X.; Asakawa, Y.; Awakawa, T.; Abe, I.; Liu, W. Org. Lett. 2012, 14, 4142–4145. doi:10.1021/ol301785x |
| 15. | van Tamelen, E. E.; Dickie, J. P.; Loomans, M. E.; Dewey, R. S.; Strong, F. M. J. Am. Chem. Soc. 1961, 83, 1639–1646. doi:10.1021/ja01468a023 |
| 16. | Imamura, N.; Nishijima, M.; Adachi, K.; Sano, H. J. Antibiot. 1993, 46, 241–246. doi:10.7164/antibiotics.46.241 |
| 17. | Hayashi, K.; Nozaki, H. J. Antibiot. 1999, 52, 325–328. doi:10.7164/antibiotics.52.325 |
| 18. | Shiomi, K.; Hatae, K.; Hatano, H.; Matsumoto, A.; Takahashi, Y.; Jiang, C.-L.; Tomoda, H.; Kobayashi, S.; Tanaka, H.; Omura, S. J. Antibiot. 2005, 58, 74–78. doi:10.1038/ja.2005.10 |
| 19. | Hosotani, N.; Kumagai, K.; Nakagawa, H.; Shimatani, T.; Saji, I. J. Antibiot. 2005, 58, 460–467. doi:10.1038/ja.2005.61 |
| 20. | Chen, G.; Lin, B.; Lin, Y.; Xie, F.; Lu, W.; Fong, W.-F. J. Antibiot. 2005, 58, 519–522. doi:10.1038/ja.2005.70 |
| 21. | Yan, L.-L.; Han, N.-N.; Zhang, Y.-Q.; Yu, L.-Y.; Chen, J.; Wei, Y.-Z.; Li, Q.-P.; Tao, L.; Zheng, G.-H.; Yang, S.-E.; Jiang, C.-X.; Zhang, X.-D.; Huang, Q.; Habdin, X.; Hu, Q.-B.; Li, Z.; Liu, S.-W.; Zhang, Z.-Z.; He, Q.-Y.; Si, S.-Y.; Sun, C.-H. J. Antibiot. 2010, 63, 259–261. doi:10.1038/ja.2010.21 |
| 22. | Strangman, W. K.; Kwon, H. C.; Broide, D.; Jensen, P. R.; Fenical, W. J. Med. Chem. 2009, 52, 2317–2327. doi:10.1021/jm801110j |
| 26. | Finlayson, B. J.; Schnick, R. A.; Cailteux, R. L.; DeMong, L.; Horton, W. D.; McClay, W.; Thompson, C. W. Fisheries 2002, 27, 10–18. doi:10.1577/1548-8446(2002)027<0010:AOAAUI>2.0.CO;2 |
| 7. | Seipke, R. F.; Barke, J.; Brearley, C.; Hill, L.; Yu, D. W.; Goss, R. J. M.; Hutchings, M. I. PLoS One 2011, 6, e22028. doi:10.1371/journal.pone.0022028 |
| 14. | Dunshee, B. R.; Leben, C.; Keitt, G. W.; Strong, F. M. J. Am. Chem. Soc. 1949, 71, 2436–2437. doi:10.1021/ja01175a057 |
| 12. | Barke, J.; Seipke, R. F.; Grüschow, S.; Heavens, D.; Drou, N.; Bibb, M. J.; Goss, R. J. M.; Yu, D. W.; Hutchings, M. I. BMC Biol. 2010, 8, No. 109. doi:10.1186/1741-7007-8-109 |
| 13. | Seipke, R. F.; Crossman, L.; Drou, N.; Heavens, D.; Bibb, M. J.; Caccamo, M.; Hutchings, M. I. J. Bacteriol. 2011, 193, 4270–4271. doi:10.1128/JB.05275-11 |
| 24. | Tappel, A. L. Biochem. Pharmacol. 1960, 3, 289–296. doi:10.1016/0006-2952(60)90094-0 |
| 31. | Kumar, S.; Kaur, N.; Singh, N. K.; Raghava, G. P. S.; Mayilraj, S. Genome Announc. 2013, 1, e00150–13. doi:10.1128/genomeA.00150-13 |
| 30. | Thomas, S.; Quinn, B. A.; Das, S. K.; Dash, R.; Emdad, L.; Dasgupta, S.; Wang, X.-Y.; Dent, P.; Reed, J. C.; Pellecchia, M.; Sarkar, D.; Fisher, P. B. Expert Opin. Ther. Targets 2013, 17, 61–75. doi:10.1517/14728222.2013.733001 |
| 25. | Yan, Y.; Zhang, L.; Ito, T.; Qu, X.; Asakawa, Y.; Awakawa, T.; Abe, I.; Liu, W. Org. Lett. 2012, 14, 4142–4145. doi:10.1021/ol301785x |
| 7. | Seipke, R. F.; Barke, J.; Brearley, C.; Hill, L.; Yu, D. W.; Goss, R. J. M.; Hutchings, M. I. PLoS One 2011, 6, e22028. doi:10.1371/journal.pone.0022028 |
| 25. | Yan, Y.; Zhang, L.; Ito, T.; Qu, X.; Asakawa, Y.; Awakawa, T.; Abe, I.; Liu, W. Org. Lett. 2012, 14, 4142–4145. doi:10.1021/ol301785x |
| 23. | Riclea, R.; Aigle, B.; Leblond, P.; Schoenian, I.; Spiteller, D.; Dickschat, J. S. ChemBioChem 2012, 13, 1635–1644. doi:10.1002/cbic.201200260 |
| 7. | Seipke, R. F.; Barke, J.; Brearley, C.; Hill, L.; Yu, D. W.; Goss, R. J. M.; Hutchings, M. I. PLoS One 2011, 6, e22028. doi:10.1371/journal.pone.0022028 |
| 7. | Seipke, R. F.; Barke, J.; Brearley, C.; Hill, L.; Yu, D. W.; Goss, R. J. M.; Hutchings, M. I. PLoS One 2011, 6, e22028. doi:10.1371/journal.pone.0022028 |
| 33. | Schoenian, I.; Paetz, C.; Dickschat, J. S.; Aigle, B.; Leblond, P.; Spiteller, D. ChemBioChem 2012, 13, 769–773. doi:10.1002/cbic.201200033 |
| 25. | Yan, Y.; Zhang, L.; Ito, T.; Qu, X.; Asakawa, Y.; Awakawa, T.; Abe, I.; Liu, W. Org. Lett. 2012, 14, 4142–4145. doi:10.1021/ol301785x |
| 32. | Hu, D.; Li, X.; Chang, Y.; He, H.; Zhang, C.; Jia, N.; Li, H.; Wang, Z. J. Bacteriol. 2012, 194, 1627. doi:10.1128/JB.06684-11 |
| 25. | Yan, Y.; Zhang, L.; Ito, T.; Qu, X.; Asakawa, Y.; Awakawa, T.; Abe, I.; Liu, W. Org. Lett. 2012, 14, 4142–4145. doi:10.1021/ol301785x |
© 2013 Seipke and Hutchings; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)