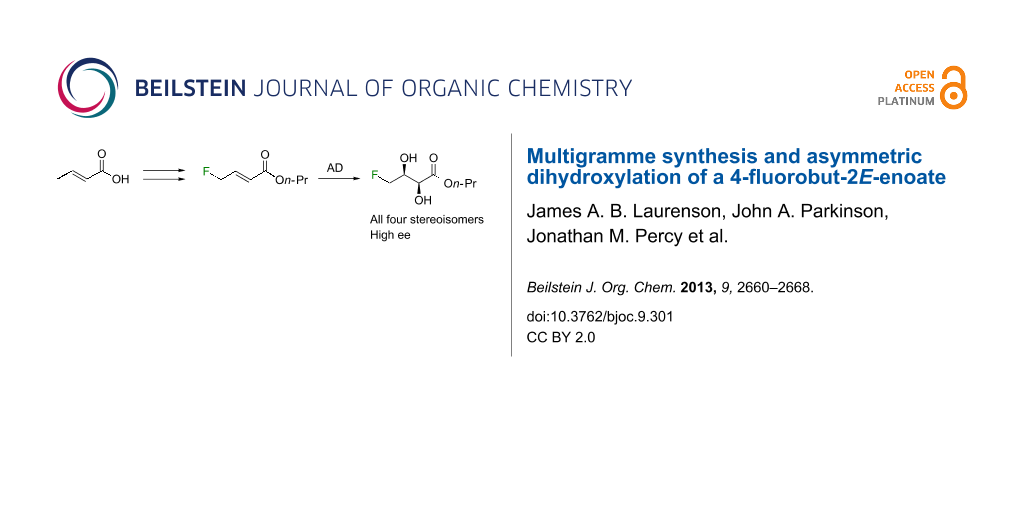
Abstract
Esters of crotonic acid were brominated on a multigramme scale using a free radical procedure. A phase transfer catalysed fluorination transformed these species to the 4-fluorobut-2E-enoates reproducibly and at scale (48–53%, ca. 300 mmol). Asymmetric dihydroxylation reactions were then used to transform the butenoate, ultimately into all four diastereoisomers of a versatile fluorinated C4 building block at high enantiomeric-enrichment. The (DHQ)2AQN and (DHQD)2AQN ligands described by Sharpless were the most effective. The development and optimisation of a new and facile method for the determination of ee is also described; 19F{1H} spectra recorded in d-chloroform/diisopropyl tartrate showed distinct baseline separated signals for different enantiomers.

Graphical Abstract
Introduction
Selective fluorination can be used to make subtle but decisive modifications of molecular properties. Sugar chemistry has proved particularly fertile ground for studies of this type; fluorine atoms can be used to replace hydroxy groups or hydrogen atoms, modifying the arrays of hydrogen bond donors and acceptors, and electron demand at the anomeric centre at minimal steric cost. Modifications of this type are sometimes accepted by sugar-processing enzymes such as the kinases and transferases involved in oligosaccharide assembly, or in antibiotic biosynthesis. Mechanistic insights, and new routes to hybrid natural products represent the rewards of this endeavour [1-10].
The synthesis of fluorinated analogues of sugars can be approached in two strategically different ways. The most common, and often most efficient approach, identifies a sugar precursor, isolates the locus for fluorination (usually an hydroxy group) by protecting all the other functional groups, and transforms it using a nucleophilic fluorinating agent [11].
The main advantages of this approach are that pre-existing stereogenic centres remain intact, while accurate inversion of configuration occurs at the locus of reaction. For one of the most common transformations, which delivers 6-deoxy-6-fluoro sugars, the locus of reaction is not even a stereogenic centre. The synthesis of 6-fluoro-D-olivose (6) in 23% overall yield from optically pure D-glucose (1) by O’Hagan and Nieschalk (Scheme 1) provides an impressive example of the approach [12].
![[1860-5397-9-301-i1]](/bjoc/content/inline/1860-5397-9-301-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Key steps from the synthesis of 6-fluoro-D-olivose (6) from D-glucose (1).
Scheme 1: Key steps from the synthesis of 6-fluoro-D-olivose (6) from D-glucose (1).
Isolation of the C-6 hydroxy group in 2 set the stage for mesylation, and conversion of 3 to fluoride 4 with an extremely economical reagent. Acetal cleavage and peracetylation released glycoside 5 which was converted to 6 via known methods. The main disadvantages of the approach are the extensive use which must be made of protection/deprotection chemistry, and in some cases, the availability of the precursor sugar. Some less common sugars are expensive and available in limited quantities.
The alternative approach involves de novo stereodivergent synthesis, which elaborates small fluorinated building blocks using the reactions of modern catalytic asymmetric chemistry; this approach still has a very restricted repertoire. Few versatile building blocks are available, particularly in supra-millimol quantities, and other disadvantages include the need to carry an expensive fluorinated material through many steps, and requirements for chromatographic separations of diastereoisomers. The costs and benefits of the de novo approach were illustrated by our recent asymmetric, stereodivergent route to selected 6-deoxy-6-fluorohexoses in which we transformed a fluorinated hexadienoate 9 into the fluorosugars 6-deoxy-6-fluoro-L-idose, 6-fluoro-L-fucose (13, shown) and 6-deoxy-6-fluoro-D-galactose (Scheme 2) [13].
![[1860-5397-9-301-i2]](/bjoc/content/inline/1860-5397-9-301-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: De novo asymmetric syntheses of 6-deoxy-6-fluorohexoses [13].
Scheme 2: De novo asymmetric syntheses of 6-deoxy-6-fluorohexoses [13].
The main challenges we faced included the synthesis of 9 and its bromide precursor 8 in acceptable yield and purity, and the unexpectedly low regioselectivity of AD reactions of the fluorinated dienoate. Methyl sorbate (7) underwent AD across the C-4/C-5 alkenyl group exclusively, but the introduction of the fluorine atom at C-6 lowered the selectivity (10:11) to 5:1 with AD-mix-α and 4:1 with AD-mix-β.
Nevertheless, de novo stereodivergent approaches are conceptually important and pave the way to wider ranges of more unnatural species. We decided to solve the problem of low regioselectivity from the hexadienoate, and to discover a more stereodivergent repertoire, by attempting to develop asymmetric chemistry based on a smaller butenoate (C4) building block, 14.
Results and Discussion
Fluorides of type 14 are uncommon in the literature (Scheme 3); silver mediated fluorination of butenoyl bromide 15 is known [14] delivering 16 in moderate yield but via a slow and expensive reaction. Wittig reaction, following in situ reduction of ethyl fluoroacetate (17) has been reported [15], while Purrington [16] prepared 19 by direct fluorination of silylketene acetal 18 with elemental fluorine.
![[1860-5397-9-301-i3]](/bjoc/content/inline/1860-5397-9-301-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Fluorobutenoate building block 14, and related species 16 and 19 from the literature [14-16].
Scheme 3: Fluorobutenoate building block 14, and related species 16 and 19 from the literature [14-16].
We decided to explore a halogen exchange approach from crotonic acid (20) which is commercially available cheaply, and in high diastereoisomeric purity (>98%). Diastereomeric purity is particularly important as the de novo syntheses must deliver the highest enantiomeric purity possible to be competitive with syntheses from enantiomerically pure natural products. n-Propyl and isopropyl esters 21 and 22 were prepared (0.5 mol scale) to moderate the volatility of intermediates, while retaining the option of distillation as a method of purification. Bromination was carried out using the method of Lester et al. [17], and while it was effective at small scales, larger scale (>150 mmol) reactions were violently exothermic. A modification of the reaction order reported earlier by Gershon and coworkers solved the problem [18]. Chlorobenzene was effective as the reaction solvent instead of carbon tetrachloride, allowing 23 and 24 to be isolated safely and reproducibly at scale (>300 mmol) in moderate yield (48–53%) after Kugelrohr distillation (Scheme 4).
![[1860-5397-9-301-i4]](/bjoc/content/inline/1860-5397-9-301-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Fluorobutenoate building blocks 25 and 26 prepared from crotonic acid.
Scheme 4: Fluorobutenoate building blocks 25 and 26 prepared from crotonic acid.
Fluorination was attempted using a range of conditions. The solvent-free reaction developed within our laboratory using commercial TBAF and KHF2 was not sufficiently effective for this substrate [13,19]. The yield of the product was moderate (37%), but the purification of the product was extremely difficult due to the complex mixture of products.
Allyl alcohol 27 (Figure 1) and starting material 23 were present and difficult to separate. During the course of this project, TBAF·(t-BuOH)4 was reported to be more effective than other fluoride sources. Kim and co-workers [20] reported that the reagent was obtained as a non-hygroscopic crystalline white solid after refluxing commercial TBAF in a mixture of hexane and t-BuOH; importantly, they claimed that it can be considered as a truly anhydrous source of the TBAF reagent. We were completely unable to reproduce the reagent preparation reported in the literature; all the materials we were able to make were extremely hygroscopic indeed, and exposure of 23 or 24 to them resulted in complete decomposition to a very complex mixture of products. However, the phase transfer catalysed procedure described by Hou and co-workers [21] which used TBAHSO4 and KF·2H2O in refluxing acetonitrile successfully effected the fluorination to allyl fluorides 25 and 26 on both small and large scales (>150 mmol). Rapid Kugelrohr distillation under reduced pressure was attempted initially but the quality of the distilled material was unsatisfactory. Fractional distillation through a Vigreux column at reduced pressure yielded the desired fluorides in an acceptable level of purity (>95% by 1H NMR) and reproducibly on a large scale (up to ~200 mmol). These outcomes represent significant practical improvements on the published methods of preparation. The subsequent transformations were carried out on the n-propyl ester 25 for two reasons; firstly, the material can be made in much higher yield, and the n-propyl ester can be cleaved under milder conditions than the isopropyl ester in 26.
![[1860-5397-9-301-1]](/bjoc/content/figures/1860-5397-9-301-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Side product 27 isolated from attempted fluorination.
Figure 1: Side product 27 isolated from attempted fluorination.
Although the commercial AD-mixes (0.4 mol % osmium/1 mol % ligand) can transform most standard substrates smoothly, osmium tetroxide is an electrophilic reagent [22], and electron deficient olefins, such as unsaturated amides and esters, react relatively slowly [23].
It was thought that the so-called “improved procedure” [24], which uses higher ligand/oxidant loadings (1 mol % osmium/5 mol % ligand) might be required to allow the reactions to proceed in acceptable yields and enantioselectivities [25]. Figure 2 shows the panel of ligands used for the asymmetric transformations. Scheme 5 shows the initial dihydroxylation carried out on 25, and Table 1 summarises the method development.
![[1860-5397-9-301-2]](/bjoc/content/figures/1860-5397-9-301-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The ligand panel used in the asymmetric dihydroxylation studies. The bold oxygen shows the point of attachment; individual ligands are represented by combinations of components, for example (DHQD)2 PHAL, present in AD-mix β.
Figure 2: The ligand panel used in the asymmetric dihydroxylation studies. The bold oxygen shows the point of...
![[1860-5397-9-301-i5]](/bjoc/content/inline/1860-5397-9-301-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Typical AD procedure; see Table 1 for outcomes.
Scheme 5: Typical AD procedure; see Table 1 for outcomes.
Table 1: Relationship between conditions, ligand and dihydroxylation ee.
| Conditions | Ligand type | DHQ/α- | DHQD/β- |
|---|---|---|---|
|
Standard
0.4 mol % osmium, 1 mol % ligand |
PHAL | 66% ee | 72% ee |
| 2 mol % osmium, 2 mol % ligand | PHAL | 80% ee | 89% ee |
|
Improved
1 mol % osmium, 5 mol % ligand |
PHAL | 83% ee | 91% ee |
| 1 mol % osmium, 10 mol % ligand | PHAL | 82% ee | 90% ee |
| 1 mol % osmium, 5 mol % ligand | AQN | 95% ee | 97% ee |
The asymmetric dihydroxylation conditions were subject to some optimization; the osmium and chiral ligand contents were varied in the first instance. While the commercial AD-mixes were used, we also carried out the dihydroxylations with 1 mol % osmium/5 mol % ligand, the so-called “improved procedure”, and with 1 mol % osmium/10 mol % ligand (results summarised in Table 1). Methyl sulfonamide which can accelerate hydrolysis and catalytic turnover was also added to the reaction mixtures [26].
Yields for the dihydroxylation chemistry were variable (44–80%); even though they are diols, these small molecules proved volatile. Reproducible yields (>55%) could be achieved if care was taken with solvent removal.
The “improved conditions” (1 mol % osmium, 5 mol % ligand) were found to give results comparable (within experimental error) to those obtained with the 2 mol % osmium/2 mol % ligand and 1 mol % osmium/10 mol % ligand conditions, suggesting the ee could not be indefinitely improved by increasing the ligand or osmium concentrations. Sharpless has reported that the (DHQ)2AQN and (DHQD)2AQN ligands based on the anthraquinone core, (Figure 2), are superior ligands for olefins bearing heteroatoms in the allylic position [27].
An asymmetric dihydroxylation reaction was performed using the improved Sharpless conditions with the newer AQN based ligands, producing excellent ee’s for both enantiomers of the diol, 95% for the enantiomer derived from AD-mix α, and 97% for the enantiomer from AD-mix β (Table 1). The corresponding isolated yields under these conditions were 54% and 56% respectively.
The ee's were measured after conversion of the diols to the dibenzoates 29 upon stirring overnight with benzoic anhydride, DMAP and polyvinylpyridine (PVP) at room temperature. The removal of the base by filtration was facile (Scheme 6). Genuine racemate 28c was synthesised via the Upjohn oxidation (catalytic osmium tetroxide, NMO aqueous t-BuOH, 83%) of 25 to avoid ambiguity, and converted to the dibenzoate 29c (not shown, 80%) as described above.
![[1860-5397-9-301-i6]](/bjoc/content/inline/1860-5397-9-301-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Conversion of enantiomerically-enriched diols to dibenzoates for HPLC analysis.
Scheme 6: Conversion of enantiomerically-enriched diols to dibenzoates for HPLC analysis.
The dibenzoates were purified by flash chromatography then examined by chiral HPLC (Chiralcel OD, 2% iPrOH in hexane). The separation of the enantiomers 29a and 29b was excellent, with over 6 minutes separating the stereoisomers in the chromatograms. Due to the robust nature of the dibenzoylation chemistry and the excellent chromatograms produced, the derivatisation/chiral HPLC assay was used routinely.
However, direct measurement of the ee's of the fluorinated diols 28a and 28b could not be achieved by the HPLC method. The very low absorbance of light at 235 nm resulted in unreliable data; small peak areas were observed for the desired compound with comparatively large peak areas for the background and trace impurities (as judged by 1H and 13C NMR spectra). Attempts to use RI detection in the chiral HPLC were no more successful. A new analytical method was therefore sought which would allow the ee’s of the diols to be measured quickly and directly using 19F{1H} NMR, avoiding the introduction of additional synthetic steps.
The determination of enantiomeric excesses using NMR is a well-established technique [28]; tactics include in situ derivatisation [29], may rely on very specific functionality [30] or may use expensive and/or structurally complex shift reagents [31]. The necessity of these reagents arises from the need to examine a single peak in a high level of detail despite the often cluttered nature of 1H (and 13C) NMR spectra, especially with large or complex structures. NMR determination of enantiomeric purity using chiral solvents though less well known has been described in the literature [32] and is particularly effective when heteroatomic NMR techniques are used [33]. For example, α-methylbenzylamine was used to resolve the components of the racemate of 2,2,2-trifluoro-1-phenylethanol in the 19F NMR spectrum (ΔδF was 0.04 ppm) [34] and in another case, a chiral liquid crystalline medium was used to resolve racemic mixtures of fluoroalkanes very effectively [35]. When solubilised in a chiral environment like diisopropyl L-tartrate (30, Figure 3), the formation of diastereoisomeric solvation complexes results in magnetic non-equivalence and hence the appearance of separate signals for the complexes in the NMR experiment.
![[1860-5397-9-301-3]](/bjoc/content/figures/1860-5397-9-301-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Diisopropyl L-tartrate (30) used as a chiral modifier for NMR determination of ee.
Figure 3: Diisopropyl L-tartrate (30) used as a chiral modifier for NMR determination of ee.
Recording the 19F{1H} NMR spectra will take advantage of the high sensitivity of 19F NMR detection and optimise S/N through the removal of splittings to protons. The NMR experiment was performed by diluting the substrate in an NMR tube with a 1:1 w/w mixture of diisopropyl L-tartrate and CDCl3. Racemic diol 28c analysed under these conditions by 19F{1H} NMR showed almost complete separation of the two enantiomers (ΔδF = 0.02 ppm). However, more complete peak separation was required before reliable integrations could be made (Figure 4).
![[1860-5397-9-301-4]](/bjoc/content/figures/1860-5397-9-301-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Partial 19F{1H} NMR spectra (376 MHz, L-(+)-DIPT/CDCl3, 300 K) spectra of (a) racemate 28c, (b) diol 28b and (c) 28a under standard acquisition parameters revealing the partial enantiomer overlap.
Figure 4: Partial 19F{1H} NMR spectra (376 MHz, L-(+)-DIPT/CDCl3, 300 K) spectra of (a) racemate 28c, (b) dio...
Alterations to the NMR acquisition parameters were made in an effort to improve the baseline resolution and separate the peaks fully.
Initial modifications caused a decrease in the quality of the spectra produced, with signal broadening and a reduction in the peak separation observed, caused by sample heating within the probe (decoupling produces heating of the sample) at the longer acquisition times. A set of experimental parameters that would allow a narrowing of the sweep width (SW), but maintain short acquisition (AQ) and relaxation times, and therefore minimise sample heating was devised; the optimised spectra are shown in Figure 5.
![[1860-5397-9-301-5]](/bjoc/content/figures/1860-5397-9-301-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Partial 19F{1H} NMR (400 MHz, L-(+)-DIPT/CDCl3, 300 K) spectra of 28b and 28a using optimised conditions: SW 40; AQ = 0.8; O1P −230; d1 = 5; 32 or 64 scans.
Figure 5: Partial 19F{1H} NMR (400 MHz, L-(+)-DIPT/CDCl3, 300 K) spectra of 28b and 28a using optimised condi...
The results obtained from integration of the signals for each enantiomer matched the chiral HPLC analysis of the derivatised dibenzoates closely; for example the ee’s for 28b and 28a, from the 1 mol % osmium, 5 mol % PHAL conditions, were 82% and 91% by NMR respectively and 83% and 91% by HPLC for the corresponding dibenzoates 29b and 29a.
The 19F{1H} NMR method uses a cheap readily available chiral solvating agent, is rapid (2 minutes per sample) and simple to perform. Although the technique is sacrificial in the sample, the quantities of sample required (<2 mg) are negligible. We make no claims for the generality of the method, but for molecules of this type, it appears highly effective.
To make our route stereodivergent, we sought access to the two anti diastereoisomers 35a and 35b via cyclic sulfate methodology (Scheme 7) [36,37].
![[1860-5397-9-301-i7]](/bjoc/content/inline/1860-5397-9-301-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Applying cyclic sulfate methodology to gain access to anti-diastereoisomers (transformations were developed from racemic diol 28c, but are shown for diol 28b only).
Scheme 7: Applying cyclic sulfate methodology to gain access to anti-diastereoisomers (transformations were d...
Cyclic sulfate 32b was prepared via literature procedures [36,37], monitoring the steps closely by 19F{1H} NMR spectroscopy which distinguishes all the species effectively. In 32b, C-3 is primed for regioselective nucleophilic attack [38]. Crude cyclic sulfate 32b was taken up in acetone, treated with solid ammonium benzoate and allowed to stir at room temperature overnight. Nucleophilic ring opening reactions were performed on the crude cyclic sulfate mixtures because avoiding column chromatography at this stage led to a vast improvement in the overall yields. After ring opening, sulfate ester cleavage was achieved by stirring the concentrated residue in acid (20% H2SO4) and ether, yielding the desired monobenzoate in moderate yield (60%) after purification. The regiochemistry of the ring opening was revealed in the HMBC spectrum of monobenzoate 33b. The 1H NMR signal corresponding to the C-2 methine proton couples (3JC-H) to both carbonyl signals in the 13C spectrum. This indicates that both carbonyl groups are within 3 bonds of the hydrogen on C-2. However, the signal from the hydrogen on C-3 couples to the carbonyl carbon of the n-propyl ester only, confirming the expected regiochemistry for structure 33b. Dibenzoate 34b was synthesised (32% overall from 28b) directly from the crude reaction mixture (Scheme 7) by treatment of the crude monobenzoate 33b with benzoic anhydride in the presence of DMAP and PVP. The syn- and anti-dibenzoates have distinct signals in the 19F NMR spectra (δF −230.3 and −231.0 ppm respectively), allowing a very high level of confidence that the ring-opening of the syn-cyclic sulfates does not produce syn-dibenzoate, and that epimerisation is not competitive with ring-opening. This was further supported by chiral HPLC analyses of the dibenzoates, which also suggests that clean conversion occurs, without epimerisation. All four dibenzoates had distinct retention times in the chiral HPLC chromatograms.
For the inversion of the diol stereochemistry to be synthetically useful, a less basic synthetic equivalent for hydroxide was required. When Mitsunobu chemistry fails, O’Doherty and co-workers have achieved hydroxy group inversion by triflation and displacement using sodium nitrite [39]. Cyclic sulfate 32b was exposed to sodium nitrite in DMF; the mixture was heated at reflux until completion of the reaction was confirmed by 19F NMR. Subsequent acid cleavage of the sulfate ester afforded the desired anti-diols in a disappointing yield (12% overall from 28b) after purification. The low yield was attributed to the small scale of the reaction and difficulty of the work-up caused by the presence of DMF. Unfortunately, attempts to carry out the reaction in acetone led to complete decomposition of the substrate.
A proof-of-concept extension sequence of the C4 building block was sought. Cyclohexylidene protection was chosen to add bulk and in aspiration to crystalline intermediates (Scheme 8).
![[1860-5397-9-301-i8]](/bjoc/content/inline/1860-5397-9-301-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Protecting and chain extending the educts of asymmetric dihydroxylation.
Scheme 8: Protecting and chain extending the educts of asymmetric dihydroxylation.
After some initial failures, cyclohexylidene 36b formed effectively in the presence of Lewis acid BF3·OEt2 in ethyl acetate [40]. Ester reduction with DIBAL-H afforded alcohol 37b; delaying purification of the products until after the reduction step increased the overall yield from butenoate 25 to 25% over 3 steps and in excellent diastereoisomeric purity. In contrast, the preparation of 37a with purifications at each stage delivered 37a in 3% overall yield. A one-pot oxidation/Wittig procedure was implemented from 37a; treatment with the Dess–Martin periodinane [41] in the presence of the stabilised ylide afforded a 4:1 E:Z mixture of the product alkene 39a in good (74%) yield. A second purification by column chromatography isolated the E-alkene diastereoisomer of 39a in 37% yield together with a mixed fraction of the E- and Z-alkenes. The E-isomer was identified by the alkene vicinal coupling values in the 1H NMR spectrum, and E:Z ratios were measured by integration of the distinct signals in the 19F{1H} NMR spectra. Analysis of the pure E-alkene using the chiral 19F{1H} NMR method revealed that the ee was unchanged from the diol 28a, confirming epimerisation was not occurring during the subsequent reactions (aldehyde 38a was of particular concern).
The synthesis of alkenes 39 is particularly significant, as at this stage the crotonic acid route overlaps with the published syntheses of 6-deoxy-6-fluorohexoses from methyl sorbate [13].
The main benefits of the crotonic acid route are the absence of regioisomers as the double bond is installed after the asymmetric oxidation and the potential to deliver all of the 6-deoxy-6-fluorohexose isomers, as the cyclic sulfate chemistry can generate the previously inaccessible anti-diol relationships, either at C2–C3, C4–C5 or both.
Conclusion
A practical route which affords 4-fluorobut-2E-enoates reproducibly and at scale (48–53%, ca. 300 mmol) has been developed, improving significantly on published methods. Catalytic asymmetric dihydroxylation can be carried out in moderate to good yields and in excellent ee using the AQN ligands. Chiral HPLC was used for ee determination of the dibenzoate derivatives, but a chiral 19F{1H} NMR method was developed to determine the enantiomeric purities of the non-chromophoric syn-diol products. Educt elaboration was achieved via cyclic sulfate methodology, leading to the stereocomplementary anti-diols, and via acetal protection, ester reduction and one-pot oxidation/Wittig reaction, re-connecting this study to the published route to 6-deoxy-6-fluorohexoses.
Experimental
A full range of experimental procedures and characterisation data is presented in Supporting Information File 1.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data. | ||
| Format: PDF | Size: 396.2 KB | Download |
Acknowledgements
This work was supported by the University of Leicester (studentship to R.R.), the Engineering and Physical Sciences Research Council (EPSRC, GR/S82053/02, fellowship to G.R., consumable support to R.R., J.A.B.L.), the University of Strathclyde Principal’s Fund (fellowship to G.R.) and WestCHEM (studentship to J.A.B.L.). We also thank the EPSRC National Mass Spectrometry Service Centre, University of Wales Swansea for accurate mass spectrometric measurements.
References
-
Tsuchiya, T. Adv. Carbohydr. Chem. Biochem. 1990, 48, 91. doi:10.1016/S0065-2318(08)60032-3
Return to citation in text: [1] -
Hartman, M. C. T.; Jiang, S.; Rush, J. S.; Waechter, C. J.; Coward, J. K. Biochemistry 2007, 46, 11630. doi:10.1021/bi700863s
Return to citation in text: [1] -
Compain, P.; Martin, O. R. Curr. Top. Med. Chem. 2003, 3, 541. doi:10.2174/1568026033452474
Return to citation in text: [1] -
Murray, B. W.; Wittmann, V.; Burkart, M. D.; Hung, S.-C.; Wong, C.-H. Biochemistry 1997, 36, 823. doi:10.1021/bi962284z
Return to citation in text: [1] -
Withers, S. G.; MacLennan, D. J.; Street, I. P. Carbohydr. Res. 1986, 154, 127. doi:10.1016/S0008-6215(00)90028-4
Return to citation in text: [1] -
Sianidis, G.; Wohlert, S.-E.; Pozidis, C.; Karamanou, S.; Luzhetskyy, A.; Vente, A.; Economou, A. J. Biotechnol. 2006, 125, 425. doi:10.1016/j.jbiotec.2006.03.035
Return to citation in text: [1] -
Burkart, M. D.; Vincent, S. P.; Düffels, A.; Murray, B. W.; Ley, S. V.; Wong, C.-H. Bioorg. Med. Chem. 2000, 8, 1937. doi:10.1016/S0968-0896(00)00139-5
Return to citation in text: [1] -
Schengrund, C.-L.; Kováč, P. Carbohydr. Res. 1999, 319, 24. doi:10.1016/S0008-6215(99)00104-4
Return to citation in text: [1] -
Ichikawa, Y.; Lin, Y. C.; Dumas, D. P.; Shen, G. J.; Garcia-Junceda, E.; Williams, M. A.; Bayer, R.; Ketcham, C.; Walker, L. E. J. Am. Chem. Soc. 1992, 114, 9283. doi:10.1021/ja00050a007
Return to citation in text: [1] -
Errey, J. C.; Mann, M. C.; Fairhurst, S. A.; Hill, L.; McNeil, M. R.; Naismith, J. H.; Percy, J. M.; Whitfield, C.; Field, R. A. Org. Biomol. Chem. 2009, 7, 1009–1016. doi:10.1039/b815549f
Return to citation in text: [1] -
Dax, K. Sci. Synth. 2006, 34, 71.
Return to citation in text: [1] -
Nieschalk, J.; O'Hagan, D. J. Fluorine Chem. 1998, 91, 159. doi:10.1016/S0022-1139(98)00220-6
Return to citation in text: [1] -
Caravano, A.; Field, R. A.; Percy, J. M.; Rinaudo, G.; Roig, R.; Singh, K. Org. Biomol. Chem. 2009, 7, 996. doi:10.1039/b815342f
Return to citation in text: [1] [2] [3] [4] -
Guha, S. K.; Shibayama, A.; Abe, D.; Sakaguchi, M.; Ukaji, Y.; Inomata, K. Bull. Chem. Soc. Jpn. 2004, 77, 2147. doi:10.1246/bcsj.77.2147
Return to citation in text: [1] [2] -
Li, Q.; Wang, W.; Berst, K. B.; Claiborne, A.; Hasvold, L.; Raye, K.; Tufano, M.; Nilius, A.; Shen, L. L.; Flamm, R.; Alder, J.; Marsh, K.; Crowell, D.; Chu, D. T. W.; Plattner, J. J. Bioorg. Med. Chem. Lett. 1998, 8, 1953. doi:10.1016/S0960-894X(98)00355-2
Return to citation in text: [1] [2] -
Purrington, S. T.; Woodard, D. L.; Cale, N. C. J. Fluorine Chem. 1990, 48, 345. doi:10.1016/S0022-1139(00)80219-5
Return to citation in text: [1] [2] -
Durrant, G.; Green, R. H.; Lambeth, P. F.; Lester, M. G.; Taylor, N. R. J. Chem. Soc., Perkin Trans. 1 1983, 2211. doi:10.1039/P19830002211
Return to citation in text: [1] -
Gershon, H.; Shanks, L.; Gawiak, D. E. J. Med. Chem. 1976, 19, 1069. doi:10.1021/jm00230a019
Return to citation in text: [1] -
Percy, J. M.; Roig, R.; Singh, K. Eur. J. Org. Chem. 2009, 1058. doi:10.1002/ejoc.200801130
Return to citation in text: [1] -
Kim, D. W.; Jeong, H.-J.; Lim, S. T.; Sohn, M.-H. Angew. Chem., Int. Ed. 2008, 47, 8404. doi:10.1002/anie.200803150
Return to citation in text: [1] -
Fan, R.-H.; Zhou, Y.-G.; Zhang, W.-X.; Hou, X.-L.; Dai, L.-X. J. Org. Chem. 2004, 69, 335. doi:10.1021/jo034895k
Return to citation in text: [1] -
Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B. Chem. Rev. 1994, 94, 2483. doi:10.1021/cr00032a009
Return to citation in text: [1] -
Walsh, P. J.; Sharpless, K. B. Synlett 1993, 605. doi:10.1055/s-1993-22548
Return to citation in text: [1] -
Blundell, P.; Ganguly, A. K.; Girijavallabhan, V. M. Synlett 1994, 263. doi:10.1055/s-1994-22820
Return to citation in text: [1] -
Körber, K.; Risch, P.; Brückner, R. Synlett 2005, 2905. doi:10.1055/s-2005-921915
Return to citation in text: [1] -
Morikawa, K.; Park, J.; Andersson, P. G.; Hashiyama, T.; Sharpless, K. B. J. Am. Chem. Soc. 1993, 115, 8463. doi:10.1021/ja00071a072
Return to citation in text: [1] -
Becker, H.; Sharpless, K. B. Angew. Chem., Int. Ed. Engl. 1996, 35, 448. doi:10.1002/anie.199604481
Return to citation in text: [1] -
Gupta, A. K.; Kazlauskas, R. J. Tetrahedron: Asymmetry 1992, 3, 243. doi:10.1016/S0957-4166(00)80201-3
Return to citation in text: [1] -
Chi, Y.; Peelen, T. J.; Gellman, S. H. Org. Lett. 2005, 7, 3469. doi:10.1021/ol051174u
Return to citation in text: [1] -
Bergmann, H.; Grosch, B.; Sitterberg, S.; Bach, T. J. Org. Chem. 2004, 69, 970. doi:10.1021/jo0354847
Return to citation in text: [1] -
Enders, D.; Thomas, C. R.; Runsink, J. Tetrahedron: Asymmetry 1999, 10, 323. doi:10.1016/S0957-4166(99)00003-8
Return to citation in text: [1] -
Bailey, D. J.; O'Hagan, D.; Tavasli, M. Tetrahedron: Asymmetry 1997, 8, 149. doi:10.1016/S0957-4166(96)00495-8
Return to citation in text: [1] -
Parker, D. Chem. Rev. 1991, 91, 1441. doi:10.1021/cr00007a009
Return to citation in text: [1] -
Pirkle, W. H. J. Am. Chem. Soc. 1966, 88, 1837. doi:10.1021/ja00960a060
Return to citation in text: [1] -
Tavasli, M.; Courtieu, J.; Goss, R. J. M.; Meddour, A.; O'Hagan, D. Chem. Commun. 2002, 844. doi:10.1039/b200992g
Return to citation in text: [1] -
Gao, Y.; Sharpless, K. B. J. Am. Chem. Soc. 1988, 110, 7538. doi:10.1021/ja00230a045
Return to citation in text: [1] [2] -
Xiong, C.; Wang, W.; Hruby, V. J. J. Org. Chem. 2002, 67, 3514. doi:10.1021/jo011172x
Return to citation in text: [1] [2] -
Byun, H.-S.; He, L.; Bittman, R. Tetrahedron 2000, 56, 7051. doi:10.1016/S0040-4020(00)00494-4
Return to citation in text: [1] -
Guo, H.; O'Doherty, G. A. J. Org. Chem. 2008, 73, 5211. doi:10.1021/jo800691v
Return to citation in text: [1] -
Gerard, B.; Sangji, S.; O'Leary, D. J.; Porco, J. A., Jr. J. Am. Chem. Soc. 2006, 128, 7754. doi:10.1021/ja062621j
Return to citation in text: [1] -
Jones, G. B.; Wright, J. M.; Rush, T. M.; Plourde, G. W., II; Kelton, T. F.; Mathews, J. E.; Huber, R. S.; Davidson, J. P. J. Org. Chem. 1997, 62, 9379. doi:10.1021/jo9716035
Return to citation in text: [1]
| 35. | Tavasli, M.; Courtieu, J.; Goss, R. J. M.; Meddour, A.; O'Hagan, D. Chem. Commun. 2002, 844. doi:10.1039/b200992g |
| 36. | Gao, Y.; Sharpless, K. B. J. Am. Chem. Soc. 1988, 110, 7538. doi:10.1021/ja00230a045 |
| 37. | Xiong, C.; Wang, W.; Hruby, V. J. J. Org. Chem. 2002, 67, 3514. doi:10.1021/jo011172x |
| 36. | Gao, Y.; Sharpless, K. B. J. Am. Chem. Soc. 1988, 110, 7538. doi:10.1021/ja00230a045 |
| 37. | Xiong, C.; Wang, W.; Hruby, V. J. J. Org. Chem. 2002, 67, 3514. doi:10.1021/jo011172x |
| 1. | Tsuchiya, T. Adv. Carbohydr. Chem. Biochem. 1990, 48, 91. doi:10.1016/S0065-2318(08)60032-3 |
| 2. | Hartman, M. C. T.; Jiang, S.; Rush, J. S.; Waechter, C. J.; Coward, J. K. Biochemistry 2007, 46, 11630. doi:10.1021/bi700863s |
| 3. | Compain, P.; Martin, O. R. Curr. Top. Med. Chem. 2003, 3, 541. doi:10.2174/1568026033452474 |
| 4. | Murray, B. W.; Wittmann, V.; Burkart, M. D.; Hung, S.-C.; Wong, C.-H. Biochemistry 1997, 36, 823. doi:10.1021/bi962284z |
| 5. | Withers, S. G.; MacLennan, D. J.; Street, I. P. Carbohydr. Res. 1986, 154, 127. doi:10.1016/S0008-6215(00)90028-4 |
| 6. | Sianidis, G.; Wohlert, S.-E.; Pozidis, C.; Karamanou, S.; Luzhetskyy, A.; Vente, A.; Economou, A. J. Biotechnol. 2006, 125, 425. doi:10.1016/j.jbiotec.2006.03.035 |
| 7. | Burkart, M. D.; Vincent, S. P.; Düffels, A.; Murray, B. W.; Ley, S. V.; Wong, C.-H. Bioorg. Med. Chem. 2000, 8, 1937. doi:10.1016/S0968-0896(00)00139-5 |
| 8. | Schengrund, C.-L.; Kováč, P. Carbohydr. Res. 1999, 319, 24. doi:10.1016/S0008-6215(99)00104-4 |
| 9. | Ichikawa, Y.; Lin, Y. C.; Dumas, D. P.; Shen, G. J.; Garcia-Junceda, E.; Williams, M. A.; Bayer, R.; Ketcham, C.; Walker, L. E. J. Am. Chem. Soc. 1992, 114, 9283. doi:10.1021/ja00050a007 |
| 10. | Errey, J. C.; Mann, M. C.; Fairhurst, S. A.; Hill, L.; McNeil, M. R.; Naismith, J. H.; Percy, J. M.; Whitfield, C.; Field, R. A. Org. Biomol. Chem. 2009, 7, 1009–1016. doi:10.1039/b815549f |
| 13. | Caravano, A.; Field, R. A.; Percy, J. M.; Rinaudo, G.; Roig, R.; Singh, K. Org. Biomol. Chem. 2009, 7, 996. doi:10.1039/b815342f |
| 22. | Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B. Chem. Rev. 1994, 94, 2483. doi:10.1021/cr00032a009 |
| 13. | Caravano, A.; Field, R. A.; Percy, J. M.; Rinaudo, G.; Roig, R.; Singh, K. Org. Biomol. Chem. 2009, 7, 996. doi:10.1039/b815342f |
| 12. | Nieschalk, J.; O'Hagan, D. J. Fluorine Chem. 1998, 91, 159. doi:10.1016/S0022-1139(98)00220-6 |
| 20. | Kim, D. W.; Jeong, H.-J.; Lim, S. T.; Sohn, M.-H. Angew. Chem., Int. Ed. 2008, 47, 8404. doi:10.1002/anie.200803150 |
| 13. | Caravano, A.; Field, R. A.; Percy, J. M.; Rinaudo, G.; Roig, R.; Singh, K. Org. Biomol. Chem. 2009, 7, 996. doi:10.1039/b815342f |
| 21. | Fan, R.-H.; Zhou, Y.-G.; Zhang, W.-X.; Hou, X.-L.; Dai, L.-X. J. Org. Chem. 2004, 69, 335. doi:10.1021/jo034895k |
| 14. | Guha, S. K.; Shibayama, A.; Abe, D.; Sakaguchi, M.; Ukaji, Y.; Inomata, K. Bull. Chem. Soc. Jpn. 2004, 77, 2147. doi:10.1246/bcsj.77.2147 |
| 15. | Li, Q.; Wang, W.; Berst, K. B.; Claiborne, A.; Hasvold, L.; Raye, K.; Tufano, M.; Nilius, A.; Shen, L. L.; Flamm, R.; Alder, J.; Marsh, K.; Crowell, D.; Chu, D. T. W.; Plattner, J. J. Bioorg. Med. Chem. Lett. 1998, 8, 1953. doi:10.1016/S0960-894X(98)00355-2 |
| 16. | Purrington, S. T.; Woodard, D. L.; Cale, N. C. J. Fluorine Chem. 1990, 48, 345. doi:10.1016/S0022-1139(00)80219-5 |
| 18. | Gershon, H.; Shanks, L.; Gawiak, D. E. J. Med. Chem. 1976, 19, 1069. doi:10.1021/jm00230a019 |
| 40. | Gerard, B.; Sangji, S.; O'Leary, D. J.; Porco, J. A., Jr. J. Am. Chem. Soc. 2006, 128, 7754. doi:10.1021/ja062621j |
| 16. | Purrington, S. T.; Woodard, D. L.; Cale, N. C. J. Fluorine Chem. 1990, 48, 345. doi:10.1016/S0022-1139(00)80219-5 |
| 13. | Caravano, A.; Field, R. A.; Percy, J. M.; Rinaudo, G.; Roig, R.; Singh, K. Org. Biomol. Chem. 2009, 7, 996. doi:10.1039/b815342f |
| 19. | Percy, J. M.; Roig, R.; Singh, K. Eur. J. Org. Chem. 2009, 1058. doi:10.1002/ejoc.200801130 |
| 41. | Jones, G. B.; Wright, J. M.; Rush, T. M.; Plourde, G. W., II; Kelton, T. F.; Mathews, J. E.; Huber, R. S.; Davidson, J. P. J. Org. Chem. 1997, 62, 9379. doi:10.1021/jo9716035 |
| 15. | Li, Q.; Wang, W.; Berst, K. B.; Claiborne, A.; Hasvold, L.; Raye, K.; Tufano, M.; Nilius, A.; Shen, L. L.; Flamm, R.; Alder, J.; Marsh, K.; Crowell, D.; Chu, D. T. W.; Plattner, J. J. Bioorg. Med. Chem. Lett. 1998, 8, 1953. doi:10.1016/S0960-894X(98)00355-2 |
| 38. | Byun, H.-S.; He, L.; Bittman, R. Tetrahedron 2000, 56, 7051. doi:10.1016/S0040-4020(00)00494-4 |
| 14. | Guha, S. K.; Shibayama, A.; Abe, D.; Sakaguchi, M.; Ukaji, Y.; Inomata, K. Bull. Chem. Soc. Jpn. 2004, 77, 2147. doi:10.1246/bcsj.77.2147 |
| 17. | Durrant, G.; Green, R. H.; Lambeth, P. F.; Lester, M. G.; Taylor, N. R. J. Chem. Soc., Perkin Trans. 1 1983, 2211. doi:10.1039/P19830002211 |
| 39. | Guo, H.; O'Doherty, G. A. J. Org. Chem. 2008, 73, 5211. doi:10.1021/jo800691v |
| 26. | Morikawa, K.; Park, J.; Andersson, P. G.; Hashiyama, T.; Sharpless, K. B. J. Am. Chem. Soc. 1993, 115, 8463. doi:10.1021/ja00071a072 |
| 24. | Blundell, P.; Ganguly, A. K.; Girijavallabhan, V. M. Synlett 1994, 263. doi:10.1055/s-1994-22820 |
| 25. | Körber, K.; Risch, P.; Brückner, R. Synlett 2005, 2905. doi:10.1055/s-2005-921915 |
| 31. | Enders, D.; Thomas, C. R.; Runsink, J. Tetrahedron: Asymmetry 1999, 10, 323. doi:10.1016/S0957-4166(99)00003-8 |
| 32. | Bailey, D. J.; O'Hagan, D.; Tavasli, M. Tetrahedron: Asymmetry 1997, 8, 149. doi:10.1016/S0957-4166(96)00495-8 |
| 29. | Chi, Y.; Peelen, T. J.; Gellman, S. H. Org. Lett. 2005, 7, 3469. doi:10.1021/ol051174u |
| 30. | Bergmann, H.; Grosch, B.; Sitterberg, S.; Bach, T. J. Org. Chem. 2004, 69, 970. doi:10.1021/jo0354847 |
| 27. | Becker, H.; Sharpless, K. B. Angew. Chem., Int. Ed. Engl. 1996, 35, 448. doi:10.1002/anie.199604481 |
| 28. | Gupta, A. K.; Kazlauskas, R. J. Tetrahedron: Asymmetry 1992, 3, 243. doi:10.1016/S0957-4166(00)80201-3 |
© 2013 Laurenson et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)