Abstract
A short synthetic approach was developed for the synthesis of a common trisaccharide core found in kankanose, kankanoside F, H1, H2, and I isolated from the medicinally active plant Cistanche tubulosa. All glycosylations were carried out under nonmetallic reaction conditions. Yields were very good in all intermediate steps.

Graphical Abstract
Introduction
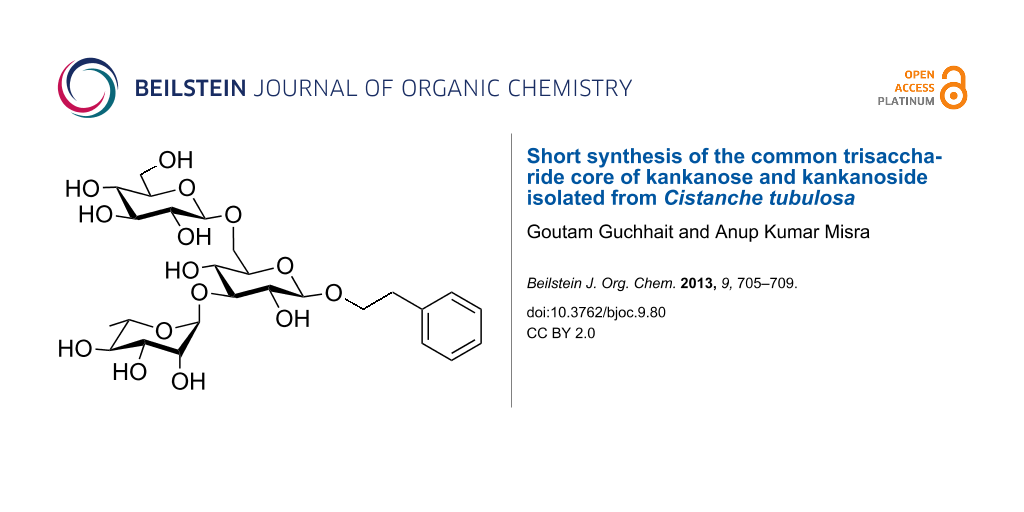
Cistanche tubulosa (C. tubulosa), an Orobanchaceae parasitic plant found in Africa, Asia and Arabia, has been traditionally used as folk medicine and tonic for the treatment of blood-circulation-related disorders, impotence, sterility and body weakness [1-3]. A significant number of bioactive compounds have been isolated from C. tubulosa and have shown promising medicinal activity such as hepatoprotective and vasorelaxant activities [4-6]. Most of the compounds isolated from C. tubulosa and related species are phenylethyl oligosaccharides, iridoids, terpenes and lignans [4-7]. Recently, Yoshikawa et al. isolated and characterized a significant number of phenylethyl oligosaccharides, which include kankanose, kankanoside F, H1, H2, I, etc. [4,5]. Since C. tubulosa has been used in the folk medicine for several years, it is beneficial to find out the biological activities of the individual compounds present in the C. tubulosa extracts. In order to establish the detailed medicinal potential of individual components, it is essential to have higher quantities of the compounds, which are difficult to isolate from the plant source. Therefore, development of concise chemical synthetic strategies would be the best option to gain access to these compounds on a large scale. A few reports are available in the literature for the synthesis of phenylethyl oligosaccharides [8,9]. In this context, we developed a synthetic strategy for the synthesis of the common trisaccharide core of kankanose, kankanoside F, H1, H2 and I isolated from C. tubulosa thereby exploiting newly developed regio- and stereoselective glycosylation conditions (Figure 1). This straightforward synthetic strategy employs a minimum number of steps.
![[1860-5397-9-80-1]](/bjoc/content/figures/1860-5397-9-80-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of the synthesized trisaccharide core found in kankanose, kankanoside F, H1, H2 and I isolated from Cistanche tubulosa.
Figure 1: Structure of the synthesized trisaccharide core found in kankanose, kankanoside F, H1, H2 and I iso...
Results and Discussion
The target trisaccharide 1 in the form of its 2-phenylethyl glycoside was synthesized from the suitably functionalized monosaccharide derivatives 2 [10], 3 [11], and 4 [12], which were prepared from the commercially available reducing sugars (Figure 1). The key features of this synthetic strategy are (a) the application of two regioselective glycosylations by using glycosyl acceptors with two hydroxy groups; (b) application of molecular iodine to the functional group transformations [13]; and (c) activation of the glycosyl trichloroacetimidate derivative by using nitrosyl tetrafluoroborate (NOBF4) [14].
The treatment of D-glucose pentaacetate with 2-phenylethanol in the presence of borontrifluoride diethyl etherate furnished 2-phenylethyl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside (2) in 84% yield [10]. Saponification of compound 2 by using 0.1 M sodium methoxide in methanol followed by benzylidene acetal formation by using benzaldehyde dimethylacetal in the presence of molecular iodine [13] furnished compound 5 in 86% yield. Regioselective 3-O-glycosylation of compound 5 with L-rhamnose derived trichloroacetimidate derivative 3 [11] in the presence of NOBF4 [14] followed by acetylation in the same pot furnished disaccharide derivative 6 in 76% yield. In this case, NOBF4 acts as a promoter for the activation of the glycosyl trichloroacetimidate derivative as well as the acetylation of the sugar derivative with acetic anhydride. The formation of compound 6 was confirmed by its spectral analysis [signals at δ 5.44 (s, PhCH), 4.79 (br s, H-1B), 4.37 (d, J = 7.5 Hz, H-1A in the 1H NMR and at δ 101.9 (PhCH), 101.3 (C-1A), 97.4 (C-1B) in the 13C NMR spectra]. Removal of the benzylidene acetal group under neutral conditions by using triethylsilane and Pd/C [15] resulted in the formation of disaccharide diol 7 in 80% yield. NOBF4 catalyzed regio- and stereoselective 6-O-glycosylation of compound 7 with D-glucose-derived trichloroacetimidate derivative 4 [12] furnished trisaccharide derivative 8 in 71% yield, which was confirmed by the spectral analysis [signals at δ 4.75 (br s, H-1B), 4.54 (d, J = 8.0 Hz, H-1C), 4.22 (d, J = 8.0 Hz, H-1A) in the 1H NMR and at δ 100.8 (C-1A), 100.5 (C-1C), 98.8 (C-1B) in the 13C NMR spectra]. Saponification of compound 8 by using 0.1 M sodium methoxide in methanol furnished compound 1 in 94% yield. Spectral analysis of compound 1 unambiguously confirmed its formation [signals at δ 5.16 (br s, H-1B), 4.37 (d, J = 7.5 Hz, H-1A), 4.32 (d, J = 8.0 Hz, H-1C) in the 1H NMR and at δ 103.5 (C-1A), 102.9 (C-1C), 101.3 (C-1B) in the 13C NMR spectra] (Scheme 1).
![[1860-5397-9-80-i1]](/bjoc/content/inline/1860-5397-9-80-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reagents: (a) 0.1 M CH3ONa, CH3OH, room temperature, 3 h, 98% for compound 5, 94% for compound 1; (b) PhCH(OCH3)2, I2, CH3CN, room temperature, 1.5 h, 86%; (c) NOBF4, CH2Cl2, −20 °C, 20 min; (d) acetic anhydride, room temperature, 1 h, 76% in two steps; (e) Et3SiH, 10% Pd/C, CH3OH/CH2Cl2 (1:1, v/v), room temperature, 30 min, 80%; (f) NOBF4, CH2Cl2, −35 °C, 30 min, 71%.
Scheme 1: Reagents: (a) 0.1 M CH3ONa, CH3OH, room temperature, 3 h, 98% for compound 5, 94% for compound 1; (...
Conclusion
In summary, a straightforward synthetic strategy was developed for the prompt synthesis of a common trisaccharide core of the kankanose, kankanoside F, H1, H2 and I isolated from the extract of C. tubulosa. All the steps are high yielding, and glycosylations were highly regio- and stereoselective. Because of the simplicity of the synthetic strategy, it can be applied in a scaled-up preparation.
Experimental
General methods
All reactions were monitored by thin-layer chromatography with silica-gel-coated TLC plates. The spots on TLC were visualized by warming ceric sulfate (2% Ce(SO4)2 in 2 N H2SO4) sprayed plates on a hot plate. Silica gel 230–400 mesh was used for column chromatography. 1H and 13C NMR spectra were recorded on a Bruker Avance 500 MHz by using CDCl3 as the solvent and TMS as the internal reference unless stated otherwise. Chemical shifts δ are expressed in parts per million (ppm). ESIMS were recorded on a Micromass mass spectrometer. Optical rotations were recorded in a Jasco P-2000 spectrometer. Commercially available grades of organic solvents of adequate purity were used in all reactions.
2-Phenylethyl 4,6-O-benzylidene-β-D-glucopyranoside (5): A solution of compound 2 (5.0 g, 11.05 mmol) in 0.1 M CH3ONa in CH3OH (70 mL) was stirred at room temperature for 3 h and was neutralized with Amberlite IR-120 (H+) resin. The reaction mixture was filtered and concentrated under reduced pressure. To a solution of the deacetylated product (3.1 g) in CH3CN (10 mL) was added benzaldehyde dimethylacetal (2.5 mL, 16.65 mmol) followed by molecular iodine (0.3 g, 1.18 mmol), and the reaction mixture was stirred at room temperature for 1.5 h. The reaction mixture was evaporated and co-evaporated with toluene (3 × 20 mL) under reduced pressure to give the crude product, which was purified over SiO2 by using hexane/EtOAc (2:1) as an eluant to give pure compound 5 (3.5 g, 86%). White solid; mp 158–160 °C (EtOH); [α]D25 −16 (c 1.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.48–7.21 (m, 10H, Ar-H), 5.50 (s, 1H, PhCH), 4.35 (d, J = 7.7 Hz, 1H, H-1), 4.32 (dd, J = 10.5, 4.9 Hz, 1H, H-6a), 4.17–4.12 (m, 1H, CH2), 3.78–3.72 (m, 3H, H-3, H-6b, CH2), 3.52 (t, J = 9.3 Hz, 1H, H-4), 3.47–3.39 (m, 2H, H-2, H-5), 2.99–2.93 (m, 2H, CH2), 2.90 (br s, 1H, OH), 2.54 (br s, 1H, OH); 13C NMR (125 MHz, CDCl3) δ 138.1–126.2 (Ar-C), 103.3 (C-1), 101.9 (PhCH), 80.5 (C-5), 74.5 (C-3), 72.9 (C-4), 71.1 (C-2), 68.6 (CH2), 66.4 (C-6), 36.1 (CH2); ESIMS: 395.1 [M + Na]+; Anal. calcd for C21H24O6: C, 67.73; H, 6.50; found: C, 67.57; H, 6.65.
2-Phenylethyl (2,3,4-tri-O-acetyl-α-L-rhamnopyranosyl)-(1→3)-2-O-acetyl-4,6-O-benzylidene-β-D-glucopyranoside (6): A solution of compound 5 (3.0 g, 8.06 mmol) and compound 3 (3.6 g, 8.28 mmol) in anhydrous CH2Cl2 (15 mL) was cooled to −20 °C under argon. To the cooled reaction mixture was added NOBF4 (1.0 g, 8.56 mmol), and the reaction mixture was stirred at the same temperature for 20 min. After consumption of the starting material (TLC; hexane/EtOAc 4:1), acetic anhydride (3 mL) was added to the reaction mixture, and the mixture was stirred at room temperature for 1 h. The reaction mixture was diluted with CH2Cl2 (100 mL) and the organic layer was washed with satd. NaHCO3 and water, dried (Na2SO4) and concentrated to a crude product, which was purified over SiO2 by using hexane/EtOAc (3:1) as an eluant to give the pure product 6 (4.2 g, 76%). Yellow oil; [α]D25 −60 (c 1.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.39–7.11 (m, 10H, Ar-H), 5.44 (s, 1H, PhCH), 5.23 (dd, J = 10.0, 3.5 Hz, 1H, H-3B), 4.97 (t, J = 8.0 Hz, 1H, H-2A), 4.89 (br s, 1H, H-2B), 4.85 (t, J = 10.0 Hz, 1H, H-4B), 4.79 (br s, 1H, H-1B), 4.37 (d, J = 7.5 Hz, 1H, H-1A), 4.27 (dd, J = 10.5, 5.0 Hz, 1H, H-6aA), 4.04–3.97 (m, 2H, H-5A, CH2), 3.78 (t, J = 9.5 Hz, 1H, H-3A), 3.69 (t, J = 10.0 Hz, 1H, H-6bA), 3.60–3.54 (m, 2H, H-4A, CH2), 3.39–3.36 (m, 1H, H-5B), 2.81–2.78 (m, 2H, CH2), 2.01, 1.91, 1.89, 1.88 (4 s, 12H, 4 COCH3), 0.59 (d, J = 6.0 Hz, 3H, CCH3); 13C NMR (125 MHz, CDCl3) δ 170.0, 169.9, 169.8, 169.4 (4 COCH3), 138.5–126.3 (Ar-C), 101.9 (PhCH), 101.3 (C-1A), 97.4 (C-1B), 79.0 (C-4A), 76.8 (C-3A), 73.3 (C-2A), 71.3 (C-4B), 70.6 (C-2B), 70.5 (CH2), 68.7 (C-6A), 68.4 (C-3B), 66.6 (C-5B), 66.2 (C-5A), 36.0 (CH2), 20.9, 20.8, 20.7, 20.6 (4 COCH3), 16.5 (CH3); ESIMS: 709.2 [M + Na]+; Anal. calcd for C35H42O14: C, 61.22; H, 6.16; found: C, 61.05; H, 6.35.
2-Phenylethyl (2,3,4-tri-O-acetyl-α-L-rhamnopyranosyl)-(1→3)-2-O-acetyl-β-D-glucopyranoside (7): To a stirred solution of compound 6 (4.0 g, 5.82 mmol) and 10% Pd/C (0.5 g) in CH3OH/CH2Cl2 (15 mL, 1:1, v/v) was added Et3SiH (2.8 mL, 17.53 mmol) dropwise, and the reaction mixture was stirred for 30 min at room temperature. The reaction mixture was filtered through a Celite® bed, and the filtering bed was washed with CH2Cl2 (50 mL). The combined filtrate was concentrated under reduced pressure to give the crude product, which was purified over SiO2 by using hexane/EtOAc (1:1) as an eluant to give pure compound 7 (2.8 g, 80%). Yellow oil; [α]D25 −30 (c 1.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.28–7.18 (m, 5H, Ar-H), 5.23 (dd, J = 10.0, 3.5 Hz, 1H, H-3B), 5.10–5.09 (m, 1H, H-2B), 5.05 (t, J = 10.0 Hz, 1H, H-4B), 4.94 (t, J = 8.0 Hz, 1H, H-2A), 4.87 (d, J = 1.8 Hz, 1H, H-1B), 4.41 (d, J = 8.0 Hz, 1H, H-1A), 4.16–4.06 (m, 2H, CH2), 3.92–3.86 (m, 1H, H-6aA), 3.82–3.78 (m, 1H, H-6bA), 3.69–3.61 (m, 2H, H-3A, H-5B), 3.58 (t, J = 9.0 Hz, 1H, H-4A), 3.54–3.31 (m, 1H, H-5A), 2.91–2.83 (m, 2H, CH2), 2.13, 2.04, 2.01, 1.98 (4 s, 12H, 4 COCH3), 1.21 (d, J = 6.0 Hz, 3H, CH3); 13C NMR (125 MHZ, CDCl3) δ 170.0, 169.9, 169.7, 169.5 (4 COCH3), 138.5–126.3 (Ar-C), 100.7 (C-1A), 98.8 (C-1B), 84.8 (C-4A), 75.2 (C-3A), 71.4 (C-2A), 70.7 (C-4B), 70.4 (C-2B), 69.9 (2C, C-5B, CH2), 68.6 (C-3B), 67.7 (C-5A), 62.2 (C-6A), 36.0 (CH2), 20.9, 20.8, 20.7, 20.6 (4 COCH3), 17.4 (CH3); ESIMS: 621.2 [M + Na]+; Anal. calcd for C28H38O14: C, 56.18; H, 6.40; found: C, 56.05; H, 6.55.
2-Phenylethyl (2,3,4-tri-O-acetyl-α-L-rhamnopyranosyl)-(1→3)-[2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)-(1→6)]-2-O-acetyl-β-D-glucopyranoside (8): A solution of compound 4 (1.7 g, 3.45 mmol) and compound 7 (2.0 g, 3.34 mmol) in anhydrous CH2Cl2 (20 mL) was cooled to −35 °C under argon. To the cooled reaction mixture was added NOBF4 (410 mg, 3.51 mmol), and the reaction mixture was stirred at the same temperature for 30 min. The reaction mixture was diluted with CH2Cl2 (100 mL), and the organic layer was washed with satd. NaHCO3 and water, dried (Na2SO4), and concentrated to a crude product, which was purified over SiO2 by using hexane/EtOAc (3:1) as an eluant to give the pure product 8 (2.2 g, 71%). Yellow oil; [α]D25 −32 (c 1.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.17–7.07 (m, 5H, Ar-H), 5.12 (dd, J = 10.0, 3.5 Hz, 1H, H-3B), 5.07 (t, J = 10.0 Hz, 1H, H-3C), 4.99–4.93 (m, 3H, H-2B, H-4B, H-4C), 4.89 (t, J = 10.0 Hz, 1H, H-2C), 4.82 (t, J = 10.0 Hz, 1H, H-2A), 4.75 (br s, 1H, H-1B), 4.54 (d, J = 8.0 Hz, 1H, H-1C), 4.22 (d, J = 8.0 Hz, 1H, H-1A), 4.16 (dd, J = 12.0, 5.0 Hz, 1H, H-6aC), 4.07–3.96 (m, H-6aA, H-6bC, CH2), 3.69–3.65 (m, 1H, H-6bA), 3.60–3.56 (m, 1H, H-5A), 3.55–3.48 (m, 1H, CH2), 3.43 (t, J = 10.0 Hz, 1H, H-3A), 3.42–3.35 (m, 1H, H-5C), 3.33–3.31 (m, 1H, H-5B), 2.77–2.74 (m, 2H, CH2), 2.08, 2.07, 2.01, 2.00, 1.99, 1.96, 1.94, 1.90 (8 s, 24H, 8 COCH3), 1.10 (d, J = 6.0 Hz, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ 170.7, 170.2, 170.0, 169.9, 169.6, 169.4, 169.3, 169.2 (8 COCH3), 138.6–126.2 (Ar-C), 100.8 (C-1A), 100.5 (C-1C), 98.8 (C-1B), 84.3 (C-5C), 74.8 (C-5B), 72.7 (C-3C), 71.9 (C-4A), 71.5 (C-2B), 71.1 (C-5A), 70.8 (C-2C), 70.3 (CH2), 70.2 (C-2A), 70.0 (C-3A), 68.7 (C-6A), 68.5 (C-4C), 68.2 (C-4B), 67.5 (C-3B), 61.7 (C-6C), 35.9 (CH2), 20.9, 20.8, 20.7 (3 C), 20.6 (3 C) (8 COCH3), 17.3 (CH3); ESIMS: 951.3 [M + Na]+; Anal. calcd for C42H56O23: C, 54.31; H, 6.08; found: C, 54.18; H, 6.25.
2-Phenylethyl (α-L-rhamnopyranosyl)-(1→3)-[β-D-glucopyranosyl)-(1→6)]-β-D-glucopyranoside (1): A solution of compound 8 (2.0 g, 2.15 mmol) in 0.1 M CH3ONa in CH3OH (50 mL) was stirred at room temperature for 3 h and neutralized with Amberlite IR-120 (H+) resin. The reaction mixture was filtered and concentrated to give the crude product, which was purified over Sephadex® LH-20 gel by using CH3OH/H2O (10:1) as an eluant to give pure compound 1 (1.2 g, 94%). White powder; [α]D25 −11 (c 1.2, CH3OH); 1H NMR (500 MHz, CD3OD) δ 7.26–7.16 (m, 5H, Ar-H), 5.16 (br s, 1H, H-1B), 4.37 (d, J = 7.5 Hz, 1H, H-1A), 4.32 (d, J = 8.0 Hz, 1H, H-1C), 4.15–4.13 (m, 1H, H-6aC), 4.10–4.04 (m, 1H, CH2), 4.03–3.98 (m, 1H, H-5B), 3.95–3.92 (m, 1H, H-2B), 3.88–3.82 (m, 1H, H-6aA), 3.81–3.73 (m, 2H, H-6bC, CH2), 3.70 (dd, J = 10.0, 3.5 Hz, 1H, H-3B), 3.68–3.64 (m, 1H, H-6bA), 3.50 (t, J = 10.0 Hz, 1H, H-4A), 3.48–3.44 (m, 2H, H-4B, H-5A), 3.39 (t, J = 10.0 Hz, 1H, H-4C), 3.30–3.35 (m, 4H, H-2A, H-3A, H-3C, H-5C), 3.21 (t, J = 9.0 Hz, 1H, H-2C), 2.94–2.90 (m, 2H, CH2), 1.25 (d, J = 6.0 Hz, 3H, CH3); 13C NMR (125 MHz, CD3OD) δ 138.6–125.8 (Ar-C), 103.5 (C-1A), 102.9 (C-1C), 101.3 (C-1B), 82.6 (C-4A), 76.6 (2C, C-3A, C-3C), 75.6 (C-5A), 74.3 (C-5C), 73.7 (C-2C), 72.6 (C-4C), 71.0 (C-4B), 70.9 (C-2A), 70.6 (CH2), 70.2 (C-2B), 68.6 (C-3B), 68.5 (C-5B), 68.3 (C-6C), 61.3 (C-6A), 35.8 (CH2), 16.5 (CH3); ESIMS: 615.2 [M + Na]+; Anal. calcd for C26H40O15: C, 52.70; H, 6.80; found: C, 52.56; H, 7.0.
Supporting Information
| Supporting Information File 1: 1H NMR and 13C NMR spectra of compounds 1, 2, 5, 6, 7 and 8. | ||
| Format: PDF | Size: 2.5 MB | Download |
References
-
Kobayashi, H.; Oguchi, H.; Takizawa, N.; Miyase, T.; Ueno, A.; Usmanghani, K.; Ahmad, M. Chem. Pharm. Bull. 1987, 35, 3309–3314. doi:10.1248/cpb.35.3309
Return to citation in text: [1] -
Xinjiang Institute of Traditional Chinese and Ethnologic Medicines., Ed. Culture Techniques of Xinjiang Staple Medicinal Plants; Xinjiang Science and Technology Press, 2004; pp 84–88.
Return to citation in text: [1] -
Muraoka, O.; Morikawa, T.; Zhang, Y.; Ninomiya, K.; Nakamura, S.; Matsuda, H.; Yoshikawa, M. Tetrahedron 2009, 65, 4142–4148. doi:10.1016/j.tet.2009.03.040
Return to citation in text: [1] -
Morikawa, T.; Pan, Y.; Ninomiya, K.; Imura, K.; Matsuda, H.; Yoshikawa, M.; Yuan, D.; Muraoka, O. Bioorg. Med. Chem. 2010, 18, 1882–1890. doi:10.1016/j.bmc.2010.01.047
Return to citation in text: [1] [2] [3] -
Yoshikawa, M.; Matsuda, H.; Morikawa, T.; Xie, H.; Nakamura, S.; Muraoka, O. Bioorg. Med. Chem. 2006, 14, 7468–7475. doi:10.1016/j.bmc.2006.07.018
Return to citation in text: [1] [2] [3] -
Xie, H.; Morikawa, T.; Matsuda, H.; Nakamura, S.; Muraoka, O.; Yoshikawa, M. Chem. Pharm. Bull. 2006, 54, 669–675. doi:10.1248/cpb.54.669
Return to citation in text: [1] [2] -
Jiménez, C.; Riguera, R. Nat. Prod. Rep. 1994, 11, 591–606. doi:10.1039/np9941100591
And references therein.
Return to citation in text: [1] -
Das, S. K.; Reddy, K. A.; Mukkanti, K. Carbohydr. Res. 2007, 342, 2309–2315. doi:10.1016/j.carres.2007.06.022
Return to citation in text: [1] -
Zhou, F.-Y.; She, J.; Wang, Y.-G. Carbohydr. Res. 2006, 341, 2469–2477. doi:10.1016/j.carres.2006.08.006
Return to citation in text: [1] -
Sinha, B.; Pansare, V. S. Indian J. Chem., Sect. B 1980, 19B, 825–826.
Return to citation in text: [1] [2] -
van Steijn, A. M. P.; Kamerling, J. P.; Vliegenthart, J. F. G. Carbohydr. Res. 1991, 211, 261–277. doi:10.1016/0008-6215(91)80096-6
Return to citation in text: [1] [2] -
Schmidt, R. R.; Michel, J.; Roos, M. Liebigs Ann. Chem. 1984, 1343–1357. doi:10.1002/jlac.198419840710
Return to citation in text: [1] [2] -
Panchadhayee, R.; Misra, A. K. J. Carbohydr. Chem. 2008, 27, 148–155. doi:10.1080/07328300802030837
Return to citation in text: [1] [2] -
Sau, A.; Santra, A.; Misra, A. K. Synlett 2012, 2341–2348. doi:10.1055/s-0032-1317135
Return to citation in text: [1] [2] -
Santra, A.; Ghosh, T.; Misra, A. K. Beilstein J. Org. Chem. 2013, 9, 74–78. doi:10.3762/bjoc.9.9
Return to citation in text: [1]
| 1. | Kobayashi, H.; Oguchi, H.; Takizawa, N.; Miyase, T.; Ueno, A.; Usmanghani, K.; Ahmad, M. Chem. Pharm. Bull. 1987, 35, 3309–3314. doi:10.1248/cpb.35.3309 |
| 2. | Xinjiang Institute of Traditional Chinese and Ethnologic Medicines., Ed. Culture Techniques of Xinjiang Staple Medicinal Plants; Xinjiang Science and Technology Press, 2004; pp 84–88. |
| 3. | Muraoka, O.; Morikawa, T.; Zhang, Y.; Ninomiya, K.; Nakamura, S.; Matsuda, H.; Yoshikawa, M. Tetrahedron 2009, 65, 4142–4148. doi:10.1016/j.tet.2009.03.040 |
| 8. | Das, S. K.; Reddy, K. A.; Mukkanti, K. Carbohydr. Res. 2007, 342, 2309–2315. doi:10.1016/j.carres.2007.06.022 |
| 9. | Zhou, F.-Y.; She, J.; Wang, Y.-G. Carbohydr. Res. 2006, 341, 2469–2477. doi:10.1016/j.carres.2006.08.006 |
| 15. | Santra, A.; Ghosh, T.; Misra, A. K. Beilstein J. Org. Chem. 2013, 9, 74–78. doi:10.3762/bjoc.9.9 |
| 4. | Morikawa, T.; Pan, Y.; Ninomiya, K.; Imura, K.; Matsuda, H.; Yoshikawa, M.; Yuan, D.; Muraoka, O. Bioorg. Med. Chem. 2010, 18, 1882–1890. doi:10.1016/j.bmc.2010.01.047 |
| 5. | Yoshikawa, M.; Matsuda, H.; Morikawa, T.; Xie, H.; Nakamura, S.; Muraoka, O. Bioorg. Med. Chem. 2006, 14, 7468–7475. doi:10.1016/j.bmc.2006.07.018 |
| 12. | Schmidt, R. R.; Michel, J.; Roos, M. Liebigs Ann. Chem. 1984, 1343–1357. doi:10.1002/jlac.198419840710 |
| 4. | Morikawa, T.; Pan, Y.; Ninomiya, K.; Imura, K.; Matsuda, H.; Yoshikawa, M.; Yuan, D.; Muraoka, O. Bioorg. Med. Chem. 2010, 18, 1882–1890. doi:10.1016/j.bmc.2010.01.047 |
| 5. | Yoshikawa, M.; Matsuda, H.; Morikawa, T.; Xie, H.; Nakamura, S.; Muraoka, O. Bioorg. Med. Chem. 2006, 14, 7468–7475. doi:10.1016/j.bmc.2006.07.018 |
| 6. | Xie, H.; Morikawa, T.; Matsuda, H.; Nakamura, S.; Muraoka, O.; Yoshikawa, M. Chem. Pharm. Bull. 2006, 54, 669–675. doi:10.1248/cpb.54.669 |
| 7. |
Jiménez, C.; Riguera, R. Nat. Prod. Rep. 1994, 11, 591–606. doi:10.1039/np9941100591
And references therein. |
| 11. | van Steijn, A. M. P.; Kamerling, J. P.; Vliegenthart, J. F. G. Carbohydr. Res. 1991, 211, 261–277. doi:10.1016/0008-6215(91)80096-6 |
| 4. | Morikawa, T.; Pan, Y.; Ninomiya, K.; Imura, K.; Matsuda, H.; Yoshikawa, M.; Yuan, D.; Muraoka, O. Bioorg. Med. Chem. 2010, 18, 1882–1890. doi:10.1016/j.bmc.2010.01.047 |
| 5. | Yoshikawa, M.; Matsuda, H.; Morikawa, T.; Xie, H.; Nakamura, S.; Muraoka, O. Bioorg. Med. Chem. 2006, 14, 7468–7475. doi:10.1016/j.bmc.2006.07.018 |
| 6. | Xie, H.; Morikawa, T.; Matsuda, H.; Nakamura, S.; Muraoka, O.; Yoshikawa, M. Chem. Pharm. Bull. 2006, 54, 669–675. doi:10.1248/cpb.54.669 |
| 14. | Sau, A.; Santra, A.; Misra, A. K. Synlett 2012, 2341–2348. doi:10.1055/s-0032-1317135 |
| 13. | Panchadhayee, R.; Misra, A. K. J. Carbohydr. Chem. 2008, 27, 148–155. doi:10.1080/07328300802030837 |
| 12. | Schmidt, R. R.; Michel, J.; Roos, M. Liebigs Ann. Chem. 1984, 1343–1357. doi:10.1002/jlac.198419840710 |
| 13. | Panchadhayee, R.; Misra, A. K. J. Carbohydr. Chem. 2008, 27, 148–155. doi:10.1080/07328300802030837 |
| 11. | van Steijn, A. M. P.; Kamerling, J. P.; Vliegenthart, J. F. G. Carbohydr. Res. 1991, 211, 261–277. doi:10.1016/0008-6215(91)80096-6 |
| 14. | Sau, A.; Santra, A.; Misra, A. K. Synlett 2012, 2341–2348. doi:10.1055/s-0032-1317135 |
© 2013 Guchhait and Misra; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)