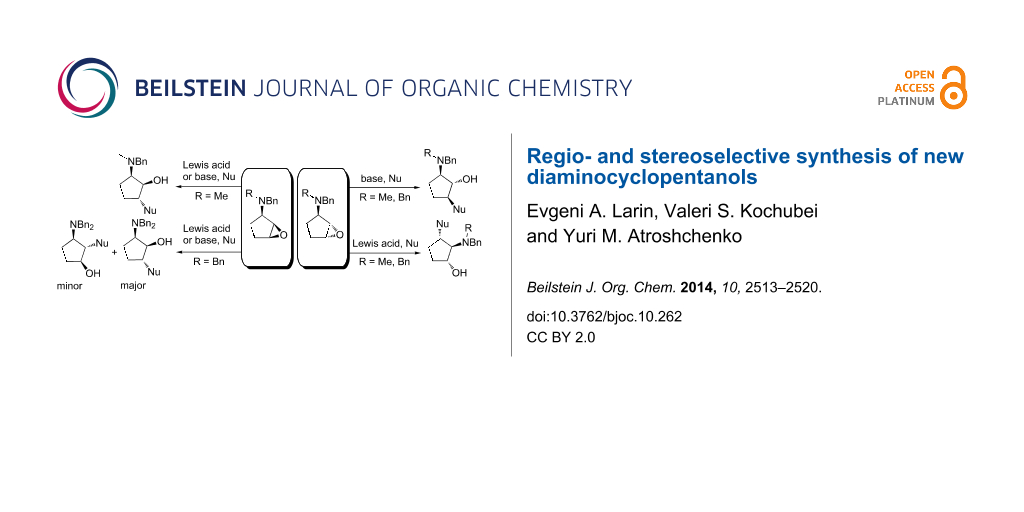
Abstract
The optimal conditions for regio- and stereoselective epoxide ring opening of N,N-disubstituted 1,2-epoxy-3-aminocyclopentanes by different nucleophilic reagents have been developed. The substituents on the nitrogen atom in the epoxide precursor and the orientation of the oxirane ring are crucial for the reaction outcome. Thus, treatment of (1RS,2SR,3SR)-1,2-epoxy-3-(N,N-dibenzylamino)cyclopentane (3b) with amines gave a mixture of C1 and C2 regioadducts, while the use of (1RS,2SR,3SR)-1,2-epoxy-3-(N-benzyl-N-methylamino)cyclopentane (3a) led ultimately to C1 adducts. Base-catalyzed aminolysis of epoxides 6a,b afforded mainly C1 adducts 13a,b arising from trans-diaxal opening of the epoxide ring. Using a Lewis acid catalyst, epoxides 6a,b were transformed into diaminocyclopentanols 14a,b via an alternative pathway involving the formation of aziridinium intermediate 17.

Graphical Abstract
Introduction
In recent years mimicry of aminoglycosides [1-7] and nucleosides [8-10] has become an important field in pharmaceutical research. Regio- and stereochemical diversities within a sugar-like moiety in those mimics may subtly influence their biological activity [11-14]. The functionalization of synthetic, unnatural aminocyclitols represents an attractive strategy towards the preparation of aminoglycoside and nucleoside mimics, and the development of common synthesis routes to various regio- and stereoisomeric aminocyclitol derivatives remains in demand. One of the optimal routes involves the stereoselective ring opening of epoxides by different nucleophiles in the presence of a variety of activators [15-19]. In this context, epoxidation of cyclic allylic amines and subsequent oxirane ring opening represent a viable approach for the development of new pharmaceutically relevant scaffolds.
As a part of our ongoing research in the development of new aminocyclitols, we exploited cyclopentane derivatives to mimic both the 2-deoxystreptamine ring, a core component in aminoglycosides [7], and nucleosides containing 9H-purin-6-amine as a nucleobase portion. High levels of stereoselectivity have been observed in substrate-controlled diastereoselective epoxidation of cyclic alkenes with O- and N-allylic directing groups [20,21]. Several 3-substituted diastereomeric epoxides have recently been synthesized via the ammonium-directed olefinic oxidation of cyclic allylic amines. It has been reported that functionalization of a range of allylic 3-(N,N-dibenzylamino)cycloalkenes with m-CPBA in the presence of trichloroacetic acid gave exclusively corresponding syn-epoxides [22]. Examples of stereoselective epoxide opening of these cyclic amine derivatives are limited to the preparation of the corresponding diols under acidic conditions [23]. Other reported strategies involve the formation of diaminocyclohexanols from epoxides under basic conditions [24] or by activating the epoxides with hydrogen bond donors [25]. Additionally, the synthesis of aminocyclitols from cyclitol epoxides has been described [26,27]. It has been shown that the reaction of cyclitol epoxides with nitrogen-containing nucleophiles in the presence of Lewis acids gave a mixture of C1 and C2 adducts. Both epoxide carbons can react with a nucleophile to produce regioisomeric aminocyclitols. Herein, we describe the regio- and stereoselective synthesis of diaminocyclopentanol derivatives from N-protected cyclopentanamine epoxides using nitrogen-containing nucleophiles.
Results and Discussion
Preparation of starting epoxides
Epoxides 3a,b (Scheme 1) were obtained by the addition of benzyl(methyl)amine or dibenzylamine to cyclopent-2-en-1-yl acetate (1) followed by epoxidation [28]. Epoxides 6a,b were synthesized from 3a,b through epoxide ring inversion using glacial acetic acid as the oxirane-cleaving agent [29].
![[1860-5397-10-262-i1]](/bjoc/content/inline/1860-5397-10-262-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of the starting materials.
Scheme 1: Preparation of the starting materials.
The treatment of the corresponding acetates 4 with mesyl chloride and subsequent transesterification of mesylated substrates 5 resulted in the formation of 6a,b. Epoxides 3 and 6 were identified by 1H NMR data [28]. Morpholine (7a), 2-methyl-1H-imidazole (7b), N-acetylpiperazine (7c) and 9H-purin-6-amine (7d) were used as nucleophiles (Figure 1). Starting amines were selected based on the fact that these motifs are common structural features in drug molecules.
![[1860-5397-10-262-1]](/bjoc/content/figures/1860-5397-10-262-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Amine-based nucleophiles used in the epoxide ring opening reaction.
Figure 1: Amine-based nucleophiles used in the epoxide ring opening reaction.
Optimization of the epoxide ring opening reaction of 3a
The opening of epoxides with nucleophiles in the presence of Lewis acid or base promoters is well documented [30-34]. We conducted a number of experiments to optimize the ring opening in 3a (Table 1). The initial catalytic epoxide ring-opening experiments of 3a in MeCN at 80 °C [35] were unsuccessful, since only starting material was recovered. A series of experiments was performed under solvent-free conditions at 100 °C. In case of morpholine (7a), the best catalytic effect was observed with LiClO4 [36] and Zn(ClO4)2·6H2O [37] affording 56 and 76% yield of 8a after isolation and purification, therefore the absence of the solvent seems crucial for the reaction outcome (Table 1, entries 2 and 3).
Table 1: Reactions of 3a with nucleophiles in the presence of various catalysts.
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-262-i3.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Nu | Catalyst | 8 | Yield (%)a,b | Yield (%)b,c |
|---|---|---|---|---|---|
| 1 | 7a | ZrCl4 | 8a | 14d | traces |
| 2 | 7a | LiClO4 | 8a | 56 | – |
| 3 | 7a | Zn(ClO4)2·6H2O | 8a | 76 | 5 |
| 4 | 7a | Bi(OTf)3 | 8a | 5 | traces |
| 5 | 7a | Cs2CO3 | 8a | – | traces |
| 6 | 7a | K2CO3 | 8a | – | -– |
| 7 | 7b | ZrCl4 | 8b | tracesd | tracesd |
| 8 | 7b | LiClO4 | 8b | 27 | – |
| 9 | 7b | Zn(ClO4)2·6H2O | 8b | 21 | – |
| 10 | 7b | Bi(OTf)3 | 8b | 53 | 24d |
| 11 | 7b | Cs2CO3 | 8b | 61 | 75 |
| 12 | 7b | K2CO3 | 8b | 38 | 16d |
| 13 | 7c | Zn(ClO4)2·6H2O | 8c | 44 | 7 |
| 14 | 7d | Cs2CO3 | 8d | – | 65 |
aReagents and conditions: 5.0 mmol epoxide, 10.0 mol % catalyst, 6.5 mmol nucleophile, neat, 100 °C, 2 h. bIsolated yield. cReagents and conditions: 5.0 mmol epoxide, 10.0 mol % catalyst, 6.5 mmol nucleophile, DMSO (10 mL), 120 °C, 2 h. dReaction time 4 h.
On the contrary, the epoxide ring opening of 3a with 2-methyl-1H-imidazole (7b) in the presence of Lewis acid catalysts produced 8b in low yields (Table 1, entries 7–9), except for the reaction in the presence of Bi(OTf)3 [38] (Table 1, entry 10). In this case, we planned to evaluate the catalytic efficiency of base promoters such as Cs2CO3 and K2CO3 [39-41]. The best result was obtained with Cs2CO3 using DMSO as a solvent (Table 1, entry 11). The catalytic effect of Cs2CO3 could be explained by its ability to increase the poor nucleophilicity of 2-methyl-1H-imidazole (7b). Eventually, Zn(ClO4)2·6H2O and Cs2CO3 were selected as the catalysts for further experiments. These conditions were applied for aminolysis of 3a with N-acetylpiperazine (7c) and 9H-purin-6-amine (7d) (Table 1, entries 13 and 14) to provide 44 and 65% yields of the corresponding aminocyclopentanols 8c and 8d (Supporting Information File 1).
In every experiment, 1,2-trans-2,3-cis-aminocyclopentanols, arising from opening of epoxide 3a at C1, were the only regioisomers isolated. The stereo- and regiochemistry of 8a and 8d were assigned by 2D NMR (HSQC-DEPT, 1H,1H COSY and NOESY experiments). The 2D carbon–proton chemical shift correlation study on 8a showed that the proton resonances at δ 2.43–2.46, 2.55 and 3.90 correspond to methylene groups, and that these resonances can be assigned by C(1)H-OH and C(2)H-NCH3 COSY correlations. The presence of the C(1)H-C(2)H correlation and the absence of the C(2)H-C(5)H correlation in the NOESY spectrum support the stereochemistry of 8a. The same relative configuration of 8d was assigned from 2D NMR analysis, and the stereochemistry of 8b and 8c was additionally confirmed by the comparison of 3J coupling constants of the resonances corresponding to C(1)H, C(2)H and C(5)H (Supporting Information File 2). Therefore, spectral data obtained for the compounds 8a–d are consistent with the acid-catalyzed trans-diaxial epoxide opening, proceeding via a late-transition state, and the N-benzyl-N-methylammonium moiety promotes the nucleophilic attack at the C1-oxirane carbon atom [29].
Synthesis of diaminocyclopentanols using epoxide 3b
Next, we explored the influence of the N,N-dibenzylamino group on the ring opening reaction using the optimized reaction conditions for 3b (Table 2). Surprisingly, the ring opening of N,N-dibenzyl derivative 4b displayed poor regioselectivity. In fact, a mixture of the separable regioisomers 9a–d and 10a–d were obtained, where the major products 9a–d were formed due to the attack of the nucleophile at the C1-oxirane carbon atom.
Table 2: Regioselectivity in the epoxide ring opening of 3b with nucleophiles.
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-262-i4.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Nu | Catalyst | Product | Yield (%)a,b | rrc (9:10) |
|---|---|---|---|---|---|
| 1 | 7a | Zn(ClO4)2·6H2O | 9a | 48 | 2:1 |
| 10a | 24 | ||||
| 2 | 7a | LiClO4 | 9a | 50 | 8:5 |
| 10a | 31 | ||||
| 3 | 7b | Cs2CO3 | 9b | 47 | 7:3 |
| 10b | 20 | ||||
| 4 | 7b | Cs2CO3 | 9b | 64d | 2:1 |
| 10b | 26d | ||||
| 5 | 7c | Zn(ClO4)2·6H2O | 9c | 43 | 2:1 |
| 10c | 21 | ||||
| 6 | 7c | LiClO4 | 9c | 45d | 9:5 |
| 10c | 25d | ||||
| 7 | 7d | Cs2CO3 | 9d | 46 | 2:1 |
| 10d | 23 | ||||
aReagents and conditions: 5.0 mmol epoxide, 10.0 mol % catalyst, 6.5 mmol nucleophile, neat, 100 °C, 2 h. bIsolated yield. cRegioisomeric ratio for separated isomers. dReactions were performed in DMSO at 120 °C.
In case of aliphatic cyclic amines (morpholine (7a) and N-acetylpiperazine (7c)), the best regioisomeric ratio (2:1) was observed using Zn(ClO4)2·6H2O as a catalyst (Table 2, entries 1 and 5). The use of LiClO4 led to lower regioselectivity (Table 2, entries 2 and 6). Apparently, the nature of the catalyst and the ability of the metal ion to coordinate with the oxirane oxygen have no significant influence on the regioisomeric ratio (rr). The reactions of 3b with 2-methyl-1H-imidazole (7b) and 9H-purin-6-amine (7d) in the presence of Cs2CO3 in DMSO showed the same regioselectivity (Table 2, entries 4 and 7), indicating that the epoxide opening reactions are not directed by the amine nucleophilicity. Moreover, there was not much effect on the outcome of the reactions conducted either under solvent-free conditions or using DMSO as a solvent.
The regioisomers 9a–d and 10a–d were isolated through column chromatographic separation and fully characterized in order to avoid ambiguity. The stereochemistry of the major regioisomers 9a–d was deduced from the analysis of 1H NMR and 2D NMR data as described for 8a–d. Assignments of CH proton resonances of 10a–d were established by 1H,13C HMBC and HSQC-DEPT experiments, and connectivity was established by the analysis of 1H,1H COSY spectra. The structures of 10a and 10c were elucidated based on COSY correlation of C(1)H-OH resonances at δ 3.88 for 10a and 3.85 for 10c. 1H NMR NOESY analyses of 10a and 10c facilitated the initial assignment of the relative configuration. Thus, the values of C(1)H-C(3)H, C(2)H-OH and C(3)H-CH2N NOEs were quite diagnostic. C(1)H proton resonances were observed at δ 3.84–3.95 for 10b and 4.53 for 10d. C(2)H and C(3)H resonances appeared as doublets of doublets at δ 4.20 (J = 9.3 and 7.8 Hz) and 3.38 (J = 17.7 and 8.6 Hz) for 10b and at δ 4.61 (J = 9.8 and 7.9 Hz) and 3.71 (J = 18.3 and 8.7 Hz) for 10d. C(2)H-C(3)H, C(1)H-OH, C(2)H-C(1)H COSY correlations and C(1)H-C(3)H, C(2)H-OH, C(2)H-CH2Ph NOEs indicated the relative 1,2-anti-2,3-anti-configurations of 10b and 10d (Supporting Information File 2).
In order to investigate the influence of different substituents on the nitrogen atom on the regioselective outcome, epoxides 3c and 3d were additionally synthesized, and the results are summarized in Table 3.
Table 3: Epoxide ring opening of 3a–d containing different substituents on the nitrogen atom.
![[Graphic 3]](/bjoc/content/inline/1860-5397-10-262-i5.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Epoxide | R1 | R2 | Reaction time, ha | Product (C1)b | Product (C2)b | rr (C1:C2) |
|---|---|---|---|---|---|---|---|
| 1 | 3a | Me | Bn | 2 | 8a | – | >19:1 |
| 2 | 3b | Bn | Bn | 2 | 9a | 10a | 2:1 |
| 3 | 3c | Ph | Bn | 6 | 11 | 12 | 5:1c |
| 4 | 3d | Ph | Ph | 6d | – | – | – |
aReagents and conditions: 5.0 mmol epoxide, 10.0 mol % Zn(ClO4)2·6H2O, 6.5 mmol morpholine (7a), neat, 100 °C. bProducts formed due to the nucleophilic attack at the C1 or C2 oxirane carbon atoms. cThe regiochemistry was established from 1H NMR analysis of the mixture. dNo reaction was observed.
It has been proposed earlier that the coordination of the Lewis acid to both oxygen atoms in 2,3-epoxy alcohols and acids leads to the formation of the intermediate complex, for which nucleophiles attack preferably the C3 position [42,43]. The electron donating methyl group in 3a seems to improve the binding of the Lewis acid to the nitrogen atom, favoring the formation of the C1-adduct, and the lower basicity of the dibenzylamino moiety in 3b may lead to the diminished coordination of Lewis acid and hence the lower regioselectivity (Table 3, entries 1 and 2). Unexpectedly, using 3c, with an electron withdrawing phenyl group on the nitrogen atom, provided higher regioselectivity towards the C1-adduct (rr 5:1) in comparison with 3b (Table 3, entries 2 and 3). The structure of the major regioisomer 11 was established by the analysis of 1H NMR and 1H,1H COSY data (Supporting Information File 2). This fact can be explained by the suggestion that despite the induced binding of the Lewis acid to the nitrogen atom due to the negative inductive effect of the phenyl substituent, the intermediate complex is likely to be stabilized by the π electrons of the phenyl ring, which leads to the formation of 11 as a major product. However, the presence of electron withdrawing substituents on the nitrogen atom and, as a result, the diminished coordination of the Lewis acid required the longer reaction time (6 h), while in case of 3d bearing two phenyl groups no epoxide ring opening was observed (Table 3, entries 3 and 4).
Ring opening reactions for epoxides 6a,b
Ring opening of epoxides 6a,b was investigated by the reaction with 9H-purin-6-amine (7d) and morpholine (7a) as the nucleophiles under the above conditions (Table 4). As it was expected, single regioisomers 13a,b with 1,2-anti-2,3-anti-configuration (Table 4, entries 1 and 2) were obtained. C(1)H-C(2)H, C(1)H-OH, C(1)H-C(5)H COSY correlations and C(2)H-C(5)H, C(2)H-OH, C(5)H-OH NOEs demonstrated that product 13b has the structure shown in Table 4. The presence of C(2)H-C(5)H and C(1)H-NCH3 NOEs were supportive of the assigned configuration of 13a (Supporting Information File 2). The ring opening reaction of epoxide 6b with 9H-purin-6-amine (7d) in the presence of Cs2CO3 (Table 4, entry 2) showed the higher level of regioselectivity in comparison with the regioselective outcome for epoxide 3b (Table 2, entry 7), which can be interpreted as a result of dominance of steric over electronic factors in the case of epoxides 6a,b.
Table 4: The epoxide ring opening reactions of 6a,b.
![[Graphic 4]](/bjoc/content/inline/1860-5397-10-262-i6.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Epoxide | Nu | Catalyst | Product | Yield (%)a |
|---|---|---|---|---|---|
| 1 | 6a | 7d | Cs2CO3 | 13a | 68b |
| 2 | 6b | 7d | Cs2CO3 | 13b | 55b |
| 3 | 6a | 7a | Zn(ClO4)2·6H2O | 14a | 84c |
| 4 | 6b | 7a | Zn(ClO4)2·6H2O | 14b | 80c |
| 15 | 6d | ||||
aIsolated yield. bReagents and conditions: 5.0 mmol epoxide, 10.0 mol % catalyst, 6.5 mmol nucleophile, DMSO (10 mL), 120 °C, 2 h. cReagents and conditions: 5.0 mmol epoxide, 10.0 mol % catalyst, 6.5 mmol nucleophile, neat, 100 °C, 2 h. dRegioisomeric ratio (14b:15) – 12:1.
A bulky N,N-disubstituted amino group is prone to adopt a pseudoequatorial orientation. In basic conditions, the nucleophile (9H-purin-6-amine (7d)) attacks the oxirane carbon atom from the side of the carbocyclic ring where the N,N-disubstituted amino group is located, and this precludes the approach of the nucleophile to the C2 carbon atom because of sterical hindrance. Thus, C1 of 6a,b is the favoured site for the nucleophilic attack, which gives rise to the formation of products 13a,b with essentially complete regioselectivity (Table 4, entries 1 and 2). Surprisingly, aminolysis of substrates 6a,b under Lewis acid-catalyzed conditions resulted mostly in the formation of regioisomers 14a,b (Table 4, entries 3 and 4), while the target isomer 15 was obtained only from epoxide 6b, as the minor product in 6% yield (Table 4, entry 4). Aminocyclopentanols 14a,b provided quite similar 1H NMR spectra, and methine protons showed similar multiplicity patterns. For example, the resonance corresponding to C(2)H of 14a appeared as a doublet of doublets (J = 7.0 and 4.4 Hz) centered at δ 2.84, while the signal corresponding to C(2)H of 14b also appeared as a doublet of doublets (J = 7.3 and 4.3 Hz) centered at δ 2.92. The structure and the relative configuration of 14a,b were unambiguously confirmed by the presence of C(1)H-C(2)H, C(1)H-OH, C(2)H-C(3)H COSY and C(1)H-C(3)H, C(2)H-OH, C(3)H-CH2Ph NOESY correlations observed in 2D spectra. The structure of 15 was determined by the analysis of 2D NMR spectra by analogy with that of 13b (Supporting Information File 2).
These results are in contrast to the outcome of the ring opening reactions of epoxides 3a,b, and this may be explained by the formation of 14a,b via the aziridimium intermediate 17. Based on earlier results [44], a mechanism of this transformation was hypothesized as shown in Scheme 2. The intermediate 17 is formed after the intramolecular rearrangement of intermediate 16 formed from Zn(ClO4)2-catalyzed C–O bond cleavage followed by the attack of the N,N-disubstituted amino moiety towards C2. The approach of the N,N-disubstituted amino group to C2 would be more favorable than that of the nucleophile (morpholine) to either oxirane carbon atoms. Therefore, the nucleophilic attack is subsequent to the formation of the aziridinium ring, which is consistent with our experimental results.
![[1860-5397-10-262-i2]](/bjoc/content/inline/1860-5397-10-262-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Postulated mechanism for the formation of 14a,b.
Scheme 2: Postulated mechanism for the formation of 14a,b.
Conclusion
In summary, we have optimized the reaction conditions of epoxide ring opening of epoxides 3a,b and 6a,b with a variety of amines to give the corresponding diaminocyclopentanols in good yields. It has been shown that using Zn(ClO4)2·6H2O under solvent-free conditions and Cs2CO3 in DMSO is preferable to the ring opening of di-N-protected cyclopentanamine epoxides. We have highlighted the influence of the nature of the N,N-disubstituted amino moiety and the orientation of the oxirane ring on the stereo- and regioselective outcomes. Aminolysis of epoxides 3a,b is mainly dictated by electronic bias to afford the corresponding C1 adducts for 3a and the mixture of C1 and C2 adducts in the ratio 2:1 for 3b. The treatment of epoxides 6a,b with 9H-purin-6-amine (7d) under base-catalyzed conditions gives C1 adducts as the sole products. Thus, the nucleophilic attack of the amine towards the C2 oxirane carbon atom can be controlled by steric constraints, and it is obvious that the bulky N,N-dibenzylamino moiety of epoxide 6b impedes the formation of the corresponding C2 adduct due to the steric hindrance. Application of Lewis acid as a catalyst for the ring opening reactions of 6a,b provides an alternative mechanism that involves the formation of aziridinium intermediate 17. As a result, regioisomers 14a,b were obtained as the major products.
References
-
Mehta, G.; Lakshminath, S.; Talukdar, P. Tetrahedron Lett. 2002, 43, 335–338. doi:10.1016/S0040-4039(01)02125-6
Return to citation in text: [1] -
Magnet, S.; Blanchard, J. S. Chem. Rev. 2005, 105, 477–498. doi:10.1021/cr0301088
Return to citation in text: [1] -
Dey, R. T.; Sarkar, T. K. J. Org. Chem. 2010, 75, 4521–4529. doi:10.1021/jo100724w
Return to citation in text: [1] -
Cong, X.; Liao, Q.-J.; Yao, Z.-J. J. Org. Chem. 2004, 69, 5314–5321. doi:10.1021/jo0496547
Return to citation in text: [1] -
Pandey, G.; Tiwari, K. N.; Puranik, V. G. Org. Lett. 2008, 10, 3611–3614. doi:10.1021/ol801381t
Return to citation in text: [1] -
Sakairi, N.; Hayashida, M.; Amano, A.; Kuzuhara, H. J. Chem. Soc., Perkin Trans. 1 1990, 1301–1313. doi:10.1039/p19900001301
Return to citation in text: [1] -
Busscher, G. F.; Rutjes, F. P. J. T.; van Delft, F. L. Chem. Rev. 2005, 105, 775–792. doi:10.1021/cr0404085
Return to citation in text: [1] [2] -
Kapeller, H.; Baumgartner, H.; Marschner, C.; Pucher, R.; Griengl, H. Monatsh. Chem. 1997, 128, 953–960. doi:10.1007/BF00807105
Return to citation in text: [1] -
Hatton, W.; Arosio, D.; Re, M.; Giudici, D.; Bernardi, A.; Seneci, P. C. R. Chim. 2010, 13, 1284–1300. doi:10.1016/j.crci.2009.11.004
Return to citation in text: [1] -
Somu, R. V.; Wilson, D. J.; Bennett, E. M.; Boshoff, H. I.; Celia, L.; Beck, B. J.; Barry, C. E., III; Aldrich, C. C. J. Med. Chem. 2006, 49, 7623–7635. doi:10.1021/jm061068d
Return to citation in text: [1] -
Bergmeier, S. C. Tetrahedron 2000, 56, 2561–2576. doi:10.1016/S0040-4020(00)00149-6
Return to citation in text: [1] -
Chattopadhyay, S. K.; Bandyopadhyay, A. Tetrahedron Lett. 2011, 52, 3942–3944. doi:10.1016/j.tetlet.2011.05.103
Return to citation in text: [1] -
Verhelst, S. H. L.; Wennekes, T.; van der Marel, G. A.; Overkleeft, H. S.; van Boeckel, C. A. A.; van Boom, J. H. Tetrahedron 2004, 60, 2813–2822. doi:10.1016/j.tet.2004.01.063
Return to citation in text: [1] -
Trapero, A.; Alfonso, I.; Butters, T. D.; Llebaria, A. J. Am. Chem. Soc. 2011, 133, 5474–5484. doi:10.1021/ja111480z
Return to citation in text: [1] -
Calvani, F.; Crotti, P.; Gardelli, C.; Pineschi, M. Tetrahedron 1994, 50, 12999–13022. doi:10.1016/S0040-4020(01)81219-9
Return to citation in text: [1] -
Chini, M.; Crotti, P.; Macchia, F. Tetrahedron Lett. 1990, 31, 4661–4664. doi:10.1016/S0040-4039(00)97701-3
Return to citation in text: [1] -
Zeynizadeh, B.; Sadighnia, L. Bull. Korean Chem. Soc. 2010, 31, 2644–2648. doi:10.5012/bkcs.2010.31.9.2644
Return to citation in text: [1] -
Fan, R.-H.; Hou, X.-L. Tetrahedron Lett. 2003, 44, 4411–4413. doi:10.1016/S0040-4039(03)00943-2
Return to citation in text: [1] -
Arbelo, D. O.; Prieto, J. A. Tetrahedron Lett. 2002, 43, 4111–4114. doi:10.1016/S0040-4039(02)00739-6
Return to citation in text: [1] -
Hoveyda, A. H.; Evans, D. A.; Fu, G. C. Chem. Rev. 1993, 93, 1307–1370. doi:10.1021/cr00020a002
Return to citation in text: [1] -
Asensio, G.; Boix-Bernardini, C.; Andreu, C.; González-Núñez, M. E.; Mello, R.; Edwards, J. O.; Carpenter, G. B. J. Org. Chem. 1999, 64, 4705–4711. doi:10.1021/jo982512q
Return to citation in text: [1] -
Davies, S. G.; Long, M. J. C.; Smith, A. D. Chem. Commun. 2005, 4536–4538. doi:10.1039/b509088a
Return to citation in text: [1] -
Aciro, C.; Claridge, T. D. W.; Davies, S. G.; Roberts, P. M.; Russell, A. J.; Thomson, J. E. Org. Biomol. Chem. 2008, 6, 3751–3761. doi:10.1039/b808811j
Return to citation in text: [1] -
Zhao, S.; Freeman, J. P.; Chidester, C. G.; VonVoigtlander, P. F.; Mizsak, S. A.; Szmuszkovicz, J. J. Org. Chem. 1993, 58, 4043–4048. doi:10.1021/jo00067a042
Return to citation in text: [1] -
Threadgill, M. D.; Webb, P. J. Chem. Soc., Chem. Commun. 1991, 269–271. doi:10.1039/c39910000269
Return to citation in text: [1] -
Serrano, P.; Llebaria, A.; Delgado, A. J. Org. Chem. 2002, 67, 7165–7167. doi:10.1021/jo0261146
Return to citation in text: [1] -
Chini, M.; Crotti, P.; Favero, L.; Macchia, F.; Pineschi, M. Tetrahedron Lett. 1994, 35, 433–436. doi:10.1016/0040-4039(94)85073-9
Return to citation in text: [1] -
Brennan, M.; Claridge, T. D. W.; Compton, R. G.; Davies, S. G.; Fletcher, A. M.; Henstridge, M. C.; Hewings, D. S.; Kurosawa, W.; Lee, J. A.; Roberts, P. M.; Schoonen, A. K.; Thomson, J. E. J. Org. Chem. 2012, 77, 7241–7261. doi:10.1021/jo3010556
Return to citation in text: [1] [2] -
Bond, C. W.; Cresswell, A. J.; Davies, S. G.; Fletcher, A. M.; Kurosawa, W.; Lee, J. A.; Roberts, P. M.; Russell, A. J.; Smith, A. D.; Thomson, J. E. J. Org. Chem. 2009, 74, 6735–6748. doi:10.1021/jo9012783
Return to citation in text: [1] [2] -
Swamy, N. R.; Kondaji, G.; Nagaiah, K. Synth. Commun. 2002, 32, 2307–2312. doi:10.1081/SCC-120006000
Return to citation in text: [1] -
Kim, B.-H.; Piao, F.; Lee, E.-J.; Kim, J.-S.; Jun, Y.-M.; Lee, B.-M. Bull. Korean Chem. Soc. 2004, 25, 881–888. doi:10.5012/bkcs.2004.25.6.881
Return to citation in text: [1] -
Pujala, B.; Rana, S.; Chakraborti, A. K. J. Org. Chem. 2011, 76, 8768–8780. doi:10.1021/jo201473f
Return to citation in text: [1] -
Sabitha, G.; Reddy, G. S. K. K.; Reddy, K. B.; Yadav, J. S. Synthesis 2003, 2298–2300. doi:10.1055/s-2003-41070
Return to citation in text: [1] -
Jacobsen, E. N. Acc. Chem. Res. 2000, 33, 421–431. doi:10.1021/ar960061v
Return to citation in text: [1] -
Chakraborti, A. K.; Kondaskar, A. Tetrahedron Lett. 2003, 44, 8315–8319. doi:10.1016/j.tetlet.2003.09.046
Return to citation in text: [1] -
Serrano, P.; Llebaria, A.; Vázquez, J.; de Pablo, J.; Anglada, J. M.; Delgado, A. Chem. – Eur. J. 2005, 11, 4465–4472. doi:10.1002/chem.200401270
Return to citation in text: [1] -
Pujala, B.; Chakraborti, A. K. J. Org. Chem. 2007, 72, 3713–3722. doi:10.1021/jo062674j
Return to citation in text: [1] -
Ollevier, T.; Nadeau, E. Tetrahedron Lett. 2008, 49, 1546–1550. doi:10.1016/j.tetlet.2007.12.100
Return to citation in text: [1] -
Flessner, T.; Doye, S. J. Prakt. Chem. 1999, 341, 186–190. doi:10.1002/(SICI)1521-3897(199902)341:2<186::AID-PRAC186>3.0.CO;2-6
Return to citation in text: [1] -
Fink, D. M. Synlett 2004, 2394–2396. doi:10.1055/s-2004-832836
Return to citation in text: [1] -
Birajdar, S. S.; Hatnapure, G. D.; Keche, A. P.; Kamble, V. M. J. Chem. Pharm. Res. 2013, 5, 583–589.
Return to citation in text: [1] -
Caron, M.; Sharpless, K. B. J. Org. Chem. 1985, 50, 1557–1560. doi:10.1021/jo00209a047
Return to citation in text: [1] -
Chong, J. M.; Sharpless, K. B. J. Org. Chem. 1985, 50, 1560–1563. doi:10.1021/jo00209a048
Return to citation in text: [1] -
Aciro, C.; Davies, S. G.; Roberts, P. M.; Russell, A. J.; Smith, A. D.; Thomson, J. E. Org. Biomol. Chem. 2008, 6, 3762–3770. doi:10.1039/b808812h
Return to citation in text: [1]
| 38. | Ollevier, T.; Nadeau, E. Tetrahedron Lett. 2008, 49, 1546–1550. doi:10.1016/j.tetlet.2007.12.100 |
| 36. | Serrano, P.; Llebaria, A.; Vázquez, J.; de Pablo, J.; Anglada, J. M.; Delgado, A. Chem. – Eur. J. 2005, 11, 4465–4472. doi:10.1002/chem.200401270 |
| 37. | Pujala, B.; Chakraborti, A. K. J. Org. Chem. 2007, 72, 3713–3722. doi:10.1021/jo062674j |
| 1. | Mehta, G.; Lakshminath, S.; Talukdar, P. Tetrahedron Lett. 2002, 43, 335–338. doi:10.1016/S0040-4039(01)02125-6 |
| 2. | Magnet, S.; Blanchard, J. S. Chem. Rev. 2005, 105, 477–498. doi:10.1021/cr0301088 |
| 3. | Dey, R. T.; Sarkar, T. K. J. Org. Chem. 2010, 75, 4521–4529. doi:10.1021/jo100724w |
| 4. | Cong, X.; Liao, Q.-J.; Yao, Z.-J. J. Org. Chem. 2004, 69, 5314–5321. doi:10.1021/jo0496547 |
| 5. | Pandey, G.; Tiwari, K. N.; Puranik, V. G. Org. Lett. 2008, 10, 3611–3614. doi:10.1021/ol801381t |
| 6. | Sakairi, N.; Hayashida, M.; Amano, A.; Kuzuhara, H. J. Chem. Soc., Perkin Trans. 1 1990, 1301–1313. doi:10.1039/p19900001301 |
| 7. | Busscher, G. F.; Rutjes, F. P. J. T.; van Delft, F. L. Chem. Rev. 2005, 105, 775–792. doi:10.1021/cr0404085 |
| 7. | Busscher, G. F.; Rutjes, F. P. J. T.; van Delft, F. L. Chem. Rev. 2005, 105, 775–792. doi:10.1021/cr0404085 |
| 30. | Swamy, N. R.; Kondaji, G.; Nagaiah, K. Synth. Commun. 2002, 32, 2307–2312. doi:10.1081/SCC-120006000 |
| 31. | Kim, B.-H.; Piao, F.; Lee, E.-J.; Kim, J.-S.; Jun, Y.-M.; Lee, B.-M. Bull. Korean Chem. Soc. 2004, 25, 881–888. doi:10.5012/bkcs.2004.25.6.881 |
| 32. | Pujala, B.; Rana, S.; Chakraborti, A. K. J. Org. Chem. 2011, 76, 8768–8780. doi:10.1021/jo201473f |
| 33. | Sabitha, G.; Reddy, G. S. K. K.; Reddy, K. B.; Yadav, J. S. Synthesis 2003, 2298–2300. doi:10.1055/s-2003-41070 |
| 34. | Jacobsen, E. N. Acc. Chem. Res. 2000, 33, 421–431. doi:10.1021/ar960061v |
| 15. | Calvani, F.; Crotti, P.; Gardelli, C.; Pineschi, M. Tetrahedron 1994, 50, 12999–13022. doi:10.1016/S0040-4020(01)81219-9 |
| 16. | Chini, M.; Crotti, P.; Macchia, F. Tetrahedron Lett. 1990, 31, 4661–4664. doi:10.1016/S0040-4039(00)97701-3 |
| 17. | Zeynizadeh, B.; Sadighnia, L. Bull. Korean Chem. Soc. 2010, 31, 2644–2648. doi:10.5012/bkcs.2010.31.9.2644 |
| 18. | Fan, R.-H.; Hou, X.-L. Tetrahedron Lett. 2003, 44, 4411–4413. doi:10.1016/S0040-4039(03)00943-2 |
| 19. | Arbelo, D. O.; Prieto, J. A. Tetrahedron Lett. 2002, 43, 4111–4114. doi:10.1016/S0040-4039(02)00739-6 |
| 35. | Chakraborti, A. K.; Kondaskar, A. Tetrahedron Lett. 2003, 44, 8315–8319. doi:10.1016/j.tetlet.2003.09.046 |
| 11. | Bergmeier, S. C. Tetrahedron 2000, 56, 2561–2576. doi:10.1016/S0040-4020(00)00149-6 |
| 12. | Chattopadhyay, S. K.; Bandyopadhyay, A. Tetrahedron Lett. 2011, 52, 3942–3944. doi:10.1016/j.tetlet.2011.05.103 |
| 13. | Verhelst, S. H. L.; Wennekes, T.; van der Marel, G. A.; Overkleeft, H. S.; van Boeckel, C. A. A.; van Boom, J. H. Tetrahedron 2004, 60, 2813–2822. doi:10.1016/j.tet.2004.01.063 |
| 14. | Trapero, A.; Alfonso, I.; Butters, T. D.; Llebaria, A. J. Am. Chem. Soc. 2011, 133, 5474–5484. doi:10.1021/ja111480z |
| 29. | Bond, C. W.; Cresswell, A. J.; Davies, S. G.; Fletcher, A. M.; Kurosawa, W.; Lee, J. A.; Roberts, P. M.; Russell, A. J.; Smith, A. D.; Thomson, J. E. J. Org. Chem. 2009, 74, 6735–6748. doi:10.1021/jo9012783 |
| 8. | Kapeller, H.; Baumgartner, H.; Marschner, C.; Pucher, R.; Griengl, H. Monatsh. Chem. 1997, 128, 953–960. doi:10.1007/BF00807105 |
| 9. | Hatton, W.; Arosio, D.; Re, M.; Giudici, D.; Bernardi, A.; Seneci, P. C. R. Chim. 2010, 13, 1284–1300. doi:10.1016/j.crci.2009.11.004 |
| 10. | Somu, R. V.; Wilson, D. J.; Bennett, E. M.; Boshoff, H. I.; Celia, L.; Beck, B. J.; Barry, C. E., III; Aldrich, C. C. J. Med. Chem. 2006, 49, 7623–7635. doi:10.1021/jm061068d |
| 28. | Brennan, M.; Claridge, T. D. W.; Compton, R. G.; Davies, S. G.; Fletcher, A. M.; Henstridge, M. C.; Hewings, D. S.; Kurosawa, W.; Lee, J. A.; Roberts, P. M.; Schoonen, A. K.; Thomson, J. E. J. Org. Chem. 2012, 77, 7241–7261. doi:10.1021/jo3010556 |
| 24. | Zhao, S.; Freeman, J. P.; Chidester, C. G.; VonVoigtlander, P. F.; Mizsak, S. A.; Szmuszkovicz, J. J. Org. Chem. 1993, 58, 4043–4048. doi:10.1021/jo00067a042 |
| 26. | Serrano, P.; Llebaria, A.; Delgado, A. J. Org. Chem. 2002, 67, 7165–7167. doi:10.1021/jo0261146 |
| 27. | Chini, M.; Crotti, P.; Favero, L.; Macchia, F.; Pineschi, M. Tetrahedron Lett. 1994, 35, 433–436. doi:10.1016/0040-4039(94)85073-9 |
| 42. | Caron, M.; Sharpless, K. B. J. Org. Chem. 1985, 50, 1557–1560. doi:10.1021/jo00209a047 |
| 43. | Chong, J. M.; Sharpless, K. B. J. Org. Chem. 1985, 50, 1560–1563. doi:10.1021/jo00209a048 |
| 23. | Aciro, C.; Claridge, T. D. W.; Davies, S. G.; Roberts, P. M.; Russell, A. J.; Thomson, J. E. Org. Biomol. Chem. 2008, 6, 3751–3761. doi:10.1039/b808811j |
| 28. | Brennan, M.; Claridge, T. D. W.; Compton, R. G.; Davies, S. G.; Fletcher, A. M.; Henstridge, M. C.; Hewings, D. S.; Kurosawa, W.; Lee, J. A.; Roberts, P. M.; Schoonen, A. K.; Thomson, J. E. J. Org. Chem. 2012, 77, 7241–7261. doi:10.1021/jo3010556 |
| 44. | Aciro, C.; Davies, S. G.; Roberts, P. M.; Russell, A. J.; Smith, A. D.; Thomson, J. E. Org. Biomol. Chem. 2008, 6, 3762–3770. doi:10.1039/b808812h |
| 22. | Davies, S. G.; Long, M. J. C.; Smith, A. D. Chem. Commun. 2005, 4536–4538. doi:10.1039/b509088a |
| 39. | Flessner, T.; Doye, S. J. Prakt. Chem. 1999, 341, 186–190. doi:10.1002/(SICI)1521-3897(199902)341:2<186::AID-PRAC186>3.0.CO;2-6 |
| 40. | Fink, D. M. Synlett 2004, 2394–2396. doi:10.1055/s-2004-832836 |
| 41. | Birajdar, S. S.; Hatnapure, G. D.; Keche, A. P.; Kamble, V. M. J. Chem. Pharm. Res. 2013, 5, 583–589. |
| 20. | Hoveyda, A. H.; Evans, D. A.; Fu, G. C. Chem. Rev. 1993, 93, 1307–1370. doi:10.1021/cr00020a002 |
| 21. | Asensio, G.; Boix-Bernardini, C.; Andreu, C.; González-Núñez, M. E.; Mello, R.; Edwards, J. O.; Carpenter, G. B. J. Org. Chem. 1999, 64, 4705–4711. doi:10.1021/jo982512q |
| 25. | Threadgill, M. D.; Webb, P. J. Chem. Soc., Chem. Commun. 1991, 269–271. doi:10.1039/c39910000269 |
| 29. | Bond, C. W.; Cresswell, A. J.; Davies, S. G.; Fletcher, A. M.; Kurosawa, W.; Lee, J. A.; Roberts, P. M.; Russell, A. J.; Smith, A. D.; Thomson, J. E. J. Org. Chem. 2009, 74, 6735–6748. doi:10.1021/jo9012783 |
© 2014 Larin et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)