Abstract
A series of azobenzene–cholesterol organogel compounds (M0–M12) with different spacers were designed and synthesized. The molecular structures were confirmed by 1H NMR and 13C NMR spectroscopy. The rapid and reversible photoresponsive properties of the compounds were investigated by UV–vis spectroscopy. Their thermal phase behaviors were studied by DSC. The length of the spacer plays a crucial role in the gelation. Compound M6 is the only one that can gelate in ethanol, isopropanol and 1-butanol and the reversible gel–sol transitions are also investigated. To obtain visual insight into the microstructure of the gels, the typical structures of the xerogels were studied by SEM. Morphologies of the aggregates change from flower-like, network and rod with different sizes. By using IR and XRD characterization, it is found that intermolecular H-bonding, the solvents and van der Waals interaction are the main contributions to the specific superstructure.

Graphical Abstract
Introduction
In the past few decades, low molecular mass organic gelators (LMOGs) have attracted increasing attention not only for basic self-assembly behavior but also for their potential application in areas such as templates [1], light harvesting [2], fluorescent scensing [3], etc. The driving force for the spontaneous formation of gel could be relatively non-covalent interactions such as π–π stacking [4-7], hydrogen bonding [7-9], dipole–dipole [10], and van der Waals interactions [11,12]. A series of cholesterol-based gelators were reported [13-15]. These new classes of organogelator architectures have been systematically studied because of their reversible gel process and unique directional self-association through weak van der Waals interactions [16,17]. Up to now, numerous attempts have been made to develop novel supramolecular architectures, which can bring about new predictable gelation abilities for the construction of novel cholesterol-based gelators.
Azobenzene is one of the smartest molecules among all known photochromic compounds because of its E/Z isomerization [18]. Based on this, photomechanical soft materials containing azobenzene have been utilized in supramolecular systems [19]. Examples of several cholesterol-linked azobenzenes with gel–sol reversible changes also have been studied [20-23]. R. Zentel found that the photoinduced reversible gel–sol changes between isotropic and anisotropic gels of a class of semicarbazide–azobenzene-based gelators [24]. A new multistimuli photoresponsive organogel containing azobenzene groups was designed and studied by D. Zhu and his co-workers [25]. Q. Zhang synthesized a series of new symmetric dicholesterol-linked gelators [26]. Upon UV–vis irradiation of the gels, a reversible gel–sol transition occurred by breaking and reforming of van der Waals interactions. The cholesterol-substituted diacetylenic polymerized gels exhibit enhanced stability upon thermo- or photo-stimuli [27]. Although similar compounds have been discussed, it is still not very clear about the relationship between the spacer length and the gelation ability of gelators. Herein we synthesized a series of compounds with different spacers between cholesteryl and azobenene units (Scheme 1). In these compounds, M6 is the only one that can gelate in ethanol, isopropanol and 1-butanol. It is found that intermolecular H-bonding, the solvents and van der Waals interaction are the main contributions to the specific superstructure.
![[1860-5397-11-122-i1]](/bjoc/content/inline/1860-5397-11-122-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
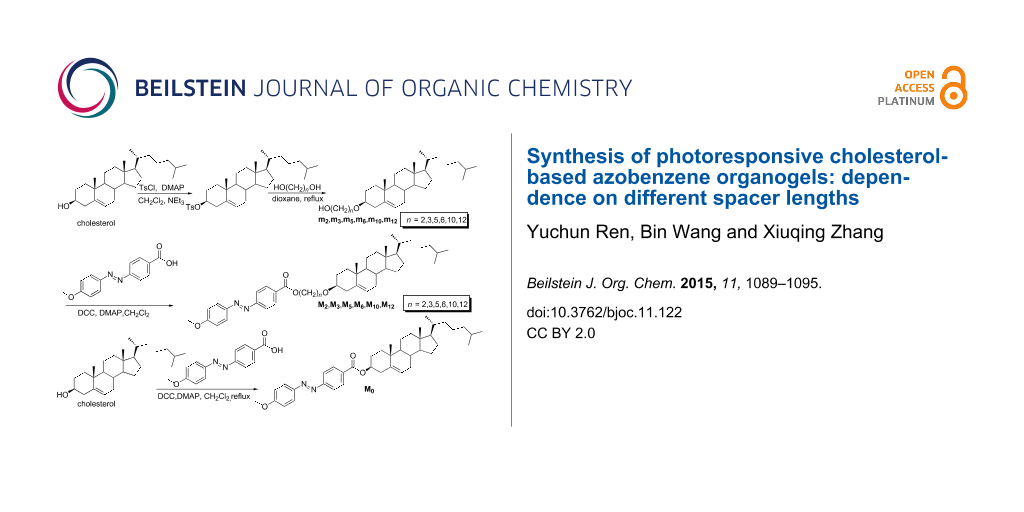
Scheme 1: Synthesis route and chemical structure of the compounds.
Scheme 1: Synthesis route and chemical structure of the compounds.
Results and Discussion
To investigate the photoresponsiveness of these compounds, the dilute THF solution of compounds were irradiated by UV and visible light. Figure 1a shows an obvious increase in the peak at 258 nm and a concurrent decrease in the strong peak at 358 nm. The photostationary state was attained after 94 min of irradiation and the spectral changes are attributable to the trans-to-cis isomerization under UV light irradiation. Further visible light (450 nm) irradiation on the cis-solution led to an increase in the absorption intensity at 358 nm and a decrease around 258 nm, and the original spectrum recovered by the reverse cis-to-trans isomerization (Figure 1b). The absorption band at 358 nm corresponding to the π–π* transition of azobenzene increased, respectively. It should be emphasized that such reversible spectral transformation could be repeated several times. The trans-to-cis isomerization of the azobenzenze unit had been demonstrated. Compounds M2, M5, M6, and M12 have the same recoverable photoresponsive properties as M0. Their UV–vis spectra were shown in Supporting Information File 1, Figures S1–S4. M3 and M10 have the photochromic properties.
![[1860-5397-11-122-1]](/bjoc/content/figures/1860-5397-11-122-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Changes in the absorption over time in the UV–vis spectra of dilute THF solution of M0: (a) upon UV-light irradiation (λ = 365 nm) and (b) upon visible-light irradiation (λ = 450 nm) of the solution obtained after irradiation of 365 nm.
Figure 1: Changes in the absorption over time in the UV–vis spectra of dilute THF solution of M0: (a) upon UV...
Liquid crystal properties of the synthesized compounds
The structures of these compounds are shown in Scheme 1. The thermal behaviors of the seven compounds were analyzed by differential scanning calorimetry (DSC). The DSC curves of the cholesterol-based supramolecular complexes are shown in Figure 2 and phase transition temperatures are listed in Table 1. The DSC thermogram of M0 presented three peaks at 158, 211 and 249 °C on heating, corresponding to the crystal to crystal transition, crystal to mesophase transition and mesophase to isotropic transition. On the cooling run, DSC curves presented isotropic phase to mesophase, mesophase to crystal and crystal to crystal at 190, 155 and 126 °C. Each curve of compounds M2, M6 and M10 displayed a single endothermic melting peak. The heating DSC curves of M3, M5 and M12 contained two endothermic peaks. The peaks at lower temperature were due to the crystal to mesophase transitions, and the second peaks displayed the mesophase to isotropic transitions. The DSC thermogram of M3, M5 and M12 were observed two exothermic peaks on cooling process, which indicated the transitions from isotropic phase to mesophase at higher temperature and mesophase to crystal transitions at lower temperature
![[1860-5397-11-122-2]](/bjoc/content/figures/1860-5397-11-122-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: DSC thermogram of the compounds M0-n obtained on (a) heating and (b) cooling.
Figure 2: DSC thermogram of the compounds M0-n obtained on (a) heating and (b) cooling.
Table 1: Phase transition temperatures of the compounds.
| sample | heating (°C) | cooling (°C) |
|---|---|---|
| M0 | K 158 K2 211 N 249 I | I 190 N 155 K1 126 K2 |
| M2 | K 92 I | I 59 K |
| M3 | K 164 N 220 I | I 224 N 213 K |
| M5 | K 120 N 197 I | I 193 N 83 K |
| M6 | K 78 I | — |
|
M10
M12 |
K 90 I
K 75 N 120 I |
I 54 K
I 120 N 42 K |
K: crystalline state, N: nematic phase, I: isotropic liquid.
Gelation behaviors
The gelation behaviors of the seven compounds were tested in 22 solvents at room temperature and the results were listed in Table 2. Among the compounds examined, M6 is the only one that forms a gel in ethanol, 1-butanol and isopropanol, while the other compounds cannot gelate in any solvent. Generally speaking, the gels were formed by a heating and cooling cycle process. Examination of the data shown in the table reveals that the change of spacer length in the molecular skeleton has a significant effect on the gelation abilities of the compounds. To investigate the photoresponsiveness of the three gels, we performed the photoirradiation of gels using a UV lamp. The formed gels at room temperature were in an opaque state. Furthermore, the gels with both solvents are very stable for months of storage in a closed container, and no significant changes can be found. However, upon UV irradiation, the gel to sol phase transition became transparent as a result of trans-to-cis isomerization. Moreover, the solution has a tendency to reverse the sol to gel phase transition under visible irradiation. The corresponding visual images are shown in Figure 3.
Table 2: Gelation performances of the seven cholesteryl derivatives at room temperature.
| Solvent | M0 | M2 | M3 | M5 | M6 | M10 | M12 |
|---|---|---|---|---|---|---|---|
| Methanol | I | I | I | I | I | I | I |
| Ethanol | I | I | I | I | G | I | I |
| 1-Propanol | I | I | I | I | I | I | I |
| 1-Butanol | I | I | I | P | G | I | I |
| 1-Pentanol | I | I | I | I | I | I | I |
| 1-Octanol | I | P | I | I | I | I | I |
| 1,4-Butanediol | I | I | I | I | I | I | I |
| Isopropanol | I | I | I | I | G | I | I |
| Acetone | I | P | I | P | S | I | P |
| DMSO | I | I | I | I | I | I | I |
| DMF | I | P | I | I | S | I | I |
| EtOAc | I | S | I | P | S | P | P |
| Kerosene | I | P | I | I | I | P | S |
| Benzene | S | S | S | S | S | S | S |
| Toluene | S | S | S | S | S | S | S |
| Xylene | S | S | S | S | S | S | S |
| THF | S | S | S | S | S | S | S |
| n-Hexane | I | P | I | P | P | P | P |
| Dichloromethane | S | S | S | S | S | S | S |
| Chloroform | S | S | S | S | S | S | S |
| Tetrachloromethane | S | S | S | S | S | S | S |
| Distilled water | I | I | I | I | I | I | I |
G: turbid gel; S: solution; P: precipitate; I: insoluble.
![[1860-5397-11-122-3]](/bjoc/content/figures/1860-5397-11-122-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Photographic images of the M6 sol-gel transition behavior after UV or visible light irradiation in different solution (a) ethanol, (b) isopropanol and (c) 1-butanol.
Figure 3: Photographic images of the M6 sol-gel transition behavior after UV or visible light irradiation in ...
Scanning electron microscopy studies
To obtain a visual insight into the morphology of the aggregation mode, the typical structures of the xerogels prepared by the freeze-drying of the three gels in different solvents were studied by SEM (Figure 4). With the reference to the images, it can be clearly seen that the structures of the three gelators are significantly different from each other. The morphologies of the aggregates change from flower-like to network and rod with different sizes. Obviously, the gel from ethanol mainly shows a similar flower-like morphology. When we focused on the isopropanol xerogel, we could see that there are many regularly three-dimensional network structures. In 1-butanol, the organogelator molecules in the gel phase were self-assembled into tightly staked rods. Different solvents caused a sharp difference between these morphologies of the xerogels.
![[1860-5397-11-122-4]](/bjoc/content/figures/1860-5397-11-122-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: SEM images of xerogels formed with M6 from solution: (a) ethanol, (b) isopropanol and (c) 1-butanol.
Figure 4: SEM images of xerogels formed with M6 from solution: (a) ethanol, (b) isopropanol and (c) 1-butanol....
The FTIR spectroscopy
It should be emphasized that the organogels achieved through strength and directionality of hydrogen bonding. Further information about the intermolecular interactions of these gels was obtained from the FTIR spectroscopy. Figure 5 shows the IR spectra of M6 powder and its gels in ethanol, isopropanol and 1-butanol. Gels show two peaks which located at 3740 and 3672 cm−1, characteristic of the hydrogen bonding O–H stretching vibration. As for the powder, the absorption bands cannot be found in the same vibrational area. The peak observed at 3437cm−1 can be assigned to the intermolecular association hydrogen stretching vibration. However, the band of ethanol, isopropanol and 1-butanol shifted to 3438, 3439 and 3440 cm−1, respectively. The comparison of the IR spectra of powder and gels indicated the hydrogen-bond interaction in the gels. The bands of the asymmetric and symmetric CH2 stretching vibration of M6 powder emerged at 2935 and 2852 cm−1, while, in the ethanol gel, they shifted to 2934 and 2858 cm−1, in isopropanol gel, they shifted to 2934 and 2862 cm−1, in 1-butanol , they shifted to 2933 and 2859 cm−1. The above mentioned FTIR spectral changes demonstrated sufficiently that van der Waals interaction between the alkyl chains plays an important role in the self-assembly process of organogels.
![[1860-5397-11-122-5]](/bjoc/content/figures/1860-5397-11-122-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: FTIR spectra of gels: (a) powder, (b–d) ethanol, isopropanol and 1-butanol xerogels, respectively.
Figure 5: FTIR spectra of gels: (a) powder, (b–d) ethanol, isopropanol and 1-butanol xerogels, respectively.
The XRD studies
To reveal the detailed changes in gel molecular of M6, XRD analyses were conducted and the results were displayed in Figure 6. The pattern of the xerogel obtained from the 1-butanol gel exhibited five peaks at 12.84, 15.38, 17.22, and 20.44°, corresponding to d-spacing of 6.90, 5.80, 5.14, 4.30, and 3.70 nm, respectively. The d-spacing almost in ratio of 7:6:5:4, may due to the molecular arrangement with order or existence layers of the stacking between molecular. The last peak may relate to the periodic intermolecular distances at different direction and intermolecular separations. Two diffraction peaks at 17.32° and 25.40° were detected for the xerogels of another two solvents, corresponding to d values of 5.10 and 3.50 nm, respectively. The different self-assembly modes of gelator in various solvents cause the values changing. The XRD results described above also demonstrated that the molecular interaction and the solvents had a significant impact on the gel formation modes.
![[1860-5397-11-122-6]](/bjoc/content/figures/1860-5397-11-122-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: XRD patterns of xerogels. (a) ethanol, (b) isopropanol and (c) 1-butanol.
Figure 6: XRD patterns of xerogels. (a) ethanol, (b) isopropanol and (c) 1-butanol.
Experimental
Materials
The starting materials, thylene glycol, 1,3-propanediol, 1,5-pentadiol, 1,6-hexanediol, 1,10-decanediol, 1,12-dodecanediol, cholesterol, 4-toluene sulfonyl chloride, phenol, 4-aminobenzoic acid, iodomethane, sodium nitrite, hydrochloric acid, sodium hydroxide, N,N'-dicyclohexylcarbodiimide (DCC), 4-dimethylaminopyridine (DMAP), all were purchased from Sigma-Aldrich Chemicals (Shanghai, China). Other used reagents were commercially available and were distilled before used. The synthesis of the 4'-carboxy-4-methoxyazobenzene ultilized in this study was described elsewhere [28].
General measurements
NMR spectra were recorded with a Bruker-400 NMR spectrometer. Powder X-ray diffraction (XRD) measurements were carried out on a Rigaku D/max 2500PC (Japan)-type single-crystal diffractometer using Cu Kα radiation to determine the crystal structure. Infrared spectroscopy (IR) was measured on a Nicolet 6700 spectrometer from 4000 to 500 cm−1 at room temperature. The samples were prepared as KBr pellets. The morphology of the gel samples were examined by scanning electron microscopy (SEM, S5200, FEI Company) under an acceleration voltage of 30 kV. UV–vis absorption spectra were recorded using a spectrophotometer (UV-3600, Shimadzu, Japan). Differential scanning calorimetry (DSC) was carried out on a thermal analysis (TA) DSC-Q30 in nitrogen at a heating rate of 10 K/min.
Synthetic procedures
Synthesis of m2–m12. 3β-[(2-hydroxyethyl)oxy]cholest-5-ene (m2) was synthesized similar to a previous report [29]. Cholesterol 15.00 g (38 mmol) in dry dichloromethane (225 mL) and dry triethylamine (16.50 mL), p-tosyl chloride 10.35 g (54 mmol) was added. A catalytic amount of DMAP was also added. The reaction mixture was then allowed to stir at 48 °C for 12 h. The reaction mixture was washed sequentially with diluted HCl aqueous solution, saturated brine solution, and water. The organic phase was dried over anhydrous Na2SO4. The solvents were removed by vacuum evaporation and recrystallized using chloroform and methanol. The residue (7.0 g, 13 mmol) was dissolved in anhydrous dioxane (60 mL) and dry ethylene glycol (20 g, 0.32 mol) was added. The mixture was refluxed at 110 °C for 4 h. The solution was cooled and solvent was removed under vacuum. The white residue was dissolved in chloroform. The organic layer washed with NaHCO3, water, and brine; and dried over anhydrous Na2SO4. Finally, the product was purified by column chromatography over silica gel eluting with petroleum ether/ethyl acetate to yield 90% of m2. The procedure used for the preparation of m3, m5, m6, m10, and m12 are similar to that for m2.
4-Methoxy-4’-((cholesteryloxy)carbonyl)azobenzene (M0)
A mixture of 4'-carboxy-4-methoxyazobenzene (0.25 g, 10 mmol), cholesterol (0.38 g, 1 mmol), and DCC (0.21 g, 1 mmol), DMAP (0.06 g, 0.5 mmol) in 25 mL CH2Cl2 was stirred at room temperature for 24 h. The solvent was evaporated under vacuum. The product was purified by column chromatography over silica gel eluting with pure CH2Cl2, yield 70%. Related compounds (M2, M3, M5, M6, M10, M12) were synthesized by a similar method.
Gelation test
The potential gelator (25 mg) in solvent (1 mL) was placed in a sealed test tube and the solution was heated in a water bath until the solid was dissolved completely. The solution was cooled to room temperature. If the gelator formed a gel at this stage, it was classified as “G”. When the solution remained all the time, they were referred to as solutions (S). The hot clear solution systems, which precipitated when they are cooled down to room temperature, are donated as P (precipitation), respectively. Insoluble systems in which the potential gelator could not be dissolved even at the boiling point of the solvent were designated as insoluble (I).
Conclusion
In summary, we have synthesized seven cholesterol-based compounds with azobenzene groups. The compounds with different flexible alkyl spacers containing two, three, five, six, ten, or twelve carbon atoms are denoted as M2–M12, respectively. Upon UV–vis irradiation, these compounds can undergo a typical reversible trans–cis and cis–trans change due to the photo-isomerization of the azobenzene units. They possess recoverable photoresponsive properties. Their thermal phase behaviors have been investigated by DSC, indicating good liquid crystal properties. The dramatic change in the gelation behavior was produced by the different spacer lengths between cholesteryl and azobenzene. Compound M6 is the only good gelator that can gelate in ethanol, isopropanol and 1-butanol. Based on the results of SEM, IR and XRD, it demonstrated that the solvents, intermolecular H-bonding, and van der Waals interaction affected the aggregation mode and morphology of gels. These results can be applied in fields of design and development of novel organogelators and soft matter.
Supporting Information
| Supporting Information File 1: Spectroscopical and analytical data. | ||
| Format: PDF | Size: 2.0 MB | Download |
Acknowledgements
The work was financially supported by the Program for Scientific and Technological Innovative Team in Sichuan Provincial Universities of China (2010008), Key Project of Natural Science of Sichuan Educational Department of China (12ZA175), Chemical synthesis and Pollution Control Key Laboratory of Sichuan Province of China (CSCP2013-2), and University research and launch projects of China West Normal University in 2014 (14C003).
References
-
van Bommel, K. J. C.; Friggeri, A.; Shinkai, S. Angew. Chem., Int. Ed. 2003, 42, 980–999. doi:10.1002/anie.200390284
Return to citation in text: [1] -
Sugiyasu, K.; Fujita, N.; Shinkai, S. Angew. Chem., Int. Ed. 2004, 43, 1229–1233. doi:10.1002/anie.200352458
Return to citation in text: [1] -
Kartha, K. K.; Babu, S. S.; Srinivasan, S.; Ajayaghosh, A. J. Am. Chem. Soc. 2012, 134, 4834–4841. doi:10.1021/ja210728c
Return to citation in text: [1] -
van Gorp, J. J.; Vekemans, J. A. J. M.; Meijer, E. W. J. Am. Chem. Soc. 2002, 124, 14759–14769. doi:10.1021/ja020984n
Return to citation in text: [1] -
An, B.-K.; Lee, D.-S.; Lee, J.-S.; Park, Y.-S.; Song, H.-S.; Park, S. Y. J. Am. Chem. Soc. 2004, 126, 10232–10233. doi:10.1021/ja046215g
Return to citation in text: [1] -
Kishimura, A.; Yamashita, T.; Aida, T. J. Am. Chem. Soc. 2005, 127, 179–183. doi:10.1021/ja0441007
Return to citation in text: [1] -
Shao, H.; Parquette, J. R. Chem. Commun. 2010, 46, 4285–4287. doi:10.1039/c0cc00701c
Return to citation in text: [1] [2] -
de Jong, J. J. D.; Lucas, L. N.; Kellogg, R. M.; van Esch, J. H.; Feringa, B. L. Science 2004, 304, 278–281. doi:10.1126/science.1095353
Return to citation in text: [1] -
Yagai, S.; Nakajima, T.; Kishikawa, K.; Kohmoto, S.; Karatsu, T.; Kitamura, A. J. Am. Chem. Soc. 2005, 127, 11134–11139. doi:10.1021/ja052645a
Return to citation in text: [1] -
Mamiya, J.-i.; Kanie, K.; Hiyama, T.; Ikeda, T.; Kato, T. Chem. Commun. 2002, 1870–1871. doi:10.1039/b205072b
Return to citation in text: [1] -
Suzuki, M.; Nigawara, T.; Yumoto, M.; Kimura, M.; Shirai, H.; Hanabusa, K. Org. Biomol. Chem. 2003, 1, 4124–4131. doi:10.1039/b308371c
Return to citation in text: [1] -
Kramb, R. C.; Zukoski, C. F. Langmuir 2008, 24, 7565–7572. doi:10.1021/la800021h
Return to citation in text: [1] -
Chen, Q.; Zhang, D.; Zhang, G.; Zhu, D. Langmuir 2009, 25, 11436–11441. doi:10.1021/la9016257
Return to citation in text: [1] -
Lee, W.-K.; Kim, K.-N.; Achard, M. F.; Jin, J.-I. J. Mater. Chem. 2006, 16, 2289–2297. doi:10.1039/b516141j
Return to citation in text: [1] -
Xu, F.; Wang, H.; Zhao, J.; Liu, X.; Li, D.; Chen, C.; Ji, J. Macromolecules 2013, 46, 4235–4246. doi:10.1021/ma400276u
Return to citation in text: [1] -
Peng, J.; Liu, K.; Liu, J.; Zhang, Q.; Feng, X.; Fang, Y. Langmuir 2008, 24, 2992–3000. doi:10.1021/la703672u
Return to citation in text: [1] -
Chen, P.; Lu, R.; Xue, P.; Xu, T.; Chen, G.; Zhao, Y. Langmuir 2009, 25, 8395–8399. doi:10.1021/la8035727
Return to citation in text: [1] -
Xie, S.; Natansohn, A.; Rochon, P. Chem. Mater. 1993, 5, 403–411. doi:10.1021/cm00028a003
Return to citation in text: [1] -
Barrett, C. J.; Mamiya, J.-i.; Yager, K. G.; Ikeda, T. Soft Matter 2007, 3, 1249–1261. doi:10.1039/b705619b
Return to citation in text: [1] -
Murata, K.; Aoki, M.; Suzuki, T.; Harada, T.; Kawabata, H.; Komori, T.; Ohseto, F.; Ueda, K.; Shinkai, S. J. Am. Chem. Soc. 1994, 116, 6664–6676. doi:10.1021/ja00094a023
Return to citation in text: [1] -
Jung, J. H.; Ono, Y.; Shinkai, S. Angew. Chem., Int. Ed. 2000, 39, 1862–1865. doi:10.1002/(SICI)1521-3773(20000515)39:10<1862::AID-ANIE1862>3.0.CO;2-3
Return to citation in text: [1] -
Jung, J. H.; Shinkai, S.; Shimizu, T. Chem. Mater. 2003, 15, 2141–2145. doi:10.1021/cm0217912
Return to citation in text: [1] -
Koumura, N.; Kudo, M.; Tamaoki, N. Langmuir 2004, 20, 9897–9900. doi:10.1021/la048334f
Return to citation in text: [1] -
Deindörfer, P.; Davis, R.; Zentel, R. Soft Matter 2007, 3, 1308–1311. doi:10.1039/b706492f
Return to citation in text: [1] -
Wang, C.; Chen, Q.; Sun, F.; Zhang, D.; Zhang, G.; Huang, Y.; Zhao, R.; Zhu, D. J. Am. Chem. Soc. 2010, 132, 3092–3096. doi:10.1021/ja910721s
Return to citation in text: [1] -
Wu, Y.; Wu, S.; Tian, X.; Wang, X.; Wu, W.; Zou, G.; Zhang, Q. Soft Matter 2011, 7, 716–721. doi:10.1039/C0SM00330A
Return to citation in text: [1] -
Wang, J.; Yang, G.; Jiang, H.; Zou, G.; Zhang, Q. Soft Matter 2013, 9, 9785–9791. doi:10.1039/c3sm51896e
Return to citation in text: [1] -
Gao, J.; He, Y.; Liu, F.; Zhang, X.; Wang, Z.; Wang, X. Chem. Mater. 2007, 19, 3877–3881. doi:10.1021/cm0707197
Return to citation in text: [1] -
Bajaj, A.; Kondaiah, P.; Bhattacharya, S. Bioconjugate Chem. 2008, 19, 1640–1651. doi:10.1021/bc700381v
Return to citation in text: [1]
| 28. | Gao, J.; He, Y.; Liu, F.; Zhang, X.; Wang, Z.; Wang, X. Chem. Mater. 2007, 19, 3877–3881. doi:10.1021/cm0707197 |
| 29. | Bajaj, A.; Kondaiah, P.; Bhattacharya, S. Bioconjugate Chem. 2008, 19, 1640–1651. doi:10.1021/bc700381v |
| 1. | van Bommel, K. J. C.; Friggeri, A.; Shinkai, S. Angew. Chem., Int. Ed. 2003, 42, 980–999. doi:10.1002/anie.200390284 |
| 7. | Shao, H.; Parquette, J. R. Chem. Commun. 2010, 46, 4285–4287. doi:10.1039/c0cc00701c |
| 8. | de Jong, J. J. D.; Lucas, L. N.; Kellogg, R. M.; van Esch, J. H.; Feringa, B. L. Science 2004, 304, 278–281. doi:10.1126/science.1095353 |
| 9. | Yagai, S.; Nakajima, T.; Kishikawa, K.; Kohmoto, S.; Karatsu, T.; Kitamura, A. J. Am. Chem. Soc. 2005, 127, 11134–11139. doi:10.1021/ja052645a |
| 26. | Wu, Y.; Wu, S.; Tian, X.; Wang, X.; Wu, W.; Zou, G.; Zhang, Q. Soft Matter 2011, 7, 716–721. doi:10.1039/C0SM00330A |
| 4. | van Gorp, J. J.; Vekemans, J. A. J. M.; Meijer, E. W. J. Am. Chem. Soc. 2002, 124, 14759–14769. doi:10.1021/ja020984n |
| 5. | An, B.-K.; Lee, D.-S.; Lee, J.-S.; Park, Y.-S.; Song, H.-S.; Park, S. Y. J. Am. Chem. Soc. 2004, 126, 10232–10233. doi:10.1021/ja046215g |
| 6. | Kishimura, A.; Yamashita, T.; Aida, T. J. Am. Chem. Soc. 2005, 127, 179–183. doi:10.1021/ja0441007 |
| 7. | Shao, H.; Parquette, J. R. Chem. Commun. 2010, 46, 4285–4287. doi:10.1039/c0cc00701c |
| 27. | Wang, J.; Yang, G.; Jiang, H.; Zou, G.; Zhang, Q. Soft Matter 2013, 9, 9785–9791. doi:10.1039/c3sm51896e |
| 3. | Kartha, K. K.; Babu, S. S.; Srinivasan, S.; Ajayaghosh, A. J. Am. Chem. Soc. 2012, 134, 4834–4841. doi:10.1021/ja210728c |
| 24. | Deindörfer, P.; Davis, R.; Zentel, R. Soft Matter 2007, 3, 1308–1311. doi:10.1039/b706492f |
| 2. | Sugiyasu, K.; Fujita, N.; Shinkai, S. Angew. Chem., Int. Ed. 2004, 43, 1229–1233. doi:10.1002/anie.200352458 |
| 25. | Wang, C.; Chen, Q.; Sun, F.; Zhang, D.; Zhang, G.; Huang, Y.; Zhao, R.; Zhu, D. J. Am. Chem. Soc. 2010, 132, 3092–3096. doi:10.1021/ja910721s |
| 16. | Peng, J.; Liu, K.; Liu, J.; Zhang, Q.; Feng, X.; Fang, Y. Langmuir 2008, 24, 2992–3000. doi:10.1021/la703672u |
| 17. | Chen, P.; Lu, R.; Xue, P.; Xu, T.; Chen, G.; Zhao, Y. Langmuir 2009, 25, 8395–8399. doi:10.1021/la8035727 |
| 19. | Barrett, C. J.; Mamiya, J.-i.; Yager, K. G.; Ikeda, T. Soft Matter 2007, 3, 1249–1261. doi:10.1039/b705619b |
| 13. | Chen, Q.; Zhang, D.; Zhang, G.; Zhu, D. Langmuir 2009, 25, 11436–11441. doi:10.1021/la9016257 |
| 14. | Lee, W.-K.; Kim, K.-N.; Achard, M. F.; Jin, J.-I. J. Mater. Chem. 2006, 16, 2289–2297. doi:10.1039/b516141j |
| 15. | Xu, F.; Wang, H.; Zhao, J.; Liu, X.; Li, D.; Chen, C.; Ji, J. Macromolecules 2013, 46, 4235–4246. doi:10.1021/ma400276u |
| 20. | Murata, K.; Aoki, M.; Suzuki, T.; Harada, T.; Kawabata, H.; Komori, T.; Ohseto, F.; Ueda, K.; Shinkai, S. J. Am. Chem. Soc. 1994, 116, 6664–6676. doi:10.1021/ja00094a023 |
| 21. | Jung, J. H.; Ono, Y.; Shinkai, S. Angew. Chem., Int. Ed. 2000, 39, 1862–1865. doi:10.1002/(SICI)1521-3773(20000515)39:10<1862::AID-ANIE1862>3.0.CO;2-3 |
| 22. | Jung, J. H.; Shinkai, S.; Shimizu, T. Chem. Mater. 2003, 15, 2141–2145. doi:10.1021/cm0217912 |
| 23. | Koumura, N.; Kudo, M.; Tamaoki, N. Langmuir 2004, 20, 9897–9900. doi:10.1021/la048334f |
| 11. | Suzuki, M.; Nigawara, T.; Yumoto, M.; Kimura, M.; Shirai, H.; Hanabusa, K. Org. Biomol. Chem. 2003, 1, 4124–4131. doi:10.1039/b308371c |
| 12. | Kramb, R. C.; Zukoski, C. F. Langmuir 2008, 24, 7565–7572. doi:10.1021/la800021h |
| 10. | Mamiya, J.-i.; Kanie, K.; Hiyama, T.; Ikeda, T.; Kato, T. Chem. Commun. 2002, 1870–1871. doi:10.1039/b205072b |
| 18. | Xie, S.; Natansohn, A.; Rochon, P. Chem. Mater. 1993, 5, 403–411. doi:10.1021/cm00028a003 |
© 2015 Ren et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)