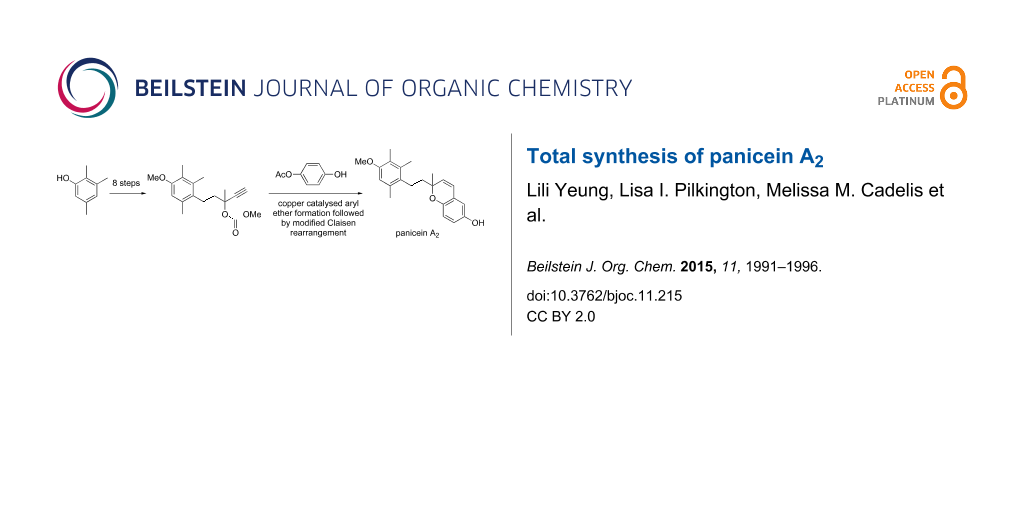
Abstract
The first total synthesis of the unusual aromatic sesquiterpene panicein A2 is reported and the structure of the natural product has been confirmed. When tested by the NCI against a range of human cancer cell lines, it was found that panicein A2 exhibits very little antiproliferative activity at 10 μM – an observation that is at odds with the earlier report that stated panicein A2 exhibits in vitro cytotoxicity against a number of tumour cell lines.

Graphical Abstract
Introduction
The panicein family is an unusual family of natural products, which generally consist of an aromatic sesquiterpene group linked to a quinone (as seen in panicein A (1), Figure 1), hydroquinone moiety (as seen in paniceins D (2), F (3) and F1 (4)) or chromenol as seen in panicein A2 (5).
![[1860-5397-11-215-1]](/bjoc/content/figures/1860-5397-11-215-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Members of the panicein family of aromatic sesquiterpenoids.
Figure 1: Members of the panicein family of aromatic sesquiterpenoids.
The first members of the panicein family were isolated by Cimino et al. in 1973 from the marine sponge Halichondria panacea [1]; members of this family have since been isolated from Reniera fulva and R. mucosa [2,3]. It has been postulated that the biosynthesis of paniceins centres around the cyclisation of a farnesyl precursor 6 [4] to an abscisane derivative 7 followed by a 1,2-methyl migration and subsequent oxidation to give panicein A (1) [1,3]. Subsequent electrocyclisation [5,6] would afford panicein A2 (5) (Figure 2). This hypothesis has been supported by the isolation of species believed to be intermediates along this proposed reaction pathway [3].
![[1860-5397-11-215-2]](/bjoc/content/figures/1860-5397-11-215-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Proposed biogenesis of panicein A2 (5).
Figure 2: Proposed biogenesis of panicein A2 (5).
Panicein A2 (5), an example of a cyclised variant of the panicein structure, is a racemic compound that was first isolated in 1994 from Reniera mucosa alongside its non-cyclised isomer 1, and ten other members of the panicein family [3]. Panicein A2 (5) and D (2) were reported to exhibit in vitro cytotoxicity against four cancer cell lines (P388 mice lymphoma, A549 human lung carcinoma, HT29 human colon carcinoma and MEL28 human melanoma) with an ED50 of 5 μg/mL [3]. Panicein F1 (4) was active against P388, A549 and MEL20 cell lines (ED50 = 2.5 μg/mL). The diversity of antiproliferative activities shown by these compounds, and particularly panicein A2 (5), presents them to be attractive synthetic targets.
We proposed that panicein A2 (5) could be synthesised from the cyclisation and subsequent deprotection of propargyl ether 8. Ether 8 could be formed through the addition of the appropriate phenol to acetylenic alcohol 9, which itself can be derived from aldehyde 10 (Figure 3).
![[1860-5397-11-215-3]](/bjoc/content/figures/1860-5397-11-215-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Retrosynthetic analysis of panicein A2 (5).
Figure 3: Retrosynthetic analysis of panicein A2 (5).
Results and Discussion
Synthesis of panicein A2
Firstly, aldehyde 10 was required; it was prepared through methylation followed by Vilsmeier–Haack reaction of 2,3,5-trimethylphenol (11), giving 10 in 51% yield over two steps (Scheme 1) [7]. Aldehyde 10 then underwent an aldol reaction according to the procedures of Samokhvalov et al. to provide ketone 12 in 66% yield [8]. The selective reduction of the olefin in α,β-unsaturated ketone 12 was then attempted using the Raney nickel catalyst under hydrogen as previously reported [8]; unfortunately no product 13 was formed. Following this, a range of conditions were employed to reduce the double bond in the presence of the α,β-unsaturated ketone, including Pd/C and H2, NaBH4 and Pd/C in the presence of acetic acid [9], and NaBH4 with CoCl2 [10]; all of these reductive conditions gave complex, inseparable mixtures of overreduction products of the ketone functionality. We therefore decided to selectively reduce the ketone to an alcohol – this would allow for the uncomplicated hydrogenolysis of the olefin. Following this, subsequent oxidation of the alcohol would give the desired ketone 13. The reduction of ketone 12 to alcohol 14 with NaBH4 was complete after a reaction time of 10 minutes, giving 14 in an excellent 94% yield. Hydrogenolysis of the alkene in 14 using Pd/C in MeOH proceeded in 2 hours, providing alcohol 15 which was then oxidised to the desired ketone 13 with Dess–Martin periodinane in 75% yield over two steps. Although this approach was ultimately two steps longer than initially planned, ketone 13 was provided in three short steps from 12, all of which were high yielding, even when conducted on a multigram scale.
![[1860-5397-11-215-i1]](/bjoc/content/inline/1860-5397-11-215-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
The addition of ethynylmagnesium bromide to ketone 13 was initially attempted using THF as the solvent, only providing alcohol 9 in 47% yield (Scheme 2). Altering the solvent system to a 1:1 mixture of THF and diethyl ether, which also improved the solubility of the Grignard reagent, increased the yield of 9 to 95%. With alcohol 9 in hand, the next step was the key coupling of alcohol 9 and phenol 16 [11] to form the required propargyl ether 8. Godfrey et al. demonstrated an efficient, one-step method to synthesise propargyl ethers bearing electron withdrawing groups under mild conditions, achieved through the in situ formation of a trifluoroacetate intermediate and subsequent addition of a phenol [12], a method that has since been utilised in a number of syntheses [13-17]. The conversion of alcohol 9 to trifluoroacetate 17 by the addition of trifluoroacetic anhydride and DBU was monitored by TLC and observed to be complete after 1 hour. The phenoxide of phenol 16, prepared by the deprotonation of 16 by DBU, was then added to intermediate 17. Unfortunately, the desired product 8 was only obtained in trace amounts.
![[1860-5397-11-215-i2]](/bjoc/content/inline/1860-5397-11-215-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of propargyl ether 8 through formation of trifluoroacetate intermediate 17.
Scheme 2: Synthesis of propargyl ether 8 through formation of trifluoroacetate intermediate 17.
Carbonate intermediates have been shown to be an effective alternative to trifluoroacetate intermediates [18], and have the added advantage to being stable to an aqueous work-up and thus can be isolated. With that knowledge, it was decided to synthesise carbonate 18 through the addition of methyl chloroformate to alcohol 9 following treatment of 9 with n-BuLi. Unfortunately, when the reaction was first attempted using 2 equivalents of methyl chloroformate, only 13% of the desired carbonate was formed, with the major product (43%) being the double-addition species 19 (Scheme 3). Lowering the amount of methyl chloroformate to 1.5 equivalents provided the desired product 18 in 51% yield. Phenol 16 was successfully added to carbonate 18 using the conditions previously attempted, giving a mixture of propargyl ether 8 and its deacetylation product 20.
![[1860-5397-11-215-i3]](/bjoc/content/inline/1860-5397-11-215-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of propargyl ether 8 through carbonate 18.
Scheme 3: Synthesis of propargyl ether 8 through carbonate 18.
The direct conversion of deacetylated product 20 to panicein A2 (5) through a modified Claisen rearrangement [19,20] was then attempted. Unfortunately, after heating 20 in toluene for 48 hours, no desired product 5 was obtained, with a complex mixture of compounds produced (Scheme 4). The reaction was then attempted with the acetyl-protected propargyl ether 8. Gratifyingly, cyclised product 21 was obtained in a 46% yield, based on returned starting material. To investigate the effect of an alternative protecting group on the cyclisation reaction, TBDMS ether 22 was synthesised from alcohol 20. Cyclisation of 22 did proceed to give the desired product 23, however in a yield of about 33%, further complicated by being inseparable from the starting material 22 (Scheme 4). Due to the poor yields of this reaction, we decided to proceed with the acetate-protected species, and thus alcohol 20 was converted to acetate 8 in 66% yield with acetic anhydride in pyridine (Scheme 3). Finally, acetate 21 was deprotected to give the natural product panicein A2 (5) in 80% yield. The NMR data for synthetic panicein A2 (5) matched literature values (see Supporting Information File 1 for data comparison tables).
![[1860-5397-11-215-i4]](/bjoc/content/inline/1860-5397-11-215-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Biological testing
When isolated, natural panicein A2 (5) exhibited activity against four cancer cell lines with ED50 = 5 μg/mL ≈ 15 μM. To further expand upon these promising results, synthetic panicein A2 (5) was tested by the NCI against 57 human cancer cell lines, through their developmental therapeutics program. Interestingly, at the tested concentration of 10 μM, panicein A2 (5) showed very little activity against most cell lines. The best performance of panicein A2 (5) was against T-47D, a breast cancer cell line, in which it showed a 43% reduction of growth when compared to a control. Two of the cell lines tested by the NCI were the same as those tested against in the original isolation study (A549 and HT29). Our results show that panicein A2 (5) only reduces growth of A549 human lung carcinoma by 14% and had no effect on the HT29 human colon cancer cell line compared to control. These results indicated panicein A2 (5) to have poor antiproliferative activity and the possibility that the originally tested natural material was contaminated with trace amounts of an even more active compound.
Conclusion
In conclusion, the first total synthesis of aromatic sesquiterpine panicein A2 (5) has been achieved. This synthesis hinges on key steps involving the addition of phenol 16 to carbonate 18 to provide propargyl ether 8 which was then cyclised through a modified Claisen rearrangement to ultimately give the desired cyclic structure of 5. The correlation of literature values for the isolated natural product and synthetic panicein A2 (5) confirm the structure of the natural product. Of particular note, when synthetic 5 was tested against a broad range of human cancer cell lines, it was found to exhibit very little activity at 10 μM – this observation is at odds with the earlier report that stated 5 exhibits in vitro cytotoxicity against a number of cell lines (ED50 = 5 μg/mL).
Supporting Information
| Supporting Information File 1: Experimental procedures, characterisation data of new compounds, NMR comparison tables of natural and synthetic 5 and NCI testing results sheet. | ||
| Format: PDF | Size: 556.4 KB | Download |
| Supporting Information File 2: 1H/13C NMR spectra of synthesised compounds. | ||
| Format: PDF | Size: 2.1 MB | Download |
References
-
Cimino, G.; De Stefano, S.; Minale, L. Tetrahedron 1973, 29, 2565–2570. doi:10.1016/0040-4020(73)80174-7
Return to citation in text: [1] [2] -
Casapullo, A.; Minale, L.; Zollo, F. J. Nat. Prod. 1993, 56, 527–533. doi:10.1021/np50094a012
Return to citation in text: [1] -
Zubía, E.; Ortega, M. J.; Carballo, J. L.; Salvá, J. Tetrahedron 1994, 50, 8153–8160. doi:10.1016/S0040-4020(01)85297-2
Return to citation in text: [1] [2] [3] [4] [5] -
Ochi, M.; Kotsuki, H.; Inoue, S.; Taniguchi, M.; Tokoroyama, T. Chem. Lett. 1979, 8, 831–832. doi:10.1246/cl.1979.831
Return to citation in text: [1] -
Chan, S. T. S.; Pullar, M. A.; Khalil, I. M.; Allouche, E.; Barker, D.; Copp, B. R. Tetrahedron Lett. 2015, 56, 1486–1488. doi:10.1016/j.tetlet.2015.02.024
Return to citation in text: [1] -
Chan, S. T. S.; Pearce, A. N.; Januario, A. H.; Page, M. J.; Kaiser, M.; McLaughlin, R. J.; Harper, J. L.; Webb, V. L.; Barker, D.; Copp, B. R. J. Org. Chem. 2011, 76, 9151–9156. doi:10.1021/jo201654h
Return to citation in text: [1] -
Andriamialisoa, Z.; Valla, A.; Cartier, D.; Labia, R. Helv. Chim. Acta 2002, 85, 2926–2929. doi:10.1002/1522-2675(200209)85:9<2926::AID-HLCA2926>3.0.CO;2-5
Return to citation in text: [1] -
Samokhvalov, G. I.; Zakharova, N. I.; Sokolova, N. N.; Filippova, T. M.; Raigorodskaya, O. I.; Murav’ev, V. V.; Tuguzova, A. M.; Khristoforov, V. L. Pharm. Chem. J. 1994, 28, 343–348. doi:10.1007/BF02218431
Return to citation in text: [1] [2] -
Russo, A. T.; Amezcua, K. L.; Huynh, V. A.; Rousslang, Z. M.; Cordes, D. B. Tetrahedron Lett. 2011, 52, 6823–6826. doi:10.1016/j.tetlet.2011.10.056
Return to citation in text: [1] -
Aramini, A.; Brinchi, L.; Germani, R.; Savelli, G. Eur. J. Org. Chem. 2000, 1793–1797. doi:10.1002/(SICI)1099-0690(200005)2000:9<1793::AID-EJOC1793>3.0.CO;2-A
Return to citation in text: [1] -
Belyanin, M. L.; Stepanova, E. V.; Ogorodnikov, V. D. Carbohydr. Res. 2012, 363, 66–72. doi:10.1016/j.carres.2012.10.006
Return to citation in text: [1] -
Godfrey, J. D., Jr.; Mueller, R. H.; Sedergran, T. C.; Soundararajan, N.; Colandrea, V. J. Tetrahedron Lett. 1994, 35, 6405–6408. doi:10.1016/S0040-4039(00)78231-1
Return to citation in text: [1] -
Gruner, K. K.; Knölker, H.-J. Org. Biomol. Chem. 2008, 6, 3902–3904. doi:10.1039/b813670j
Return to citation in text: [1] -
Allan, A. C.; Billinton, A.; Brown, S. H.; Chowdhury, A.; Eatherton, A. J.; Fieldhouse, C.; Giblin, G. M. P.; Goldsmith, P.; Hall, A.; Hurst, D. N.; Naylor, A.; Rawlings, D. A.; Sime, M.; Scoccitti, T.; Theobald, P. J. Bioorg. Med. Chem. Lett. 2011, 21, 4343–4348. doi:10.1016/j.bmcl.2011.05.047
Return to citation in text: [1] -
Hesse, R.; Gruner, K. K.; Kataeva, O.; Schmidt, A. W.; Knölker, H.-J. Chem. – Eur. J. 2013, 19, 14098–14111. doi:10.1002/chem.201301792
Return to citation in text: [1] -
Hou, S.; Liu, Y.; Kong, Y.; Brown, M. L. Org. Lett. 2015, 17, 2298–2301. doi:10.1021/acs.orglett.5b00422
Return to citation in text: [1] -
Nicolaou, K. C.; Xu, H.; Wartmann, M. Angew. Chem., Int. Ed. 2005, 44, 756–761. doi:10.1002/anie.200462211
Return to citation in text: [1] -
Davis, C. J.; Hurst, T. E.; Jacob, A. M.; Moody, C. J. J. Org. Chem. 2005, 70, 4414–4422. doi:10.1021/jo050336x
Return to citation in text: [1] -
Brown, P. E.; Lewis, R. A.; Waring, M. A. J. Chem. Soc., Perkin Trans. 1 1990, 2979–2988. doi:10.1039/p19900002979
Return to citation in text: [1] -
Zsindely, J.; Schmid, H. Helv. Chim. Acta 1968, 51, 1510–1514. doi:10.1002/hlca.19680510703
Return to citation in text: [1]
| 18. | Davis, C. J.; Hurst, T. E.; Jacob, A. M.; Moody, C. J. J. Org. Chem. 2005, 70, 4414–4422. doi:10.1021/jo050336x |
| 19. | Brown, P. E.; Lewis, R. A.; Waring, M. A. J. Chem. Soc., Perkin Trans. 1 1990, 2979–2988. doi:10.1039/p19900002979 |
| 20. | Zsindely, J.; Schmid, H. Helv. Chim. Acta 1968, 51, 1510–1514. doi:10.1002/hlca.19680510703 |
| 1. | Cimino, G.; De Stefano, S.; Minale, L. Tetrahedron 1973, 29, 2565–2570. doi:10.1016/0040-4020(73)80174-7 |
| 5. | Chan, S. T. S.; Pullar, M. A.; Khalil, I. M.; Allouche, E.; Barker, D.; Copp, B. R. Tetrahedron Lett. 2015, 56, 1486–1488. doi:10.1016/j.tetlet.2015.02.024 |
| 6. | Chan, S. T. S.; Pearce, A. N.; Januario, A. H.; Page, M. J.; Kaiser, M.; McLaughlin, R. J.; Harper, J. L.; Webb, V. L.; Barker, D.; Copp, B. R. J. Org. Chem. 2011, 76, 9151–9156. doi:10.1021/jo201654h |
| 12. | Godfrey, J. D., Jr.; Mueller, R. H.; Sedergran, T. C.; Soundararajan, N.; Colandrea, V. J. Tetrahedron Lett. 1994, 35, 6405–6408. doi:10.1016/S0040-4039(00)78231-1 |
| 1. | Cimino, G.; De Stefano, S.; Minale, L. Tetrahedron 1973, 29, 2565–2570. doi:10.1016/0040-4020(73)80174-7 |
| 3. | Zubía, E.; Ortega, M. J.; Carballo, J. L.; Salvá, J. Tetrahedron 1994, 50, 8153–8160. doi:10.1016/S0040-4020(01)85297-2 |
| 13. | Gruner, K. K.; Knölker, H.-J. Org. Biomol. Chem. 2008, 6, 3902–3904. doi:10.1039/b813670j |
| 14. | Allan, A. C.; Billinton, A.; Brown, S. H.; Chowdhury, A.; Eatherton, A. J.; Fieldhouse, C.; Giblin, G. M. P.; Goldsmith, P.; Hall, A.; Hurst, D. N.; Naylor, A.; Rawlings, D. A.; Sime, M.; Scoccitti, T.; Theobald, P. J. Bioorg. Med. Chem. Lett. 2011, 21, 4343–4348. doi:10.1016/j.bmcl.2011.05.047 |
| 15. | Hesse, R.; Gruner, K. K.; Kataeva, O.; Schmidt, A. W.; Knölker, H.-J. Chem. – Eur. J. 2013, 19, 14098–14111. doi:10.1002/chem.201301792 |
| 16. | Hou, S.; Liu, Y.; Kong, Y.; Brown, M. L. Org. Lett. 2015, 17, 2298–2301. doi:10.1021/acs.orglett.5b00422 |
| 17. | Nicolaou, K. C.; Xu, H.; Wartmann, M. Angew. Chem., Int. Ed. 2005, 44, 756–761. doi:10.1002/anie.200462211 |
| 4. | Ochi, M.; Kotsuki, H.; Inoue, S.; Taniguchi, M.; Tokoroyama, T. Chem. Lett. 1979, 8, 831–832. doi:10.1246/cl.1979.831 |
| 10. | Aramini, A.; Brinchi, L.; Germani, R.; Savelli, G. Eur. J. Org. Chem. 2000, 1793–1797. doi:10.1002/(SICI)1099-0690(200005)2000:9<1793::AID-EJOC1793>3.0.CO;2-A |
| 2. | Casapullo, A.; Minale, L.; Zollo, F. J. Nat. Prod. 1993, 56, 527–533. doi:10.1021/np50094a012 |
| 3. | Zubía, E.; Ortega, M. J.; Carballo, J. L.; Salvá, J. Tetrahedron 1994, 50, 8153–8160. doi:10.1016/S0040-4020(01)85297-2 |
| 11. | Belyanin, M. L.; Stepanova, E. V.; Ogorodnikov, V. D. Carbohydr. Res. 2012, 363, 66–72. doi:10.1016/j.carres.2012.10.006 |
| 7. | Andriamialisoa, Z.; Valla, A.; Cartier, D.; Labia, R. Helv. Chim. Acta 2002, 85, 2926–2929. doi:10.1002/1522-2675(200209)85:9<2926::AID-HLCA2926>3.0.CO;2-5 |
| 8. | Samokhvalov, G. I.; Zakharova, N. I.; Sokolova, N. N.; Filippova, T. M.; Raigorodskaya, O. I.; Murav’ev, V. V.; Tuguzova, A. M.; Khristoforov, V. L. Pharm. Chem. J. 1994, 28, 343–348. doi:10.1007/BF02218431 |
| 3. | Zubía, E.; Ortega, M. J.; Carballo, J. L.; Salvá, J. Tetrahedron 1994, 50, 8153–8160. doi:10.1016/S0040-4020(01)85297-2 |
| 9. | Russo, A. T.; Amezcua, K. L.; Huynh, V. A.; Rousslang, Z. M.; Cordes, D. B. Tetrahedron Lett. 2011, 52, 6823–6826. doi:10.1016/j.tetlet.2011.10.056 |
| 3. | Zubía, E.; Ortega, M. J.; Carballo, J. L.; Salvá, J. Tetrahedron 1994, 50, 8153–8160. doi:10.1016/S0040-4020(01)85297-2 |
| 3. | Zubía, E.; Ortega, M. J.; Carballo, J. L.; Salvá, J. Tetrahedron 1994, 50, 8153–8160. doi:10.1016/S0040-4020(01)85297-2 |
| 8. | Samokhvalov, G. I.; Zakharova, N. I.; Sokolova, N. N.; Filippova, T. M.; Raigorodskaya, O. I.; Murav’ev, V. V.; Tuguzova, A. M.; Khristoforov, V. L. Pharm. Chem. J. 1994, 28, 343–348. doi:10.1007/BF02218431 |
© 2015 Yeung et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)