
Abstract
Biocatalysts, capable of efficiently transforming CO2 into other more reduced forms of carbon, offer sustainable alternatives to current oxidative technologies that rely on diminishing natural fossil-fuel deposits. Enzymes that catalyse CO2 fixation steps in carbon assimilation pathways are promising catalysts for the sustainable transformation of this safe and renewable feedstock into central metabolites. These may be further converted into a wide range of fuels and commodity chemicals, through the multitude of known enzymatic reactions. The required reducing equivalents for the net carbon reductions may be drawn from solar energy, electricity or chemical oxidation, and delivered in vitro or through cellular mechanisms, while enzyme catalysis lowers the activation barriers of the CO2 transformations to make them more energy efficient. The development of technologies that treat CO2-transforming enzymes and other cellular components as modules that may be assembled into synthetic reaction circuits will facilitate the use of CO2 as a renewable chemical feedstock, greatly enabling a sustainable carbon bio-economy.
Graphical Abstract
Introduction
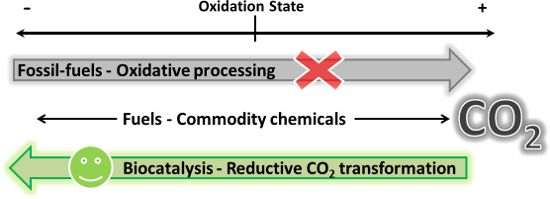
Depletion of fossil-fuel feedstocks and pollution resulting from their unsustainable processing and use constitute challenging global issues [1,2]. Catalysis has an important role to play in addressing these challenges through the generation of fuels and commodity chemicals from renewable sources in a sustainable manner [3]. In this context, CO2 has become a compound of key interest as it is one of the main contributors to fossil-fuel pollution [4,5]. As a result, decreasing CO2 emissions and CO2 sequestration technologies are subjects of intense research. In addition, CO2 may hold an even more important role in a sustainable future, as a readily available and renewable material that may be utilised as an alternative feedstock for the production of many of the chemicals we have come to rely on [6-11]. Chemical processes that employ CO2 as a synthon for the production of commodity chemicals may form the basis of a sustainable carbon economy.
The benefits notwhithstanding, chemical conversion of CO2 into other forms of carbon remains challenging because the transformations typically have high activation barriers and are therefore very energy intensive [12]. Catalysis will therefore play a critical role in the development of viable solutions for the transformation of CO2. Biocatalysts are very likely to contribute towards this end due to their ability to efficiently catalyse processes under mild conditions with limited byproduct formation [13,14]. These catalysts have been developed by nature to utilise diverse substrates including simple compounds such as CO2. Indeed, life itself depends on the ability of autotrophic organisms to convert CO2 into other materials, and these are therefore a valuable source of the required biocatalysts.
The development of methodologies for expression, characterisation, engineering and optimisation of CO2-transforming enzymes will form the basis of any future biotechnology that aims to use CO2 as a feedstock for the generation of other materials. Here we provide an overview of the biocatalysts that have already been applied to relevant technologies and are set to play an important role in future bioprocesses for the transformation of CO2 into fuels and commodity chemicals. As well as reviewing applications of these biocatalysts, we highlight the chemical, biochemical and biological contexts in which they operate, the understanding of which is critical for effective application. As commodity chemicals contain carbon at lower oxidation states than CO2, only enzymes that involve CO2 reduction will be covered here and not carbonic anhydrase for the conversion to HCO3−, which is extensively covered in other reviews on carbon-capture technology [15].
Review
Biotechnological transformation of CO2
Synthesis of commercial materials through the biological transformation of CO2 is the basis of all agriculture. Through the cultivation of crops, CO2 is converted into more useful forms of carbon, such as starch and lignocellulosic materials. In turn, these materials have been employed as carbon sources for fermentative processes, and more recently in first and second generation biofuel production processes. In this way, the carbon fixed by plants (biomass) is further transformed into a wide array of products through microbial processing [16]. Genetically engineered plants and algae have been employed to divert carbon flux in planta towards other metabolic products of interest, as an alternative to microbial processes [17,18]. Yet another alternative approach is to directly fix the CO2 with microorganisms, circumventing the intermediacy of crop derived biomass [19,20]. This can be done with autotrophic microbes, though these are generally poorly understood, and the genetic tools required to divert carbon flux towards useful products are still under-developed with these species. Alternatively, as discussed in detail below, well understood microbes for which genetic modification methodologies are widely available, such as E. coli, have been used as hosts for heterologous CO2 fixation reactions [21], that may then be coupled to an extensive array of metabolic pathways for the delivery of target compounds.
Biological strategies to increase CO2 reactivity
Energetic demand of CO2 transformation
Most of the carbon associated with fossil-fuel based technologies will eventually be converted to CO2 through combustion or oxidative degradation [12,19,22], because this is the most oxidised and stable state of carbon (+4). Converting CO2 into other more reduced forms of carbon, as found in organic commodity chemicals, requires large energy inputs. As a result, there are only a limited number of examples of industrial chemical processes which use CO2 as feedstock, and those that do, such as the Bosch–Meiser process [7], are very energy intensive.
Associated with the dependence of autotrophic organisms on CO2 as a carbon source, biological systems have developed various strategies to avoid energy constraints, and as a result there are several metabolic pathways for reductive transformation of CO2 (Figure 1) [23,24]. Generally, such processes are driven by coupling the CO2 transformations with oxidations that generate reducing equivalents, sometimes in conjunction with the hydrolysis of phosphoanhydride bonds [25-27]. For reductases or dehydrogenases operating in reverse, the electrons required to reduce CO2 are provided through oxidation of reduced forms of redox cofactors, either directly or through electron driving protein mediation (NAD(P)H or equivalents). For a number of carboxylases, phosphoanhydride bonds in ATP are hydrolysed to drive CO2 transformation through various molecular mechanisms. For example, biotin carboxylase catalysed reactions proceed through electrophilic activation of CO2 to carboxyphosphate to facilitate an attack by a nucleophile [28].
![[1860-5397-11-259-1]](/bjoc/content/figures/1860-5397-11-259-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Biocatalytic routes for conversion of CO2 into compounds with carbon in the reduced oxidation states indicated at the top. FDH: formate dehydrogenase, FaldDH: formaldehyde dehydrogenase, ADH: alcohol dehydrogenase, CODH: carbon monoxide dehydrogenase, RuBisCO: ribulose-1,5-bisphosphate carboxylase oxygenase, CA: carbonic anhydrase, R: H, CH3.
Figure 1: Biocatalytic routes for conversion of CO2 into compounds with carbon in the reduced oxidation state...
In all known natural CO2 fixation pathways, ATP and NADH or their equivalents are consumed in order to generate the intermediates that may feed into central metabolism [23]. This consumption is used as a measure of pathway efficiency for CO2-fixation, and pathways are considered most efficient when it is minimised [25,27]. By balancing thermodynamic feasibililty and a low requirement in NADH and ATP or equivalents, Milo and coworkers [25] were able to computationally predict the most efficient synthetic CO2 fixation pathways, using all known natural enzymes.
Aqueous solubility and hydration of CO2
A particular limitation for aqueous CO2 transformations stems from the low concentration of dissolved CO2 at saturation. At physiological pH, CO2 is hydrated and exists predominantly as the bicarbonate anion (HCO3−) [29]. Within cells, CO2 and HCO3− rapidly interconvert through catalysis by carbonic anhydrase, the archetypal super-enzyme for which catalytic rates reach the limits of diffusion [30,31]. CO2 consumed by enzymes is therefore efficiently replenished through rapid HCO3− dehydration (Figure 2). Living organisms have developed various mechanisms to increase the effective concentration of CO2, ranging from the use of carboxylated cofactors [28,32] to complex extended metabolic pathways in C4 and CAM plants [17,33,34] and substrate channelling. In addition, a number of enzymes accept HCO3− as a substrate, which is converted to CO2 close to the active site before the reductive step [26,28].
![[1860-5397-11-259-2]](/bjoc/content/figures/1860-5397-11-259-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Carbonic anhydrase-catalysed rapid interconversion of CO2 and HCO3− in living systems.
Figure 2: Carbonic anhydrase-catalysed rapid interconversion of CO2 and HCO3− in living systems.
In photoautotrophic bacteria (cyanobacteria) micro-compartmentalisation of the CO2-fixing reactions increases reaction rates [35,36]. The bacterial micro-compartments, called carboxysomes, are highly elaborate proteinic structures that usually also incorporate carbonic anhydrase [36,37]. Carboxysomes have been the subject of studies on increasing the efficiency of C3 carbon fixation in plants [38-40]. The recent production of a transgenic tobacco plant, expressing bacterial carboxysome proteins and able to photosynthesise at an increased rate, was a significant breakthrough in this field [39]. Carboxysomal proteins have also been expressed in E. coli yielding a highly organised structure [41]. Use of carboxysomes for micro-compartmentalisation of CO2 biotransformation may therefore become a viable strategy in a range of synthetic biology applications, because not only CO2-transforming enzymes, but also the cohort of supporting cellular equipment and mechanisms that living systems employ, may be used to drive these processes.
Sources of CO2 transforming enzymes
Emergence of CO2 transforming enzymes
Autotrophic enzymes have evolved to promote and control CO2 fixation and are an obvious starting point for the biotechnological transformation of CO2 [42].
To understand the properties and distribution of these CO2-assimilating enzymes, it is important to consider the geochemical context in which they have evolved as there appears to be a strong link with atmospheric concentrations of CO2. The environment from which life emerged is thought to have been anoxic with high concentrations of CO2 [43]. In this environment, the first CO2-fixing enzymes evolved to take advantage of the most readily available carbon source. Through the action of these enzymes and geological processes for CO2 sequestration, CO2 concentrations steadily decreased, leading to average atmospheric concentrations of 200 ppm over the last 400,000 years [44]. During this time, oxygen levels steadily increased through the action of photosynthetic organisms that oxidise water to produce molecular oxygen [43]. Consequently many CO2-assimiliating enzymes evolved to be strictly anaerobic, and are limited to specific environments, while others tolerate O2 [45]. As a result, the environmental [CO2]/[O2] ratio is an important effector of enzymatic properties.
RuBisCO and the Calvin cycle
For many years, the Calvin cycle for C3 carbon fixation was thought to be the only important biological process for CO2 assimilation, as a result of its prevalence in our immediate environment. It is found in photosynthetic organisms, predominantly in plants on land and algae in water, and photosynthetic prokaryotes (cyanobacteria). This carbon fixation pathway forms part of photosynthesis and the required reducing equivalents are generated through electron gradients initiated by photons and generated through the splitting of water [46]. However, a number of autotrophic bacteria fix carbon through the Calvin cycle with electrons generated through oxidation of inorganic chemicals (chemoautotrophs) [47]. As detailed in Scheme 1, the carbon fixation step entails the carboxylation of ribulose-1,5-bisphosphate (1), generating two equivalents of 3-phosphoglycerate (2) and is catalysed by ribulose-1,5-bisphosphate carboxylase oxygenase (RuBisCO). The glycerate 2 is subsequently phosphorylated with ATP for the production of 1,3-bisphosphoglycerate (3), which is in turn reduced with NADPH to 3-phosphoglyceraldehyde (4). For every six equivalents of the aldehyde 4, one is diverted to carbohydrate biosynthesis, while the other five are used to produce the RuBisCO substrate 1.
![[1860-5397-11-259-i1]](/bjoc/content/inline/1860-5397-11-259-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The Calvin cycle for fixation of CO2 with RuBisCO.
Scheme 1: The Calvin cycle for fixation of CO2 with RuBisCO.
A property of RuBisCO with great implications is that it may also accept O2 instead of CO2 as an electrophile in the addition step, thus catalysing a counter-productive reaction, which reduces the photosynthetic output of plants using the Calvin cycle by 25% [48]. In the O2-rich environments in which it operates, this property makes RuBisCO a particularly inefficient biocatalyst and a major bottleneck to C3 carbon fixation. Through evolution, RuBisCO has adapted to rising oxygen concentrations by developing higher specifities for CO2 at the expense of catalytic turnover, making it a particularly slow enzyme [49]. As a result, evolutionary bias from limited nutrient availability has driven some plants to develop more elaborate carbon assimilation mechanisms (C4 and CAM plants) [48]. These involve an initial temporary carbon fixation step with phosphoenolpyruvate carboxylase (PEPC), followed by transport and release as CO2 in the vicinity of RuBisCO within cellular compartments with low O2 concentrations [48]. RuBisCO variants from these plants display higher turnover rates, and lower specificities for CO2 over O2. This apparent trade-off between CO2 specificity and catalytic activity greatly influences efforts towards RuBisCO biotechnological applications.
The Calvin cycle is not the only carbon fixation pathway, and at least five alternative pathways have been elucidated in recent years [24]. It is now thought that some of these alternative pathways contribute significantly to the global carbon cycle, particularly with regard to the oceanic section [50,51]. This is due to the extensive global distribution of many oceanic chemolithoautotrophic organisms, and the estimated carbon fixation in deep-sea hydrothermal vents, the meso- and bathy-pelagic ocean, and in oxygen-deficiency zones [50].
Reductive tricarboxylic acid cycle
The tricarboxylic acid (TCA) cycle is used by all aerobic organisms to generate NADH through the oxidation of small organic metabolites. For pyruvate (11), isocitrate (7) and 2-oxoglutarate (6), oxidation occurs together with a decarboxylation. In some autotrophs this pathway is known to operate in the reverse (reductive) direction resulting in CO2 fixation through carboxylation [52]. Autotrophic fixation through the reductive TCA cycle was first described by Arnon and Buchanan [53], and hence is also referred to as the Arnon–Buchanan cycle. It is considered the most efficient CO2 fixation pathway as it requires the lowest amount of reducing equivalents per carbon fixed [23,26]. This is mainly due to the fact that CO2 fixation occurs through three efficient reductive carboxylations, of which two are coupled to oxidation of the low-potential electron donor ferredoxin [26], with a requirement for strict anaerobicity, thus limiting the distribution of the reductive TCA cycle.
As detailed in Scheme 2, the reductive TCA cycle contains three CO2 fixation steps [24]. Succinyl-CoA (5) is carboxylated by ferredoxin-dependent 2-oxoglutarate synthase to produce 2-oxoglutarate (6), which is subsequently transformed to isocitrate (7) through a second CO2 fixation catalysed by isocitrate dehydrogenase. Isocitrate (7) is concominantly isomerised to citrate (8) and lysed to oxaloacetate (9) which remains in the cycle and regenerates succinyl-CoA through three catalytic steps, and acetyl-CoA (10) which enters central metabolism through a third CO2-fixation step, carried out by ferredoxin-dependent pyruvate synthase to produce pyruvate (11). The carboxylating enzymes are mechanistically complex and highly adapted to the cellular conditions in which they operate, and as a result there has been little development of their use in synthetic processes.
![[1860-5397-11-259-i2]](/bjoc/content/inline/1860-5397-11-259-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The reductive TCA cycle with CO2 fixation enzymes designated.
Scheme 2: The reductive TCA cycle with CO2 fixation enzymes designated.
Wood–Ljungdahl pathway
The Wood–Ljungdahl pathway, or reductive acetyl-CoA pathway, is used by acetogenic bacteria to reduce CO2 to either formate with formate dehydrogenase (FDH) or CO with CO dehydrogenase (CODH) [54,55]. As presented in Scheme 3, these initial steps of two separate branches of the pathway meet to produce a unit of acetyl-CoA (10) which is then incorporated into central metabolic processes [56-59]. FDHs are widely distributed enzymes, discussed in more detail below. The formate (12) produced through FDH activity is incorporated onto a tetrahydrofolate (14) and reduced to an activated methyl group (13), which is then utilised as a substrate by acetyl-CoA synthase together with the CO produced by CODH. The acetyl-CoA synthase forms a complex with CODH, to channel CO through a molecular tunnel [60]. This enzyme has been the focus of much interest due to its unusual reactivity, however, it remains poorly understood [61].
![[1860-5397-11-259-i3]](/bjoc/content/inline/1860-5397-11-259-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The Wood–Ljungdahl pathway for generation of acetyl-CoA through reduction of CO2 to formate and CO. FDH: formate dehydrogenase, CODH: CO dehydrogenase, ACS: acetyl-CoA synthase, FH4: tetrahydrofolate.
Scheme 3: The Wood–Ljungdahl pathway for generation of acetyl-CoA through reduction of CO2 to formate and CO....
Formate dehydrogenases are an extremely heterogeneous enzyme family, most commonly found to physiologically catalyse formate oxidation and release of CO2. Autotrophic acetogen FDHs are usually bound to metallo-pterin cofactors, with either a Mo or W centre [55,62,63], coordinated to a SeCys or Cys ligand. These features are not limited to acetogenic FDHs, and Mo and W FDHs are broadly distributed throughout the bacterial kingdom [63-68]. In addition, various types of Fe–S clusters are observed in FDHs, through which electrons are transported to other protein domains or to other oxidoreductases altogether [63,64,68]. Due to the presence of oxidisable cofactors, metallo-FDHs are most commonly found in anaerobic organisms. Another large family of FDHs do not contain metal cofactors, and catalyse a direct hydride transfer from formate to a nicotinamide cofactor [69]. They are commonly found in aerobic species, are generally robust and amenable to recombinant expression, but have high catalytic preferences for formate oxidation to CO2.
Acyl-CoA pathways
A number of recently elucidated cyclic pathways that exist primarily in archaea initiate through the fixation of CO2 onto acetyl-CoA (10) [51,70], and end with the generation of two equivalents of the starting substrate 10 (Scheme 4). One equivalent of the CoA thioester 10 is fed to central metabolism while the other is used in a subsequent cycle. As seen in Scheme 4, acetyl-CoA (10) is carboxylated by a bifunctional acetyl-CoA/propionyl-CoA carboxylase to malonyl-CoA (15) with HCO3− and hydrolysis of ATP. The malonate 15 is reduced to 3-hydroxypropionate (16), in two steps catalysed by NAD(P)H dependent dehydrogenases. Later in the pathway, propionyl-CoA (17) is the substrate for a second carboxylation with HCO3− to methylmalonyl-CoA (18), performed by the same ATP-dependent bifunctional carboxylase that carries out the first step [71,72]. Succinyl-CoA (5) is formed through isomerisation and recycled into two equivalents of acetyl-CoA (10). This route is encountered in two separate pathways, namely the 3-hydroxypropionate/4-hydroxybutyrate cycle and the 3-hydroxypropionate bicycle. An alternative CO2 fixation route is found in the dicarboxylate/4-hydroxybutyrate cycle [73]. Here acetyl-CoA (10) is initially reductively carboxylated to pyruvate (11), as in the reductive TCA cycle. The pyruvate 11 is phosphorylated with ATP to generate phosphoenolpyruvate (19), followed by a second carboxylation with HCO3− to oxaloacetate by PEPC.
![[1860-5397-11-259-i4]](/bjoc/content/inline/1860-5397-11-259-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: The acyl-CoA carboxylase pathways for autotrophic CO2 fixation. ACC: acetyl-CoA/propionyl-CoA carboxylase, PEPC: phosphoenolpyruvate carboxylase.
Scheme 4: The acyl-CoA carboxylase pathways for autotrophic CO2 fixation. ACC: acetyl-CoA/propionyl-CoA carbo...
Non-autotrophic CO2 fixation
A large number of enzymes use CO2 (or HCO3−) as a substrate without taking part in autotrophic pathways [26]. These are predominantly found in assimilatory pathways where small organic molecules are used as carbon sources, and anaplerosis through which intermediate metabolites in central pathways (e.g., the TCA cycle) are replenished.
These enzymes also represent interesting targets for use in CO2 transforming processes, particularly when involved in the production of TCA cycle dicarboxylates that constitute target platform chemicals, as in the cases of pyruvate carboxylase and PEPC. Enzymes that catalyse CO2 fixation in autotrophic pathways are also found in non-autotrophic pathways operating either in the same direction (PEPC), or in the reverse direction for CO2 production (FDH). However, these enzymes are still suitable targets and have been used in vitro for CO2 fixation. Finally degradative pathways contain enzymes capable of working in both carboxylating or decarboxylating direction depending on reaction conditions [12]. These, have also attracted some attention as a source for relevant biocatalysts.
Biotechnological CO2 transformation
CO2-transforming enzymes sourced from natural metabolic pathways have been utilised in biotechnological applications for the conversion of CO2, through either direct reduction of CO2 or carboxylation of another substrate.
CO2 transformation with RuBisCO
As the most well studied and best characterised autotrophic CO2-fixation enyzme, RuBisCO has received much attention for application in biotechnology for CO2-fixation, particularly using engineered photosynthetic hosts, such as plants and algae. The inefficiency of RuBisCO and promiscuity towards oxygen have directed efforts in protein engineering towards the generation of optimised mutants that overcome these limitations [74,75]. Though these studies have resulted in the recombinant expression of RuBisCO in useful hosts such as E. coli [75], development of improved selection systems for directed evolution [74], and further elucidation of RuBisCO properties [76], little progress has been made toward expression of an enzyme which is more efficient and less promiscuous. A possible explanation for this was provided by Tlusty, Milo and coworkers [77]. By processing kinetic data from various RuBisCO enzymes, it was found that variations in enzyme specificity and velocity are mutually constrained. Within this limited space, it appears that the various wild-type enzymes have been optimised through evolution to operate within their respective environments. Point-mutations in the protein itself are therefore unable to lead to great improvements in enzyme efficiency. A more promising strategy may be to employ outlying natural variants of RuBisCO that display the best properties, such as those from red algae, in combination with other components of the Calvin cycle carbon assimilation mechanism [39]. Long et al. [78] estimated that incorporation of wild-type enzymes, with higher CO2 specificity or higher catalytic activity, into C3 plants could potentially raise crop yields by more than 25%. Furthermore, incorporation of cyanobacterial carbon concentration mechanisms such as carboxysomes, combined with RuBisCO variants adapted to higher CO2 concentrations, could result in a 36% to 60% crop yield increase [79].
The main difficulties of heterologous expression of RuBisCO for CO2 fixation relate to the poorly understood post-translational steps for production of the fully active enzyme that require the action of specific chaperones as well as a separate enzymatic species, RuBisCO activase. In some cases these have to also be incorporated into the host organism in order to obtain an active enzyme.
Recently there have been two important breakthroughs on carbon assimilation in plants through RuBisCO, relating to alternative components of the RuBisCO catalytic system. Whitney et al. [80] increased the expression levels of a heterologous RuBisCO in tobacco plants, through co-expression of a RuBisCO chaperone that facilitates the assembly of the active multimeric enzyme. This resulted in two-fold increases in CO2 assimilation rate and plant growth. Hanson, Parry and co-workers [39] were able to prepare tobacco plants that expressed cyanobacterial RuBisCO together with a protein that forms part of the carboxysomal structure, which led to the generation of macromolecular complexes that are observed early in the carboxysomal biogenesis in cyanobacteria. In addition, the engineered plants were photosynthetically active, and the RuBisCO complex showed higher specific activities than the enzyme in the control tobacco line.
Algae that utilise efficient variants of RuBisCO for fixation of CO2 have been targeted as a biomass source for a third generation of biofuels, due to their lack of requirement for arable land [81]. In addition, microalgae have been employed for the production of chemicals as a metabolic end-product of the fixed carbon, with particular emphasis on oils for use as biodiesel feedstock [82]. Cyanobacteria, mainly Synechocystis spp., have proven easier to engineer than their algal and plant counterparts and have also been applied to generate higher titres of oils and alcohols [18].
Despite difficulties related to heterologous expression, recently there have also been reports of succesful use of RuBisCO in non-photosynthetic host organisms (Figure 3). In E. coli it was possible to incorporate a CO2-fixing bypass in central metabolism through expression of phosphoribulosekinase to produce ribulose-1,5-bisphosphate (1) (Scheme 1), and a cyanobacterial RuBisCO along with a RuBisCO-folding chaperone from the same source [21]. It was found that the main limiting factor to carbon fixation was the availability of CO2 in E. coli, and the yield could be increased through incorporation of a cyanobacterial carbonic anhydrase. In the yeast Saccharomyces cerevisiae, spinach phosphoribulosekinase was able to provide the bisphosphate 1 to a prokaryotic RuBisCO from Hydrogenovibrio marinus, which folded with the aid of E. coli protein chaperones (GroEL/GroES) [83]. This resulted in catalysis of CO2 fixation and increase of carbon flux towards the ethanol product and away from glycerol, a major fermentation byproduct (Figure 3).
![[1860-5397-11-259-3]](/bjoc/content/figures/1860-5397-11-259-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: RuBisCO CO2-fixing bypass installed in E. coli and S. cerevisiae to increase carbon flux toward products of interest. PRK: phosphoribulosekinase.
Figure 3: RuBisCO CO2-fixing bypass installed in E. coli and S. cerevisiae to increase carbon flux toward pro...
Synthesis of dicarboxylates through pyruvate carboxylation
Enzymatic carboxylation of a pyruvate backbone offers an avenue to dicarboxylates, which are important biotechnological targets, through the use of CO2 as feedstock. As seen, this may be carried out by pyruvate carboxylase or PEPC which acts on phosphoenolpyruvate (19). Purified PEPC has been used in an integrated system with carbonic anhydrase for in vitro carbon capture and transformation to oxaloacetate (9) (Scheme 5) [84]. This system has been further optimised with engineered variants of PEPC leading to increased rates and yields of CO2 transformation [85].
![[1860-5397-11-259-i5]](/bjoc/content/inline/1860-5397-11-259-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Integrated biocatalytic system for carboxylation of phosphoenolpyruvate (19), using PEPC and carbonic anhydrase.
Scheme 5: Integrated biocatalytic system for carboxylation of phosphoenolpyruvate (19), using PEPC and carbon...
In E. coli fermentative processes, as presented in Scheme 6, PEPC is used to produce oxaloacetate (9) directly from phosphoenolpyruvate (19) from glycolysis, through carboxylation with HCO3−. This may then be further transformed, by reversal of the activity of native oxidative TCA cycle enzymes, to produce malate (20), fumarate (21) and succinate (22), all of which have been listed in the top twelve target platform chemicals from biomass, by the US Department of Energy [86]. In this way, overexpression of Sorghum vulgare PEPC in E. coli resulted in higher fermentative yields of succinate (22) [87]. Recombinant co-expression of cyanobacterial carbonic anhydrase in E. coli BL21(DE3) increased available HCO3− resulting in a higher than five-fold increase in the observed activity of endogenous PEPC [88]. Similarly, strains with overexpressed PEPC have been engineered for the production of high yields of fumaric acid (21) [89].
![[1860-5397-11-259-i6]](/bjoc/content/inline/1860-5397-11-259-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: PEPC and pyruvate carboxylase catalysed carboxylation of pyruvate backbone for the generation of oxaloacetate (9) and other dicarboxylates.
Scheme 6: PEPC and pyruvate carboxylase catalysed carboxylation of pyruvate backbone for the generation of ox...
As some phosphoenolpyruvate (19) is lost to pyruvate (11), pyruvate carboxylase, not present naturally in E. coli, was used to increase the carbon flux to the desired products, by providing a secondary oxaloacetate (9) production route through CO2-fixation. In this way, E. coli strains overexpressing pyruvate carboxylase have been applied to CO2 fixation with the production of equimolar succinate (22) [90]. In addition the succinate yields were found to strongly depend on CO2 availability and increased by up to four-fold under increased CO2 partial pressures. Such engineered E. coli strains were also able to utilise CO2 generated during ethanol fermentation with Saccharomyces cerevisiae as the substrate for succinate production, through an integrated bioprocess [91]. Through gene deletion, other undesirable pyruvate consumption reactions such as lysis to acetyl-CoA (10) with liberation of CO2 could be blocked, allowing improved yields of dicarboxylates [92]. The carbon from CO2 was also directed to other products through the use of other types of host organisms. Overexpression of E. coli PEPC in Propionibacteria resulted in increased rates of propionic acid production as well as increased rates of carbon fixation under higher CO2 partial pressures [93,94].
Acyl-CoA carboxylases
Though acetyl-CoA carboxylases are widely distributed in living organisms, the existence of bifunctional variants with a role in autotrophy has attracted further interest for their biotechnological applications in CO2 transformation technologies. The autotrophic enzymes from Metallosphaera sedula and Acidianus berleyi have been purified and found to be catalytically active in vitro for the production of malonyl-CoA through acetyl-CoA carboxylation [71,95]. As seen in Scheme 4, two subsequent steps in the 3-hydroxypropionate/4-hydroxybutyrate cycle lead to further reduction of the fixed carbon for the generation of 3-hydroxypropionate (16), a platform chemical also in the US Department of Energy top twelve [86,96]. Archaeal thermoacidophilic Metallosphaera sedula genes were utilised in the hyperthermophilic archaeon Pyrococcus furiosus to express the first three steps of the autotrophic 3-hydroxypropionic/4-hydroxybutyrate cycle for the synthesis of 3-hydroxypropionate (16) [97,98]. This was carried out at 70 °C, where the Metallosphaera enzymes show optimal activity and background metabolism of Pyrococcus furiosus does not interfere.
Decarboxylases
A number of enzymes are capable of catalysing the reversible interconversion of lipophilic aromatics and the more polar respective carboxylates [12]. It is thought these reactions may proceed in the carboxylation direction as a detoxification mechanism under anaerobic conditions, where oxidative degradation is not possible. In work pioneered by Nagasawa and coworkers [99-102], Kirimura and coworkers [103,104], and Faber and coworkers [105-109], these enzymes have been successfully applied in vitro under conditions that drive the equilibrium toward carboxylation, such as high CO2 concentration. Successful examples include the carboxylation of phenol and hydroxystyrene derivatives including catechol [102], guaiacol [110], indole [101] and pyrrole [100] (Scheme 7).
![[1860-5397-11-259-i7]](/bjoc/content/inline/1860-5397-11-259-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Decarboxylase catalysed carboxylation of (a) phenol derivatives, (b) indole and (c) pyrrole.
Scheme 7: Decarboxylase catalysed carboxylation of (a) phenol derivatives, (b) indole and (c) pyrrole.
Isocitrate dehydrogenase
As discussed above, as part of the reductive TCA cycle (Scheme 2) isocitrate dehydrogenase catalyses the carboxylation of 2-oxoglutarate (6) to produce isocitrate (7). Exploitation in biotechnological applications has been challenging due to the unfavourable thermodynamics of the carboxylation. Recently, the use of purified isocitrate dehydrogenase for CO2 fixation was reported [111]. Carbon fixation was driven thermodynamically by maintaining a low pH, where CO2 concentrations are highest, and coupling the reaction to aconitase catalysed removal of isocitrate (7) to produce aconitate. Switching the pH allowed for subsequent release of the captured CO2 and regeneration of the carbon-capture substrate 2-oxoglutarate (6). Though this application is aimed at CO2 sequestration rather than transformation, it shows that the reductive TCA cycle isocitrate dehydrogenase may be used in vitro to fix CO2 to a species that may be further transformed enzymatically.
FDH catalysed formate production
Due to the direct CO2 reduction to a C1 species, as opposed to carboxylation of a secondary substrate catalysed by most other enzymes, FDHs have attracted more widespread attention as catalysts for the transformation of CO2 with numerous examples in recent literature. Applications span all aspects of enzyme technology including isolated biocatalysts, immobilised biocatalysts, whole-cell catalysts and bioelectrocatalytic systems. Theoretical studies modelling potential formatotrophic organisms showed significant promise for such systems [112].
Isolated FDH. Enzymes from acetogenic sources have been characterised and found to be capable of catalysing CO2 reduction in vitro under thermodynamically favourable conditions. Acetogenic FDH from Clostridium thermoaceticum (now Moorella thermoacetica) was reported by Wood and Ljundahl in 1966 [113], where an exchange between 14CO2 and formate was observed, though no net synthesis of formate. Thauer [114] was the first to observe a net CO2 reduction to formate for the acetogenic FDH by recycling of the reduced cofactor, and prove that this enzyme utilised NADPH for the reduction of CO2 as the first step in one branch of the Wood–Ljungdahl pathway. Similarly, FDH in cell-free lysate of Clostridium acidiurici catalysed CO2 reduction to formate with reduced ferredoxin and NADH [115]. Earlier, it had been shown that it was possible for an enzyme found in the related non-acetogenic Clostridium pasteurianum to carry out direct reduction of CO2 to formate with reduced ferredoxin alone, rather than through a two-step process involving acetyl-CoA as a CO2 acceptor, disproving the established view at that time that biological CO2 reduction may only proceed indirectly [116]. Furthermore, Thauer et al. [117] were able to prove that this FDH utilises CO2, rather than HCO3−, as the active species, through experiments carried out at low temperatures where CO2 hydration is slow. The enzyme from Clostridium carboxidivorans was recombinantly expressed in E. coli and shown to display higher CO2 reducing activity and poorer affinity for formate, as compared to a non-acetogenic Candida boidinii FDH prepared in parallel, known to efficiently oxidise formate [62]. This suggests that weak formate binding contributes toward the catalytic preference of the acetogenic enzyme. Clostridium autoethanogenum was purified as a complex with an electron bifurcating hydrogenase that is NADPH and ferredoxin dependent, and found to catalyse reduction of CO2 with NADPH and reduced ferredoxin or H2 [63]. An FDH was also purified as a complex with hydrogenase from the acetogen Acetobacterium woodii and found to directly utilise H2 as an electron donor for the reduction of CO2 [118].
Furthermore, there is a growing list of examples of non-acetogenic metallo-FDHs, naturally catalysing formate oxidation, found to also be capable of catalysing CO2 reduction in vitro. FDH from Pseudomonas oxalaticus was the first isolated enzyme reported to catalyse both formate oxidation and CO2 reduction under appropriate conditions, using substrate amounts of NAD+/NADH [119]. This enzyme was later used in the seminal work of Parkinson and Weaver [120], where electrons were supplied through a semiconductor photoelectrode using light in the visible spectrum (>1.35 eV) and coupled to FDH activity through a mediator to drive CO2 reduction. Two W-dependent FDHs, isolated from the syntrophic bacterium Syntrophobacter fumaroxidans, showed high catalytic activity for CO2 reduction, using reduced methyl viologen as the electron donor. Later, one of these was immobilised onto an electrode and shown to reduce CO2 electrochemically through direct use of the electrons provided [121]. In this way the reaction could be electrochemically driven in either direction. Recently the Mo-dependent FDH from E. coli was shown to be capable of catalysing CO2 reduction employing a similar approach [122]. An oxygen-tolerant Mo-dependent FDH from Rhodobacter capsulatus was reported to catalyse the reduction of CO2 with NADH [123].
FDHs without metal cofactors have also been employed to reduce CO2 in vitro. Despite interest in application of Candida boidinii FDH due to its stability, the observed turnover for this enzyme is generally low. However, application of a bioelectrochemical system allowed production of formate from CO2 with proton transfer from an electrical source through NAD+ to this FDH [124]. Choe et al. [125,126] showed that a series of robust acidophilic nonmetallo-FDHs were particularly useful in the catalysis of CO2 reduction. As these enzymes are stable at the lower pH ranges where the concentration of solvated CO2 is highest, improved formate yields were obtained.
All this suggests that the ability of FDHs to reversibly catalyse formate and CO2 interconversion is broadly distributed in nature, irrespective of metabolic directionality, however, catalytic properties vary greatly depending on the source organism. As expected, the enzymes that naturally catalyse CO2 reduction and highly homologous FDHs from formate oxidation pathways display higher reduction activities than FDHs of lower homology. The possibility of recycling the reduced electron donating cofactor, through the action of a second enzyme such as hydrogenase, or through direct or mediated electron delivery from an electrode, greatly enhance the potential of FDHs for application in large-scale biotechnological processes (Figure 4).
![[1860-5397-11-259-4]](/bjoc/content/figures/1860-5397-11-259-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Formate dehydrogenase (FDH) catalysed reversible reduction of CO2 to formate with electron donor regeneration through hydrogenase-catalysed H2 oxidation (red arrow) or electrochemical reduction at a cathode (blue arrow).
Figure 4: Formate dehydrogenase (FDH) catalysed reversible reduction of CO2 to formate with electron donor re...
Whole-cell FDH application. In addition to the examples already mentioned for in vitro enzymatic production of formate, this has also been achieved using resting or immobilised cells as catalysts. Due to the ease of combination of multiple enzymatic activities in whole-cell applications, there has been a particular focus on coupling FDH activity to a hydrogenase, with which it is commonly found as the formate hydrogen lyase complex in nature. To this effect, immobilised Alcaligenes eutrophus (reclassified as Ralstonia eutropha) whole-cell catalysts were able to catalyse hydrogenation of CO2 to similar levels as Pd adsorbed on activated carbon [127]. In the previously mentioned work [118] on the purified acetogenic FDH-hydrogenase complex from Acetobacterium woodii, a whole-cell biocatalyst was also reported, generating high yields of formate from CO2 and H2. Resting cells from the common biotechnological host E. coli have been known to generate modest yields of formate from CO2 hydrogenation, when grown on formate for induction of the native enzymes [128]. More recently, by overexpressing suitable recombinant FDHs in E. coli JM109(DE3), high formate yields were obtained from CO2 hydrogenation, without need for cellular growth on formate for induction [129]. An alternative whole-cell system was later reported, using an electrochemical cell, where the reducing equivalents are generated by an electrode, rather than H2 oxidation, as has been done for purified enzymes [130].
Methanol production through formate. Due to the advantages of direct formatogenesis from CO2, there has been a number of investigations into further biocatalytic conversion of formate into other desirable chemicals. A possibility that has gathered much attention is the consecutive reduction to formaldehyde and methanol, first described by Kuwabata and co-workers [131,132]. This is of particular interest due to the potential use of methanol as a fuel. Methanol production has been achieved in vitro utilising FDH in series with formaldehyde dehydrogenase (FaldDH) and alcohol dehydrogenase (ADH) [133-136]. One of the main hurdles to the utilisation of this process relates to the requirement for the additional two enzymes to work in the reverse to physiological direction, as well as the generally unfavourable thermodynamic equilibria. An attractive approach utilised photocatalysts to generate electrons from solar energy, which in turn were donated for the production of methanol [137]. Though methanol yields and catalyst efficiencies are low, these results are highly promising for the future development of biochemical systems for the solar-driven generation of formate, formaldehyde and methanol from CO2 (Figure 5).
![[1860-5397-11-259-5]](/bjoc/content/figures/1860-5397-11-259-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Sequential generation of formate, formaldehyde and methanol from CO2 using reducing equivalents sourced through electrochemical cells or photocatalysts. ED: electron donor, FDH: formate dehydrogenase, FladDH: formaldehyde dehydrogenase, ADH: alcohol dehydrogenase.
Figure 5: Sequential generation of formate, formaldehyde and methanol from CO2 using reducing equivalents sou...
FDHs for hydrogen storage. The significance of biocatalytic systems for the production of formate with reducing equivalents from H2 extends beyond the generation of a platform chemical. Formate has also been targeted as a form of chemical storage of hydrogen fuel, due to energetic demands and hazards associated with H2 liquefaction, transport and storage [138,139]. Effective use of CO2 to store H2 would enable a sustainable hydrogen based economy, through carbon neutral technologies. Formate in particular, due to its chemical properties and the atom efficiency in complete stoichiometric retention of hydrogen, has been touted as a very promising reduced form of CO2 [138,140,141]. Consequently, many catalytic systems working in the reverse direction have also been investigated, for the regeneration of H2, along with CO2.
Many organisms, including E. coli, naturally produce H2 as an electron sink for oxidative pathways [142]. As a result, whole-cell systems have been described that work efficiently toward formate oxidation and direct electron delivery to a hydrogenase [143,144]. However, biocatalytic systems are unlikely to become suitable for decentralised H2 release, as for example will be required in hydrogen fuelled transportation vehicles. Transition metal catalysts have been reported to reach desired turnovers [138], however, in these cases cost and metal availability become hurdles in sustaining a hydrogen economy. Zeolite systems utilising Ge or Si, recently described, were able to efficiently dehydrogenate formic acid. This was guided through computational calculations, allowing the design of a zeolite catalyst displaying over 94% selectivity over the counter-productive formate dehydration reaction [145]. The combination of biological systems for centralised hydrogen storage through CO2 reduction as formate, with cheap zeolite catalysts for decentralised on demand hydrogen regeneration appears a very promising sustainable approach toward a hydrogen economy (Figure 6).
![[1860-5397-11-259-6]](/bjoc/content/figures/1860-5397-11-259-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Hydrogen storage as formic acid through biocatalytic hydrogenation of CO2 and subsequent on-demand release through zeolite catalysed dehydrogenation.
Figure 6: Hydrogen storage as formic acid through biocatalytic hydrogenation of CO2 and subsequent on-demand ...
In vitro production of CO with CODH
Reduction of CO2 to CO through in vitro application of CODH has been of interest as the enzymatic product may be further converted into hydrocarbons through the Fischer–Tropsch process [146]. In work carried out by Armstrong, Ragsdale and coworkers [147,148], metal oxide nanoparticles were functionalised with CODH and photosensitised with a Ru dye to catalyse the reduction of CO2 using visible light. Further to this, the reported ability of a V-dependent nitrogenase to slowly reduce CO to various small-chain hydrocarbons holds much promise for the development of enzymatic processes to further transform CO into products of interest [149].
Prospects and challenges for future biotechnological applications
In order for CO2 biotransformation to target a broad range of commodity chemicals, the CO2-fixing enzymes must be used as part of multi-enzymatic cascades that convert CO2 through multiple steps [111,150]. Such reactions may be performed in vitro, where the relative amounts of each biocatalyst and the intermediate concentrations during the reaction can be closely monitored and controlled. However, this is accompanied by a requirement in cost related to enzyme purification, proportional to the number of enzymes used. The application of enzymes within whole-cells allows their production and utilisation with minimal processing and circumvents biocatalyst purification, though in this case there are limitations related to substrate/product diffusion and background metabolic activity. The optimal approach in each case, as for any multi-enzymatic synthesis, will depend on a combination of factors such as the number of enzymes to be utilised, the ease of substrate and product diffusion through the cell membrane, and the presence of unwanted background reactions.
Within a well-understood cellular chassis, the heterologous expression of a CO2 fixing enzyme allows its use as a module that may be matched with other modules of choice, for the assembly of synthetic pathways [150,151]. In a CO2 transforming modular process, the CO2 fixing modules will play a central role, much like CO2 fixing enzymes do in a carbon assimilation pathway. However, the assembly will also include other genes that allow process control or express desirable features such as acid tolerance [152,153]. For these modules to be easily applied, the enzymes must be easy to express in heterologous hosts. This is greatly complicated by requirements for specialised cofactors or maturation and folding processes.
RuBisCO presents significant challenges for use in modular synthetic biology approaches, due to the observed inefficiency and requirement for expression of large amounts of protein. The difficulty of expression in hosts that do not naturally contain RuBisCO, such as E. coli, and the complicated nature of the heterologous RuBisCO systems currently developed in transgenic plants [80], means that the Calvin cycle is a challenging target for synthetic biology in non-photosynthetic microorganisms. Efforts focusing on increasing carbon fixation yields through optimisation of RuBisCO expression and activity may lead to optimised plant based synthetic systems [18,40].
In microbial systems carboxylases are promising candidates for modular design, due to their broad distribution in living organisms and lack of particular requirements in cofactors. Also the great variety of carboxylases found in nature represents a very large library from which suitable modules may be sourced that introduce carbon into metabolic pathways [26,154]. On the other hand, in order to generate a synthetic pathway where the only carbon input is CO2, these enzymes would also require the co-expression of cyclic pathways to recycle the co-substrates that are carboxylated. This may greatly hinder the overall process, as the metabolic pathways that have been developed by nature to carry out these tasks contain many steps and a number of unfavourable reactions. Indeed, attempts to transfer entire autotrophic CO2 fixation pathways into E. coli have been unsuccesful [155].
Dehydrogenases used in the reductive acetyl-CoA pathway, do not present this complication as the CO2 is reduced directly to another species, either formate or CO, with no other reactant other than a source of electrons. This means that a single enzymatic module is able to catalyse the incorporation of CO2 as a C1 species, with no other carbon requirement. In this case, the difficulties associated with expression of enzymes from niche organisms in heterologous hosts, such as requirement for particular metal cofactors and oxygen stability, complicate use in modular approaches. Also, the metabolic product must be efficiently transformed into other species in order to drive this energetically uphill carbon fixation process. Finally, as formate and CO are not metabolites in central anabolic pathways, it may be challenging to find suitable pathways that allow access to the variety of chemicals that may be produced through metabolism. This will inevitably require heterologous expression of the full reductive pathway, for production of acetyl-CoA, which however is extremely challenging due to the requirement for use of poorly understood enzymes and unusual cofactors. A recent breakthrough came with the production of a computationally designed enzyme, catalysing the carboligation of three formaldehyde units into dihydroxyacetone, thus providing direct access to central carbon metabolism through formate [156].
Sourcing of reducing equivalents. As mentioned above, any process that transforms CO2 into other chemicals, where the carbon is in a more reduced state, represents a net reduction. Therefore there is a requirement for reductive potential in the form of electrons, and the method used to source these will greatly define the utility of the overall process (Figure 7). The ATP required to drive CO2 fixation processes within living systems will be mainly produced using reducing equivalents through the complicated mechanism of oxidative phosphorylation.
![[1860-5397-11-259-7]](/bjoc/content/figures/1860-5397-11-259-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Schematic showing required flow of reducing equivalents for CO2 fixation through biotechnological applications.
Figure 7: Schematic showing required flow of reducing equivalents for CO2 fixation through biotechnological a...
Ultimately the most sustainable source of reducing equivalents is sunlight [20]. Solar energy may be directly utilised through the application of photosynthetic machinery employed by photoautotrophs to carry out the “light reactions” of photosynthesis. This will require technological advances, such as the development of bioreactors capable of maximising exposure to sunlight [157]. Another limitation to any approach relying on photosynthesis to harvest solar energy is the inherently poor efficiency and sensitivity of photosynthetic pigments and reaction centres, as highlighted by Michel [158]. An alternative approach is to convert solar energy into electricity for use as a source of electrons [20,159]. As seen, a number of enzymes and organisms are indeed capable of directly accepting electrons from electrodes in bioelectrochemical systems [160-162]. The use of electricity generated through photovoltaics allows the mediated application of solar energy for the fixation of CO2. Finally, electrons can be stored within chemical species that may then be oxidised by organisms to regenerate the electrons on-demand [20]. Hydrogen and formic acid appear most suited for such applications, due to their chemical properties, and the existence of efficient biological tools for electron regeneration through oxidation.
Conclusion
It is evident that the use of biological catalysts for CO2 fixation and conversion to a variety of chemicals is a promising approach, not limited by the availability of natural enzymes. However, in order for these to be employed in suitable bioprocesses, where they may be assembled into multi-enzymatic synthetic cascades, suitable methodologies for facile recombinant expression need to be developed further. This will extend beyond simple expression of a single gene, and may require simultaneous expression of multiple subunits, expression of seleno-proteins, proteins that deliver particular cofactors, as well as chaperones and maturation proteins that allow the production of the final active biocatalyst. Furthermore, the various biological mechanisms used in nature to improve the activity of these enzymes must be fully understood, in order to be suitably harnessed for application in synthetic processes. Host organisms must be developed with features geared towards the fixation of CO2 and its transformation through multiple enzymatic steps. Finally the reducing equivalents required for the carbon fixation step, as well as subsequent transformations, must be harnessed efficiently. Suitable technological platforms are yet to be developed.
Though there is much progress to be made before CO2 fixing enzymes may be readily used as modules in designer synthetic pathways, the rapid progress that is being made in the fields of genetic engineering, bioinformatics and synthetic biology, as well as renewable electricity generation and bioelectrochemical engineering hold much promise for the development of the biotechnological platforms that will support a future carbon bio-economy.
References
-
Capellan-Pérez, I.; Mediavilla, M.; de Castro, C.; Carpintero, Ó.; Javier Miguel, L. Energy 2014, 77, 641. doi:10.1016/j.energy.2014.09.063
Return to citation in text: [1] -
Höök, M.; Tang, X. Energy Policy 2013, 52, 797. doi:10.1016/j.enpol.2012.10.046
Return to citation in text: [1] -
Christensen, C. H.; Rass-Hansen, J.; Marsden, C. C.; Taarning, E.; Egeblad, K. ChemSusChem 2008, 1, 283. doi:10.1002/cssc.200700168
Return to citation in text: [1] -
Stocker, T. F.; Qin, D.; Plattner, G.-K.; Tignor, M.; Allen, S. K.; Boschung, J.; Nauels, A.; Xia, Y.; Bex, V.; Midgley, P. M. Climate change 2013: The Physical Science Basis. Working Group I Contribution to the IPCC 5th Assessment Report of the Intergovernmental Panel on Climate Change. 2013; http://www.ipcc.ch/report/ar5/wg1 (accessed Nov 11, 2015).
Return to citation in text: [1] -
Wei, T.; Yang, S.; Moore, J. C.; Shi, P.; Cui, X.; Duan, Q.; Xu, B.; Dai, Y.; Yuan, W.; Wei, X.; Yang, Z.; Wen, T.; Teng, F.; Gao, Y.; Chou, J.; Yan, X.; Wei, Z.; Guo, Y.; Jiang, Y.; Gao, X.; Wang, K.; Zheng, X.; Reng, F.; Lv, S.; Yu, Y.; Liu, B.; Luo, Y.; Li, W.; Ji, D.; Feng, J.; Wu, Q.; Cheng, H.; He, J.; Fu, C.; Ye, D.; Xu, G.; Dong, W. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 12911. doi:10.1073/pnas.1203282109
Return to citation in text: [1] -
Aresta, M.; Dibenedetto, A. Dalton Trans. 2007, 2975. doi:10.1039/b700658f
Return to citation in text: [1] -
Barzagli, F.; Mani, F.; Peruzzini, M. Green Chem. 2011, 13, 1267. doi:10.1039/c0gc00674b
Return to citation in text: [1] [2] -
Demirel, Y.; Matzen, M.; Winters, C.; Gao, X. Int. J. Energy Res. 2015, 39, 1011. doi:10.1002/er.3277
Return to citation in text: [1] -
Langanke, J.; Wolf, A.; Hofmann, J.; Böhm, K.; Subhani, M. A.; Müller, T. E.; Leitner, W.; Gürtler, C. Green Chem. 2014, 16, 1865. doi:10.1039/C3GC41788C
Return to citation in text: [1] -
Lively, R. P.; Sharma, P.; McCool, B. A.; Beaudry-Losique, J.; Luo, D.; Thomas, V. M.; Realff, M.; Chance, R. R. Biofuels, Bioprod. Biorefin. 2015, 9, 72. doi:10.1002/bbb.1505
Return to citation in text: [1] -
Yang, Z.-Z.; He, L.-N.; Gao, J.; Liu, A.-H.; Yu, B. Energy Environ. Sci. 2012, 5, 6602. doi:10.1039/c2ee02774g
Return to citation in text: [1] -
Glueck, S. M.; Gümüs, S.; Fabian, W. M. F.; Faber, K. Chem. Soc. Rev. 2010, 39, 313. doi:10.1039/B807875K
Return to citation in text: [1] [2] [3] [4] -
Bornscheuer, U. T.; Huisman, G. W.; Kazlauskas, R. J.; Lutz, S.; Moore, J. C.; Robins, K. Nature 2012, 485, 185. doi:10.1038/nature11117
Return to citation in text: [1] -
Wohlgemuth, R. Curr. Opin. Biotechnol. 2010, 21, 713. doi:10.1016/j.copbio.2010.09.016
Return to citation in text: [1] -
Savile, C. K.; Lalonde, J. J. Curr. Opin. Biotechnol. 2011, 22, 818. doi:10.1016/j.copbio.2011.06.006
Return to citation in text: [1] -
Philbrook, A.; Alissandratos, A.; Easton, C. J. In Environmental Biotechnology - New Approaches and Prospective Applications; Petre, M., Ed.; InTech: Rijeka, 2013; p 39.
Return to citation in text: [1] -
Ort, D. R.; Merchant, S. S.; Alric, J.; Barkan, A.; Blankenship, R. E.; Bock, R.; Croce, R.; Hanson, M. R.; Hibberd, J. M.; Long, S. P.; Moore, T. A.; Moroney, J.; Niyogi, K. K.; Parry, M. A. J.; Peralta-Yahya, P. P.; Prince, R. C.; Redding, K. E.; Spalding, M. H.; van Wijk, K. J.; Vermaas, W. F. J.; von Caemmerer, S.; Weber, A. P. M.; Yeates, T. O.; Yuan, J. S.; Zhu, X. G. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 8529. doi:10.1073/pnas.1424031112
Return to citation in text: [1] [2] -
Rosgaard, L.; de Porcellinis, A. J.; Jacobsen, J. H.; Frigaard, N.-U.; Sakuragi, Y. J. Biotechnol. 2012, 162, 134. doi:10.1016/j.jbiotec.2012.05.006
Return to citation in text: [1] [2] [3] -
Hawkins, A. S.; McTernan, P. M.; Lian, H.; Kelly, R. M.; Adams, M. W. W. Curr. Opin. Biotechnol. 2013, 24, 376. doi:10.1016/j.copbio.2013.02.017
Return to citation in text: [1] [2] -
Li, H.; Liao, J. C. Energy Environ. Sci. 2013, 6, 2892. doi:10.1039/c3ee41847b
Return to citation in text: [1] [2] [3] [4] -
Gong, F.; Liu, G.; Zhai, X.; Zhou, J.; Cai, Z.; Li, Y. Biotechnol. Biofuels 2015, 8, 86. doi:10.1186/s13068-015-0268-1
Return to citation in text: [1] [2] -
Matthessen, R.; Fransaer, J.; Binnemans, K.; De Vos, D. E. Beilstein J. Org. Chem. 2014, 10, 2484. doi:10.3762/bjoc.10.260
Return to citation in text: [1] -
Ducat, D. C.; Silver, P. A. Curr. Opin. Chem. Biol. 2012, 16, 337. doi:10.1016/j.cbpa.2012.05.002
Return to citation in text: [1] [2] [3] -
Fuchs, G. Annu. Rev. Microbiol. 2011, 65, 631. doi:10.1146/annurev-micro-090110-102801
Return to citation in text: [1] [2] [3] -
Bar-Even, A.; Noor, E.; Lewis, N. E.; Milo, R. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 8889. doi:10.1073/pnas.0907176107
Return to citation in text: [1] [2] [3] -
Erb, T. J. Appl. Environ. Microbiol. 2011, 77, 8466. doi:10.1128/AEM.05702-11
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Bar-Even, A.; Noor, E.; Milo, R. J. Exp. Bot. 2012, 63, 2325. doi:10.1093/jxb/err417
Return to citation in text: [1] [2] -
Knowles, J. R. Annu. Rev. Biochem. 1989, 58, 195. doi:10.1146/annurev.bi.58.070189.001211
Return to citation in text: [1] [2] [3] -
Pierre, A. C. ISRN Chem. Eng. 2012, 2012, 753687. doi:10.5402/2012/753687
Return to citation in text: [1] -
Moroney, J. V.; Ma, Y.; Frey, W. D.; Fusilier, K. A.; Pham, T. T.; Simms, T. A.; DiMario, R. J.; Yang, J.; Mukherjee, B. Photosynth. Res. 2011, 109, 133. doi:10.1007/s11120-011-9635-3
Return to citation in text: [1] -
Khalifah, R. G. Proc. Natl. Acad. Sci. U. S. A. 1973, 70, 1986. doi:10.1073/pnas.70.7.1986
Return to citation in text: [1] -
Tong, L. Cell. Mol. Life Sci. 2013, 70, 863. doi:10.1007/s00018-012-1096-0
Return to citation in text: [1] -
Christin, P.-A.; Arakaki, M.; Osborne, C. P.; Bräutigam, A.; Sage, R. F.; Hibberd, J. M.; Kelly, S.; Covshoff, S.; Wong, G. K.-S.; Hancock, L.; Edwards, E. J. J. Exp. Bot. 2014, 65, 3609. doi:10.1093/jxb/eru087
Return to citation in text: [1] -
Winter, K.; Holtum, J. A. M. J. Exp. Bot. 2014, 65, 3425. doi:10.1093/jxb/eru063
Return to citation in text: [1] -
Chen, A. H.; Robinson-Mosher, A.; Savage, D. F.; Silver, P. A.; Polka, J. K. PLoS One 2013, 8, e76127. doi:10.1371/journal.pone.0076127
Return to citation in text: [1] -
Long, B. M.; Badger, M. R.; Whitney, S. M.; Price, G. D. J. Biol. Chem. 2007, 282, 29323. doi:10.1074/jbc.M703896200
Return to citation in text: [1] [2] -
Chen, A. H.; Robinson-Mosher, A.; Savage, D. F.; Silver, P. A.; Polka, J. K. PLoS One 2013, 8, e76127. doi:10.1371/journal.pone.0076127
Return to citation in text: [1] -
Lin, M. T.; Occhialini, A.; Andralojc, P. J.; Devonshire, J.; Hines, K. M.; Parry, M. A. J.; Hanson, M. R. Plant J. 2014, 79, 1. doi:10.1111/tpj.12536
Return to citation in text: [1] -
Lin, M. T.; Occhialini, A.; Andralojc, P. J.; Parry, M. A. J.; Hanson, M. R. Nature 2014, 513, 547. doi:10.1038/nature13776
Return to citation in text: [1] [2] [3] [4] -
Price, G. D.; Pengelly, J. J. L.; Forster, B.; Du, J.; Whitney, S. M.; von Caemmerer, S.; Badger, M. R.; Howitt, S. M.; Evans, J. R. J. Exp. Bot. 2013, 64, 753. doi:10.1093/jxb/ers257
Return to citation in text: [1] [2] -
Bonacci, W.; Teng, P. K.; Afonso, B.; Niederholtmeyer, H.; Grob, P.; Silver, P. A.; Savage, D. F. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 478. doi:10.1073/pnas.1108557109
Return to citation in text: [1] -
Berg, I. A.; Kockelkorn, D.; Ramos-Vera, W. H.; Say, R. F.; Zarzycki, J.; Hügler, M.; Alber, B. E.; Fuchs, G. Nat. Rev. Microbiol. 2010, 8, 447. doi:10.1038/nrmicro2365
Return to citation in text: [1] -
Nisbet, E. G.; Grassineau, N. V.; Howe, C. J.; Abell, P. I.; Regelous, M.; Nisbet, R. E. R. Geobiology 2007, 5, 311. doi:10.1111/j.1472-4669.2007.00127.x
Return to citation in text: [1] [2] -
Galmés, J.; Kapralov, M. V.; Andralojc, P. J.; Conesa, M. À.; Keys, A. J.; Parry, M. A. J.; Flexas, J. Plant, Cell Environ. 2014, 37, 1989. doi:10.1111/pce.12335
Return to citation in text: [1] -
Berg, I. A. Appl. Environ. Microbiol. 2011, 77, 1925. doi:10.1128/AEM.02473-10
Return to citation in text: [1] -
Zhu, X.-G.; Long, S. P.; Ort, D. R. Annu. Rev. Plant Biol. 2010, 61, 235. doi:10.1146/annurev-arplant-042809-112206
Return to citation in text: [1] -
Shively, J. M.; van Keulen, G.; Meijer, W. G. Annu. Rev. Microbiol. 1998, 52, 191. doi:10.1146/annurev.micro.52.1.191
Return to citation in text: [1] -
Singh, J.; Pandey, P.; James, D.; Chandrasekhar, K.; Achary, V. M. M.; Kaul, T.; Tripathy, B. C.; Reddy, M. K. Plant Biotechnol. J. 2014, 12, 1217. doi:10.1111/pbi.12246
Return to citation in text: [1] [2] [3] -
Studer, R. A.; Christin, P.-A.; Williams, M. A.; Orengo, C. A. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 2223. doi:10.1073/pnas.1310811111
Return to citation in text: [1] -
Hügler, M.; Sievert, S. M. Annu. Rev. Mar. Sci. 2011, 3, 261. doi:10.1146/annurev-marine-120709-142712
Return to citation in text: [1] [2] -
Hugler, M.; Huber, H.; Stetter, K. O.; Fuchs, G. Arch. Microbiol. 2003, 179, 160.
Return to citation in text: [1] [2] -
Buchanan, B. B.; Arnon, D. I. Photosynth. Res. 1990, 24, 47. doi:10.1007/BF00032643
Return to citation in text: [1] -
Evans, M. C. W.; Buchanan, B. B.; Arnon, D. I. Proc. Natl. Acad. Sci. U. S. A. 1966, 55, 928. doi:10.1073/pnas.55.4.928
Return to citation in text: [1] -
Ljungdahl, L.; Wood, H. G. Annu. Rev. Microbiol. 1969, 23, 515. doi:10.1146/annurev.mi.23.100169.002503
Return to citation in text: [1] -
Ragsdale, S. W.; Pierce, E. Biochim. Biophys. Acta, Proteins Proteomics 2008, 1784, 1873. doi:10.1016/j.bbapap.2008.08.012
Return to citation in text: [1] [2] -
Clark, J. E.; Ragsdale, S. W.; Ljungdahl, L. G.; Wiegel, J. J. Bacteriol. 1982, 151, 507.
Return to citation in text: [1] -
Ragsdale, S. W. Crit. Rev. Biochem. Mol. Biol. 1991, 26, 261. doi:10.3109/10409239109114070
Return to citation in text: [1] -
Eden, G.; Fuchs, G. Arch. Microbiol. 1982, 133, 66. doi:10.1007/BF00943772
Return to citation in text: [1] -
Eden, G.; Fuchs, G. Arch. Microbiol. 1983, 135, 68. doi:10.1007/BF00419485
Return to citation in text: [1] -
Maynard, E. L.; Lindahl, P. A. J. Inorg. Biochem. 1999, 74, 227.
Return to citation in text: [1] -
Fesseler, J.; Jeoung, J.-H.; Dobbek, H. Angew. Chem., Int. Ed. 2015, 54, 8560. doi:10.1002/anie.201501778
Return to citation in text: [1] -
Alissandratos, A.; Kim, H.-K.; Matthews, H.; Hennessy, J. E.; Philbrook, A.; Easton, C. J. Appl. Environ. Microbiol. 2013, 79, 741. doi:10.1128/AEM.02886-12
Return to citation in text: [1] [2] -
Wang, S.; Huang, H.; Kahnt, J.; Mueller, A. P.; Köpke, M.; Thauer, R. K. J. Bacteriol. 2013, 195, 4373. doi:10.1128/JB.00678-13
Return to citation in text: [1] [2] [3] [4] -
Almendra, M. J.; Brondino, C. D.; Gavel, O.; Pereira, A. S.; Tavares, P.; Bursakov, S.; Duarte, R.; Caldeira, J.; Moura, J. J. G.; Moura, I. Biochemistry 1999, 38, 16366. doi:10.1021/bi990069n
Return to citation in text: [1] [2] -
Khangulov, S. V.; Gladyshev, V. N.; Dismukes, G. C.; Stadtman, T. C. Biochemistry 1998, 37, 3518. doi:10.1021/bi972177k
Return to citation in text: [1] -
Maia, L. B.; Moura, J. J. G.; Moura, I. J. Biol. Inorg. Chem. 2015, 20, 287. doi:10.1007/s00775-014-1218-2
Return to citation in text: [1] -
de Bok, F. A. M.; Hagedoorn, P.-L.; Silva, P. J.; Hagen, W. R.; Schiltz, E.; Fritsche, K.; Stams, A. J. M. Eur. J. Biochem. 2003, 270, 2476. doi:10.1046/j.1432-1033.2003.03619.x
Return to citation in text: [1] -
Seol, E.; Jang, Y.; Kim, S.; Oh, Y.-K.; Park, S. Int. J. Hydrogen Energy 2012, 37, 15045. doi:10.1016/j.ijhydene.2012.07.095
Return to citation in text: [1] [2] -
Tishkov, V. I.; Popov, V. O. Biochemistry (Moscow) 2004, 69, 1252. doi:10.1007/s10541-005-0071-x
Return to citation in text: [1] -
Berg, I. A.; Kockelkorn, D.; Buckel, W.; Fuchs, G. Science 2007, 318, 1782. doi:10.1126/science.1149976
Return to citation in text: [1] -
Chuakrut, S.; Arai, H.; Ishii, M.; Igarashi, Y. J. Bacteriol. 2003, 185, 938. doi:10.1128/JB.185.3.938-947.2003
Return to citation in text: [1] [2] -
Menendez, C.; Bauer, Z.; Huber, H.; Gad'on, N.; Stetter, K.-O.; Fuchs, G. J. Bacteriol. 1999, 181, 1088.
Return to citation in text: [1] -
Ramos-Vera, W. H.; Berg, I. A.; Fuchs, G. J. Bacteriol. 2009, 191, 4286. doi:10.1128/JB.00145-09
Return to citation in text: [1] -
Mueller-Cajar, O.; Whitney, S. M. Photosynth. Res. 2008, 98, 667. doi:10.1007/s11120-008-9324-z
Return to citation in text: [1] [2] -
Mueller-Cajar, O.; Whitney, S. M. Biochem. J. 2008, 414, 205. doi:10.1042/BJ20080668
Return to citation in text: [1] [2] -
Mueller-Cajar, O.; Morell, M.; Whitney, S. M. Biochemistry 2007, 46, 14067. doi:10.1021/bi700820a
Return to citation in text: [1] -
Savir, Y.; Noor, E.; Milo, R.; Tlusty, T. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 3475. doi:10.1073/pnas.0911663107
Return to citation in text: [1] -
Long, S. P.; Zhu, X.-G.; Naidu, S. L.; Ort, D. R. Plant, Cell Environ. 2006, 29, 315. doi:10.1111/j.1365-3040.2005.01493.x
Return to citation in text: [1] -
McGrath, J. M.; Long, S. P. Plant Physiol. 2014, 164, 2247. doi:10.1104/pp.113.232611
Return to citation in text: [1] -
Whitney, S. M.; Birch, R.; Kelso, C.; Beck, J. L.; Kapralov, M. V. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 3564. doi:10.1073/pnas.1420536112
Return to citation in text: [1] [2] -
Singh, A.; Nigam, P. S.; Murphy, J. D. Bioresour. Technol. 2011, 102, 10. doi:10.1016/j.biortech.2010.06.032
Return to citation in text: [1] -
Lue, J.; Sheahan, C.; Fu, P. Energy Environ. Sci. 2011, 4, 2451. doi:10.1039/c0ee00593b
Return to citation in text: [1] -
Guadalupe-Medina, V.; Wisselink, H. W.; Luttik, M. A. H.; de Hulster, E.; Daran, J.-M.; Pronk, J. T.; van Maris, A. J. A. Biotechnol. Biofuels 2013, 6, 125. doi:10.1186/1754-6834-6-125
Return to citation in text: [1] -
Chang, K. S.; Jeon, H.; Gu, M. B.; Pack, S. P.; Jin, E. Bioprocess Biosyst. Eng. 2013, 36, 1923. doi:10.1007/s00449-013-0968-5
Return to citation in text: [1] -
Chang, K. S.; Jeon, H.; Seo, S.; Lee, Y.; Jin, E. Enzyme Microb. Technol. 2014, 60, 64. doi:10.1016/j.enzmictec.2014.04.007
Return to citation in text: [1] -
Werpy, T.; Peterson, G. Top value added chemicals from Biomass; National Renewable Energy Laboratory, US Department of Energy, 2004.
Return to citation in text: [1] [2] -
Lin, H.; San, K.-Y.; Bennett, G. N. Appl. Microbiol. Biotechnol. 2005, 67, 515. doi:10.1007/s00253-004-1789-x
Return to citation in text: [1] -
Wang, D.; Li, Q.; Li, W.; Xing, J.; Su, Z. Enzyme Microb. Technol. 2009, 45, 491. doi:10.1016/j.enzmictec.2009.08.003
Return to citation in text: [1] -
Song, C. W.; Kim, D. I.; Choi, S.; Jang, J. W.; Lee, S. Y. Biotechnol. Bioeng. 2013, 110, 2025. doi:10.1002/bit.24868
Return to citation in text: [1] -
Liu, R.; Liang, L.; Wu, M.; Chen, K.; Jiang, M.; Ma, J.; Wei, P.; Ouyang, P. Biochem. Eng. J. 2013, 79, 77. doi:10.1016/j.bej.2013.07.004
Return to citation in text: [1] -
Wu, H.; Li, Q.; Li, Z.-m.; Ye, Q. Bioresour. Technol. 2012, 107, 376. doi:10.1016/j.biortech.2011.12.043
Return to citation in text: [1] -
Liang, L.; Liu, R.; Wang, G.; Gou, D.; Ma, J.; Chen, K.; Jiang, M.; Wei, P.; Ouyang, P. Enzyme Microb. Technol. 2012, 51, 286. doi:10.1016/j.enzmictec.2012.07.011
Return to citation in text: [1] -
Wang, Z.; Lin, M.; Wang, L.; Ammar, E. M.; Yang, S.-T. Process Biochem. 2015, 50, 194. doi:10.1016/j.procbio.2014.11.012
Return to citation in text: [1] -
Ammar, E. M.; Jin, Y.; Wang, Z.; Yang, S.-T. Appl. Microbiol. Biotechnol. 2014, 98, 7761. doi:10.1007/s00253-014-5836-y
Return to citation in text: [1] -
Hügler, M.; Krieger, R. S.; Jahn, M.; Fuchs, G. Eur. J. Biochem. 2003, 270, 736. doi:10.1046/j.1432-1033.2003.03434.x
Return to citation in text: [1] -
Ishii, M.; Chuakrut, S.; Arai, H.; Igarashi, Y. Appl. Microbiol. Biotechnol. 2004, 64, 605. doi:10.1007/s00253-003-1540-z
Return to citation in text: [1] -
Thorgersen, M. P.; Lipscomb, G. L.; Schut, G. J.; Kelly, R. M.; Adams, M. W. W. Metab. Eng. 2014, 22, 83. doi:10.1016/j.ymben.2013.12.006
Return to citation in text: [1] -
Keller, M. W.; Schut, G. J.; Lipscomb, G. L.; Menon, A. L.; Iwuchukwu, I. J.; Leuko, T. T.; Thorgersen, M. P.; Nixon, W. J.; Hawkins, A. S.; Kelly, R. M.; Adams, M. W. W. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 5840. doi:10.1073/pnas.1222607110
Return to citation in text: [1] -
Yoshida, T.; Nagasawa, T. J. Biosci. Bioeng. 2000, 89, 111. doi:10.1016/S1389-1723(00)88723-X
Return to citation in text: [1] -
Wieser, M.; Yoshida, T.; Nagasawa, T. J. Mol. Catal. B: Enzym. 2001, 11, 179. doi:10.1016/S1381-1177(00)00038-2
Return to citation in text: [1] [2] -
Yoshida, T.; Fujita, K.; Nagasawa, T. Biosci., Biotechnol., Biochem. 2002, 66, 2388. doi:10.1271/bbb.66.2388
Return to citation in text: [1] [2] -
Yoshida, T.; Inami, Y.; Matsui, T.; Nagasawa, T. Biotechnol. Lett. 2010, 32, 701. doi:10.1007/s10529-010-0210-3
Return to citation in text: [1] [2] -
Kirimura, K.; Gunji, H.; Wakayama, R.; Hattori, T.; Ishii, Y. Biochem. Biophys. Res. Commun. 2010, 394, 279. doi:10.1016/j.bbrc.2010.02.154
Return to citation in text: [1] -
Kirimura, K.; Yanaso, S.; Kosaka, S.; Koyama, K.; Hattori, T.; Ishii, Y. Chem. Lett. 2011, 40, 206. doi:10.1246/cl.2011.206
Return to citation in text: [1] -
Wuensch, C.; Pavkov-Keller, T.; Steinkellner, G.; Gross, J.; Fuchs, M.; Hromic, A.; Lyskowski, A.; Fauland, K.; Gruber, K.; Glueck, S. M.; Faber, K. Adv. Synth. Catal. 2015, 357, 1909. doi:10.1002/adsc.201401028
Return to citation in text: [1] -
Pesci, L.; Glueck, S. M.; Gurikov, P.; Smirnova, I.; Faber, K.; Liese, A. FEBS J. 2015, 282, 1334. doi:10.1111/febs.13225
Return to citation in text: [1] -
Wuensch, C.; Gross, J.; Steinkellner, G.; Lyskowski, A.; Gruber, K.; Glueck, S. M.; Faber, K. RSC Adv. 2014, 4, 9673. doi:10.1039/c3ra47719c
Return to citation in text: [1] -
Wuensch, C.; Glueck, S. M.; Gross, J.; Koszelewski, D.; Schober, M.; Faber, K. Org. Lett. 2012, 14, 1974. doi:10.1021/ol300385k
Return to citation in text: [1] -
Wuensch, C.; Schmidt, N.; Gross, J.; Grischek, B.; Glueck, S. M.; Faber, K. J. Biotechnol. 2013, 168, 264. doi:10.1016/j.jbiotec.2013.07.017
Return to citation in text: [1] -
Lupa, B.; Lyon, D.; Shaw, L. N.; Sieprawska-Lupa, M.; Wiegel, J. Can. J. Microbiol. 2008, 54, 75. doi:10.1139/W07-113
Return to citation in text: [1] -
Xia, S.; Zhao, X.; Frigo-Vaz, B.; Zheng, W.; Kim, J.; Wang, P. Bioresour. Technol. 2015, 182, 368. doi:10.1016/j.biortech.2015.01.093
Return to citation in text: [1] [2] -
Bar-Even, A.; Noor, E.; Flamholz, A.; Milo, R. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 1039. doi:10.1016/j.bbabio.2012.10.013
Return to citation in text: [1] -
Li, L. F.; Ljungdah, L.; Wood, H. G. J. Bacteriol. 1966, 92, 405.
Return to citation in text: [1] -
Thauer, R. K. FEBS Lett. 1972, 27, 111. doi:10.1016/0014-5793(72)80421-6
Return to citation in text: [1] -
Thauer, R. K. J. Bacteriol. 1973, 114, 443.
Return to citation in text: [1] -
Jungermann, K.; Kirchniawy, H.; Thauer, R. K. Biochem. Biophys. Res. Commun. 1970, 41, 682. doi:10.1016/0006-291X(70)90067-7
Return to citation in text: [1] -
Thauer, R. K.; Käufer, B.; Fuchs, G. Eur. J. Biochem. 1975, 55, 111. doi:10.1111/j.1432-1033.1975.tb02143.x
Return to citation in text: [1] -
Schuchmann, K.; Müller, V. Science 2013, 342, 1382. doi:10.1126/science.1244758
Return to citation in text: [1] [2] -
Ruschig, U.; Müller, U.; Willnow, P.; Höpner, T. Eur. J. Biochem. 1976, 70, 325. doi:10.1111/j.1432-1033.1976.tb11021.x
Return to citation in text: [1] -
Parkinson, B. A.; Weaver, P. F. Nature 1984, 309, 148. doi:10.1038/309148a0
Return to citation in text: [1] -
Reda, T.; Plugge, C. M.; Abram, N. J.; Hirst, J. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 10654. doi:10.1073/pnas.0801290105
Return to citation in text: [1] -
Bassegoda, A.; Madden, C.; Wakerley, D. W.; Reisner, E.; Hirst, J. J. Am. Chem. Soc. 2014, 136, 15473. doi:10.1021/ja508647u
Return to citation in text: [1] -
Hartmann, T.; Leimkühler, S. FEBS J. 2013, 280, 6083. doi:10.1111/febs.12528
Return to citation in text: [1] -
Srikanth, S.; Maesen, M.; Dominguez-Benetton, X.; Vanbroekhoven, K.; Pant, D. Bioresour. Technol. 2014, 165, 350. doi:10.1016/j.biortech.2014.01.129
Return to citation in text: [1] -
Choe, H.; Joo, J. C.; Cho, D. H.; Kim, M. H.; Lee, S. H.; Jung, K. D.; Kim, Y. H. PLoS One 2014, 9, e103111. doi:10.1371/journal.pone.0103111
Return to citation in text: [1] -
Choe, H.; Ha, J. M.; Joo, J. C.; Kim, H.; Yoon, H.-J.; Kim, S.; Son, S. H.; Gengan, R. M.; Jeon, S. T.; Chang, R.; Jung, K. D.; Kim, Y. H.; Lee, H. H. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2015, 71, 313. doi:10.1107/S1399004714025474
Return to citation in text: [1] -
Klibanov, A. M.; Alberti, B. N.; Zale, S. E. Biotechnol. Bioeng. 1982, 24, 25. doi:10.1002/bit.260240104
Return to citation in text: [1] -
Woods, D. D. Biochem. J. 1936, 30, 515. doi:10.1042/bj0300515
Return to citation in text: [1] -
Alissandratos, A.; Kim, H.-K.; Easton, C. J. Bioresour. Technol. 2014, 164, 7. doi:10.1016/j.biortech.2014.04.064
Return to citation in text: [1] -
Hwang, H.; Yeon, Y. J.; Lee, S.; Choe, H.; Jang, M. G.; Cho, D. H.; Park, S.; Kim, Y. H. Bioresour. Technol. 2015, 185, 35. doi:10.1016/j.biortech.2015.02.086
Return to citation in text: [1] -
Kuwabata, S.; Tsuda, R.; Nishida, K.; Yoneyama, H. Chem. Lett. 1993, 22, 1631. doi:10.1246/cl.1993.1631
Return to citation in text: [1] -
Kuwabata, S.; Tsuda, R.; Yoneyama, H. J. Am. Chem. Soc. 1994, 116, 5437. doi:10.1021/ja00091a056
Return to citation in text: [1] -
Cazelles, R.; Drone, J.; Fajula, F.; Ersen, O.; Moldovan, S.; Galarneau, A. New J. Chem. 2013, 37, 3721. doi:10.1039/c3nj00688c
Return to citation in text: [1] -
Luo, J.; Meyer, A. S.; Mateiu, R. V.; Pinelo, M. New Biotechnol. 2015, 32, 319. doi:10.1016/j.nbt.2015.02.006
Return to citation in text: [1] -
Amao, Y.; Watanabe, T. Chem. Lett. 2004, 33, 1544. doi:10.1246/cl.2004.1544
Return to citation in text: [1] -
Amao, Y.; Watanabe, T. Appl. Catal., B 2009, 86, 109. doi:10.1016/j.apcatb.2008.08.008
Return to citation in text: [1] -
Yadav, R. K.; Oh, G. H.; Park, N.-J.; Kumar, A.; Kong, K.-j.; Baeg, J.-O. J. Am. Chem. Soc. 2014, 136, 16728. doi:10.1021/ja509650r
Return to citation in text: [1] -
Enthaler, S.; von Langermann, J.; Schmidt, T. Energy Environ. Sci. 2010, 3, 1207. doi:10.1039/b907569k
Return to citation in text: [1] [2] [3] -
Joo, F. ChemSusChem 2008, 1, 805. doi:10.1002/cssc.200800133
Return to citation in text: [1] -
Boddien, A.; Gärtner, F.; Federsel, C.; Sponholz, P.; Mellmann, D.; Jackstell, R.; Junge, H.; Beller, M. Angew. Chem., Int. Ed. 2011, 50, 6411. doi:10.1002/anie.201101995
Return to citation in text: [1] -
Enthaler, S. ChemSusChem 2008, 1, 801. doi:10.1002/cssc.200800101
Return to citation in text: [1] -
Sinha, P.; Roy, S.; Das, D. Int. J. Hydrogen Energy 2015, 40, 8806. doi:10.1016/j.ijhydene.2015.05.076
Return to citation in text: [1] -
Lipscomb, G. L.; Schut, G. J.; Thorgersen, M. P.; Nixon, W. J.; Kelly, R. M.; Adams, M. W. W. J. Biol. Chem. 2014, 289, 2873. doi:10.1074/jbc.M113.530725
Return to citation in text: [1] -
Hu, H.; Wood, T. K. Biochem. Biophys. Res. Commun. 2010, 391, 1033. doi:10.1016/j.bbrc.2009.12.013
Return to citation in text: [1] -
Amos, R. I. J.; Heinroth, F.; Chan, B.; Zheng, S.; Haynes, B. S.; Easton, C. J.; Masters, A. F.; Radom, L.; Maschmeyer, T. Angew. Chem., Int. Ed. 2014, 53, 11275. doi:10.1002/anie.201405360
Return to citation in text: [1] -
Bezemer, G. L.; Bitter, J. H.; Kuipers, H.; Oosterbeek, H.; Holewijn, J. E.; Xu, X. D.; Kapteijn, F.; van Dillen, A. J.; de Jong, K. P. J. Am. Chem. Soc. 2006, 128, 3956. doi:10.1021/ja058282w
Return to citation in text: [1] -
Woolerton, T. W.; Sheard, S.; Reisner, E.; Pierce, E.; Ragsdale, S. W.; Armstrong, F. A. J. Am. Chem. Soc. 2010, 132, 2132. doi:10.1021/ja910091z
Return to citation in text: [1] -
Woolerton, T. W.; Sheard, S.; Pierce, E.; Ragsdale, S. W.; Armstrong, F. A. Energy Environ. Sci. 2011, 4, 2393. doi:10.1039/c0ee00780c
Return to citation in text: [1] -
Hu, Y.; Lee, C. C.; Ribbe, M. W. Science 2011, 333, 753. doi:10.1126/science.1206883
Return to citation in text: [1] -
Muschiol, J.; Peters, C.; Oberleitner, N.; Mihovilovic, M. D.; Bornscheuer, U. T.; Rudroff, F. Chem. Commun. 2015, 51, 5798. doi:10.1039/C4CC08752F
Return to citation in text: [1] [2] -
Ford, T. J.; Silver, P. A. Curr. Opin. Chem. Biol. 2015, 28, 20. doi:10.1016/j.cbpa.2015.05.012
Return to citation in text: [1] -
Gaida, S. M.; Al-Hinai, M. A.; Indurthi, D. C.; Nicolaou, S. A.; Papoutsakis, E. T. Nucleic Acids Res. 2013, 41, 8726. doi:10.1093/nar/gkt651
Return to citation in text: [1] -
Zingaro, K. A.; Nicolaou, S. A.; Papoutsakis, E. T. Trends Biotechnol. 2013, 31, 643. doi:10.1016/j.tibtech.2013.08.005
Return to citation in text: [1] -
Fuchs, G.; Berg, I. A. J. Biotechnol. 2014, 192, 314. doi:10.1016/j.jbiotec.2014.02.015
Return to citation in text: [1] -
Mattozzi, M. D.; Ziesack, M.; Voges, M. J.; Silver, P. A.; Way, J. C. Metab. Eng. 2013, 16, 130. doi:10.1016/j.ymben.2013.01.005
Return to citation in text: [1] -
Siegel, J. B.; Smith, A. L.; Poust, S.; Wargacki, A. J.; Bar-Even, A.; Louw, C.; Shen, B. W.; Eiben, C. B.; Tran, H. M.; Noor, E.; Gallaher, J. L.; Bale, J.; Yoshikuni, Y.; Gelb, M. H.; Keasling, J. D.; Stoddard, B. L.; Lidstrom, M. E.; Baker, D. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 3704. doi:10.1073/pnas.1500545112
Return to citation in text: [1] -
Cuaresma, M.; Janssen, M.; Vilchez, C.; Wijffels, R. H. Bioresour. Technol. 2011, 102, 5129. doi:10.1016/j.biortech.2011.01.078
Return to citation in text: [1] -
Michel, H. Angew. Chem., Int. Ed. 2012, 51, 2516. doi:10.1002/anie.201200218
Return to citation in text: [1] -
Hawkins, A. S.; Han, Y.; Lian, H.; Loder, A. J.; Menon, A. L.; Iwuchukwu, I. J.; Keller, M.; Leuko, T. T.; Adams, M. W. W.; Kelly, R. M. ACS Catal. 2011, 1, 1043. doi:10.1021/cs2003017
Return to citation in text: [1] -
Guo, K.; Prévoteau, A.; Patil, S. A.; Rabaey, K. Curr. Opin. Biotechnol. 2015, 33, 149. doi:10.1016/j.copbio.2015.02.014
Return to citation in text: [1] -
Freguia, S.; Teh, E. H.; Boon, N.; Leung, K. M.; Keller, J.; Rabaey, K. Bioresour. Technol. 2010, 101, 1233. doi:10.1016/j.biortech.2009.09.054
Return to citation in text: [1] -
Rabaey, K.; Rodríguez, J.; Blackall, L. L.; Keller, J.; Gross, P.; Batstone, D.; Verstraete, W.; Nealson, K. H. ISME J. 2007, 1, 9. doi:10.1038/ismej.2007.4
Return to citation in text: [1]
| 82. | Lue, J.; Sheahan, C.; Fu, P. Energy Environ. Sci. 2011, 4, 2451. doi:10.1039/c0ee00593b |
| 18. | Rosgaard, L.; de Porcellinis, A. J.; Jacobsen, J. H.; Frigaard, N.-U.; Sakuragi, Y. J. Biotechnol. 2012, 162, 134. doi:10.1016/j.jbiotec.2012.05.006 |
| 39. | Lin, M. T.; Occhialini, A.; Andralojc, P. J.; Parry, M. A. J.; Hanson, M. R. Nature 2014, 513, 547. doi:10.1038/nature13776 |
| 81. | Singh, A.; Nigam, P. S.; Murphy, J. D. Bioresour. Technol. 2011, 102, 10. doi:10.1016/j.biortech.2010.06.032 |
| 80. | Whitney, S. M.; Birch, R.; Kelso, C.; Beck, J. L.; Kapralov, M. V. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 3564. doi:10.1073/pnas.1420536112 |
| 86. | Werpy, T.; Peterson, G. Top value added chemicals from Biomass; National Renewable Energy Laboratory, US Department of Energy, 2004. |
| 84. | Chang, K. S.; Jeon, H.; Gu, M. B.; Pack, S. P.; Jin, E. Bioprocess Biosyst. Eng. 2013, 36, 1923. doi:10.1007/s00449-013-0968-5 |
| 85. | Chang, K. S.; Jeon, H.; Seo, S.; Lee, Y.; Jin, E. Enzyme Microb. Technol. 2014, 60, 64. doi:10.1016/j.enzmictec.2014.04.007 |
| 21. | Gong, F.; Liu, G.; Zhai, X.; Zhou, J.; Cai, Z.; Li, Y. Biotechnol. Biofuels 2015, 8, 86. doi:10.1186/s13068-015-0268-1 |
| 83. | Guadalupe-Medina, V.; Wisselink, H. W.; Luttik, M. A. H.; de Hulster, E.; Daran, J.-M.; Pronk, J. T.; van Maris, A. J. A. Biotechnol. Biofuels 2013, 6, 125. doi:10.1186/1754-6834-6-125 |
| 91. | Wu, H.; Li, Q.; Li, Z.-m.; Ye, Q. Bioresour. Technol. 2012, 107, 376. doi:10.1016/j.biortech.2011.12.043 |
| 63. | Wang, S.; Huang, H.; Kahnt, J.; Mueller, A. P.; Köpke, M.; Thauer, R. K. J. Bacteriol. 2013, 195, 4373. doi:10.1128/JB.00678-13 |
| 92. | Liang, L.; Liu, R.; Wang, G.; Gou, D.; Ma, J.; Chen, K.; Jiang, M.; Wei, P.; Ouyang, P. Enzyme Microb. Technol. 2012, 51, 286. doi:10.1016/j.enzmictec.2012.07.011 |
| 62. | Alissandratos, A.; Kim, H.-K.; Matthews, H.; Hennessy, J. E.; Philbrook, A.; Easton, C. J. Appl. Environ. Microbiol. 2013, 79, 741. doi:10.1128/AEM.02886-12 |
| 89. | Song, C. W.; Kim, D. I.; Choi, S.; Jang, J. W.; Lee, S. Y. Biotechnol. Bioeng. 2013, 110, 2025. doi:10.1002/bit.24868 |
| 119. | Ruschig, U.; Müller, U.; Willnow, P.; Höpner, T. Eur. J. Biochem. 1976, 70, 325. doi:10.1111/j.1432-1033.1976.tb11021.x |
| 90. | Liu, R.; Liang, L.; Wu, M.; Chen, K.; Jiang, M.; Ma, J.; Wei, P.; Ouyang, P. Biochem. Eng. J. 2013, 79, 77. doi:10.1016/j.bej.2013.07.004 |
| 118. | Schuchmann, K.; Müller, V. Science 2013, 342, 1382. doi:10.1126/science.1244758 |
| 87. | Lin, H.; San, K.-Y.; Bennett, G. N. Appl. Microbiol. Biotechnol. 2005, 67, 515. doi:10.1007/s00253-004-1789-x |
| 88. | Wang, D.; Li, Q.; Li, W.; Xing, J.; Su, Z. Enzyme Microb. Technol. 2009, 45, 491. doi:10.1016/j.enzmictec.2009.08.003 |
| 117. | Thauer, R. K.; Käufer, B.; Fuchs, G. Eur. J. Biochem. 1975, 55, 111. doi:10.1111/j.1432-1033.1975.tb02143.x |
| 116. | Jungermann, K.; Kirchniawy, H.; Thauer, R. K. Biochem. Biophys. Res. Commun. 1970, 41, 682. doi:10.1016/0006-291X(70)90067-7 |
| 86. | Werpy, T.; Peterson, G. Top value added chemicals from Biomass; National Renewable Energy Laboratory, US Department of Energy, 2004. |
| 96. | Ishii, M.; Chuakrut, S.; Arai, H.; Igarashi, Y. Appl. Microbiol. Biotechnol. 2004, 64, 605. doi:10.1007/s00253-003-1540-z |
| 97. | Thorgersen, M. P.; Lipscomb, G. L.; Schut, G. J.; Kelly, R. M.; Adams, M. W. W. Metab. Eng. 2014, 22, 83. doi:10.1016/j.ymben.2013.12.006 |
| 98. | Keller, M. W.; Schut, G. J.; Lipscomb, G. L.; Menon, A. L.; Iwuchukwu, I. J.; Leuko, T. T.; Thorgersen, M. P.; Nixon, W. J.; Hawkins, A. S.; Kelly, R. M.; Adams, M. W. W. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 5840. doi:10.1073/pnas.1222607110 |
| 93. | Wang, Z.; Lin, M.; Wang, L.; Ammar, E. M.; Yang, S.-T. Process Biochem. 2015, 50, 194. doi:10.1016/j.procbio.2014.11.012 |
| 94. | Ammar, E. M.; Jin, Y.; Wang, Z.; Yang, S.-T. Appl. Microbiol. Biotechnol. 2014, 98, 7761. doi:10.1007/s00253-014-5836-y |
| 71. | Chuakrut, S.; Arai, H.; Ishii, M.; Igarashi, Y. J. Bacteriol. 2003, 185, 938. doi:10.1128/JB.185.3.938-947.2003 |
| 95. | Hügler, M.; Krieger, R. S.; Jahn, M.; Fuchs, G. Eur. J. Biochem. 2003, 270, 736. doi:10.1046/j.1432-1033.2003.03434.x |
| 120. | Parkinson, B. A.; Weaver, P. F. Nature 1984, 309, 148. doi:10.1038/309148a0 |
| 61. | Fesseler, J.; Jeoung, J.-H.; Dobbek, H. Angew. Chem., Int. Ed. 2015, 54, 8560. doi:10.1002/anie.201501778 |
| 118. | Schuchmann, K.; Müller, V. Science 2013, 342, 1382. doi:10.1126/science.1244758 |
| 130. | Hwang, H.; Yeon, Y. J.; Lee, S.; Choe, H.; Jang, M. G.; Cho, D. H.; Park, S.; Kim, Y. H. Bioresour. Technol. 2015, 185, 35. doi:10.1016/j.biortech.2015.02.086 |
| 56. | Clark, J. E.; Ragsdale, S. W.; Ljungdahl, L. G.; Wiegel, J. J. Bacteriol. 1982, 151, 507. |
| 57. | Ragsdale, S. W. Crit. Rev. Biochem. Mol. Biol. 1991, 26, 261. doi:10.3109/10409239109114070 |
| 58. | Eden, G.; Fuchs, G. Arch. Microbiol. 1982, 133, 66. doi:10.1007/BF00943772 |
| 59. | Eden, G.; Fuchs, G. Arch. Microbiol. 1983, 135, 68. doi:10.1007/BF00419485 |
| 129. | Alissandratos, A.; Kim, H.-K.; Easton, C. J. Bioresour. Technol. 2014, 164, 7. doi:10.1016/j.biortech.2014.04.064 |
| 1. | Capellan-Pérez, I.; Mediavilla, M.; de Castro, C.; Carpintero, Ó.; Javier Miguel, L. Energy 2014, 77, 641. doi:10.1016/j.energy.2014.09.063 |
| 2. | Höök, M.; Tang, X. Energy Policy 2013, 52, 797. doi:10.1016/j.enpol.2012.10.046 |
| 124. | Srikanth, S.; Maesen, M.; Dominguez-Benetton, X.; Vanbroekhoven, K.; Pant, D. Bioresour. Technol. 2014, 165, 350. doi:10.1016/j.biortech.2014.01.129 |
| 123. | Hartmann, T.; Leimkühler, S. FEBS J. 2013, 280, 6083. doi:10.1111/febs.12528 |
| 127. | Klibanov, A. M.; Alberti, B. N.; Zale, S. E. Biotechnol. Bioeng. 1982, 24, 25. doi:10.1002/bit.260240104 |
| 125. | Choe, H.; Joo, J. C.; Cho, D. H.; Kim, M. H.; Lee, S. H.; Jung, K. D.; Kim, Y. H. PLoS One 2014, 9, e103111. doi:10.1371/journal.pone.0103111 |
| 126. | Choe, H.; Ha, J. M.; Joo, J. C.; Kim, H.; Yoon, H.-J.; Kim, S.; Son, S. H.; Gengan, R. M.; Jeon, S. T.; Chang, R.; Jung, K. D.; Kim, Y. H.; Lee, H. H. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2015, 71, 313. doi:10.1107/S1399004714025474 |
| 12. | Glueck, S. M.; Gümüs, S.; Fabian, W. M. F.; Faber, K. Chem. Soc. Rev. 2010, 39, 313. doi:10.1039/B807875K |
| 73. | Ramos-Vera, W. H.; Berg, I. A.; Fuchs, G. J. Bacteriol. 2009, 191, 4286. doi:10.1128/JB.00145-09 |
| 6. | Aresta, M.; Dibenedetto, A. Dalton Trans. 2007, 2975. doi:10.1039/b700658f |
| 7. | Barzagli, F.; Mani, F.; Peruzzini, M. Green Chem. 2011, 13, 1267. doi:10.1039/c0gc00674b |
| 8. | Demirel, Y.; Matzen, M.; Winters, C.; Gao, X. Int. J. Energy Res. 2015, 39, 1011. doi:10.1002/er.3277 |
| 9. | Langanke, J.; Wolf, A.; Hofmann, J.; Böhm, K.; Subhani, M. A.; Müller, T. E.; Leitner, W.; Gürtler, C. Green Chem. 2014, 16, 1865. doi:10.1039/C3GC41788C |
| 10. | Lively, R. P.; Sharma, P.; McCool, B. A.; Beaudry-Losique, J.; Luo, D.; Thomas, V. M.; Realff, M.; Chance, R. R. Biofuels, Bioprod. Biorefin. 2015, 9, 72. doi:10.1002/bbb.1505 |
| 11. | Yang, Z.-Z.; He, L.-N.; Gao, J.; Liu, A.-H.; Yu, B. Energy Environ. Sci. 2012, 5, 6602. doi:10.1039/c2ee02774g |
| 4. | Stocker, T. F.; Qin, D.; Plattner, G.-K.; Tignor, M.; Allen, S. K.; Boschung, J.; Nauels, A.; Xia, Y.; Bex, V.; Midgley, P. M. Climate change 2013: The Physical Science Basis. Working Group I Contribution to the IPCC 5th Assessment Report of the Intergovernmental Panel on Climate Change. 2013; http://www.ipcc.ch/report/ar5/wg1 (accessed Nov 11, 2015). |
| 5. | Wei, T.; Yang, S.; Moore, J. C.; Shi, P.; Cui, X.; Duan, Q.; Xu, B.; Dai, Y.; Yuan, W.; Wei, X.; Yang, Z.; Wen, T.; Teng, F.; Gao, Y.; Chou, J.; Yan, X.; Wei, Z.; Guo, Y.; Jiang, Y.; Gao, X.; Wang, K.; Zheng, X.; Reng, F.; Lv, S.; Yu, Y.; Liu, B.; Luo, Y.; Li, W.; Ji, D.; Feng, J.; Wu, Q.; Cheng, H.; He, J.; Fu, C.; Ye, D.; Xu, G.; Dong, W. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 12911. doi:10.1073/pnas.1203282109 |
| 51. | Hugler, M.; Huber, H.; Stetter, K. O.; Fuchs, G. Arch. Microbiol. 2003, 179, 160. |
| 70. | Berg, I. A.; Kockelkorn, D.; Buckel, W.; Fuchs, G. Science 2007, 318, 1782. doi:10.1126/science.1149976 |
| 122. | Bassegoda, A.; Madden, C.; Wakerley, D. W.; Reisner, E.; Hirst, J. J. Am. Chem. Soc. 2014, 136, 15473. doi:10.1021/ja508647u |
| 3. | Christensen, C. H.; Rass-Hansen, J.; Marsden, C. C.; Taarning, E.; Egeblad, K. ChemSusChem 2008, 1, 283. doi:10.1002/cssc.200700168 |
| 71. | Chuakrut, S.; Arai, H.; Ishii, M.; Igarashi, Y. J. Bacteriol. 2003, 185, 938. doi:10.1128/JB.185.3.938-947.2003 |
| 72. | Menendez, C.; Bauer, Z.; Huber, H.; Gad'on, N.; Stetter, K.-O.; Fuchs, G. J. Bacteriol. 1999, 181, 1088. |
| 121. | Reda, T.; Plugge, C. M.; Abram, N. J.; Hirst, J. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 10654. doi:10.1073/pnas.0801290105 |
| 17. | Ort, D. R.; Merchant, S. S.; Alric, J.; Barkan, A.; Blankenship, R. E.; Bock, R.; Croce, R.; Hanson, M. R.; Hibberd, J. M.; Long, S. P.; Moore, T. A.; Moroney, J.; Niyogi, K. K.; Parry, M. A. J.; Peralta-Yahya, P. P.; Prince, R. C.; Redding, K. E.; Spalding, M. H.; van Wijk, K. J.; Vermaas, W. F. J.; von Caemmerer, S.; Weber, A. P. M.; Yeates, T. O.; Yuan, J. S.; Zhu, X. G. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 8529. doi:10.1073/pnas.1424031112 |
| 18. | Rosgaard, L.; de Porcellinis, A. J.; Jacobsen, J. H.; Frigaard, N.-U.; Sakuragi, Y. J. Biotechnol. 2012, 162, 134. doi:10.1016/j.jbiotec.2012.05.006 |
| 63. | Wang, S.; Huang, H.; Kahnt, J.; Mueller, A. P.; Köpke, M.; Thauer, R. K. J. Bacteriol. 2013, 195, 4373. doi:10.1128/JB.00678-13 |
| 64. | Almendra, M. J.; Brondino, C. D.; Gavel, O.; Pereira, A. S.; Tavares, P.; Bursakov, S.; Duarte, R.; Caldeira, J.; Moura, J. J. G.; Moura, I. Biochemistry 1999, 38, 16366. doi:10.1021/bi990069n |
| 68. | Seol, E.; Jang, Y.; Kim, S.; Oh, Y.-K.; Park, S. Int. J. Hydrogen Energy 2012, 37, 15045. doi:10.1016/j.ijhydene.2012.07.095 |
| 16. | Philbrook, A.; Alissandratos, A.; Easton, C. J. In Environmental Biotechnology - New Approaches and Prospective Applications; Petre, M., Ed.; InTech: Rijeka, 2013; p 39. |
| 69. | Tishkov, V. I.; Popov, V. O. Biochemistry (Moscow) 2004, 69, 1252. doi:10.1007/s10541-005-0071-x |
| 15. | Savile, C. K.; Lalonde, J. J. Curr. Opin. Biotechnol. 2011, 22, 818. doi:10.1016/j.copbio.2011.06.006 |
| 55. | Ragsdale, S. W.; Pierce, E. Biochim. Biophys. Acta, Proteins Proteomics 2008, 1784, 1873. doi:10.1016/j.bbapap.2008.08.012 |
| 62. | Alissandratos, A.; Kim, H.-K.; Matthews, H.; Hennessy, J. E.; Philbrook, A.; Easton, C. J. Appl. Environ. Microbiol. 2013, 79, 741. doi:10.1128/AEM.02886-12 |
| 63. | Wang, S.; Huang, H.; Kahnt, J.; Mueller, A. P.; Köpke, M.; Thauer, R. K. J. Bacteriol. 2013, 195, 4373. doi:10.1128/JB.00678-13 |
| 13. | Bornscheuer, U. T.; Huisman, G. W.; Kazlauskas, R. J.; Lutz, S.; Moore, J. C.; Robins, K. Nature 2012, 485, 185. doi:10.1038/nature11117 |
| 14. | Wohlgemuth, R. Curr. Opin. Biotechnol. 2010, 21, 713. doi:10.1016/j.copbio.2010.09.016 |
| 63. | Wang, S.; Huang, H.; Kahnt, J.; Mueller, A. P.; Köpke, M.; Thauer, R. K. J. Bacteriol. 2013, 195, 4373. doi:10.1128/JB.00678-13 |
| 64. | Almendra, M. J.; Brondino, C. D.; Gavel, O.; Pereira, A. S.; Tavares, P.; Bursakov, S.; Duarte, R.; Caldeira, J.; Moura, J. J. G.; Moura, I. Biochemistry 1999, 38, 16366. doi:10.1021/bi990069n |
| 65. | Khangulov, S. V.; Gladyshev, V. N.; Dismukes, G. C.; Stadtman, T. C. Biochemistry 1998, 37, 3518. doi:10.1021/bi972177k |
| 66. | Maia, L. B.; Moura, J. J. G.; Moura, I. J. Biol. Inorg. Chem. 2015, 20, 287. doi:10.1007/s00775-014-1218-2 |
| 67. | de Bok, F. A. M.; Hagedoorn, P.-L.; Silva, P. J.; Hagen, W. R.; Schiltz, E.; Fritsche, K.; Stams, A. J. M. Eur. J. Biochem. 2003, 270, 2476. doi:10.1046/j.1432-1033.2003.03619.x |
| 68. | Seol, E.; Jang, Y.; Kim, S.; Oh, Y.-K.; Park, S. Int. J. Hydrogen Energy 2012, 37, 15045. doi:10.1016/j.ijhydene.2012.07.095 |
| 74. | Mueller-Cajar, O.; Whitney, S. M. Photosynth. Res. 2008, 98, 667. doi:10.1007/s11120-008-9324-z |
| 75. | Mueller-Cajar, O.; Whitney, S. M. Biochem. J. 2008, 414, 205. doi:10.1042/BJ20080668 |
| 145. | Amos, R. I. J.; Heinroth, F.; Chan, B.; Zheng, S.; Haynes, B. S.; Easton, C. J.; Masters, A. F.; Radom, L.; Maschmeyer, T. Angew. Chem., Int. Ed. 2014, 53, 11275. doi:10.1002/anie.201405360 |
| 75. | Mueller-Cajar, O.; Whitney, S. M. Biochem. J. 2008, 414, 205. doi:10.1042/BJ20080668 |
| 138. | Enthaler, S.; von Langermann, J.; Schmidt, T. Energy Environ. Sci. 2010, 3, 1207. doi:10.1039/b907569k |
| 26. | Erb, T. J. Appl. Environ. Microbiol. 2011, 77, 8466. doi:10.1128/AEM.05702-11 |
| 12. | Glueck, S. M.; Gümüs, S.; Fabian, W. M. F.; Faber, K. Chem. Soc. Rev. 2010, 39, 313. doi:10.1039/B807875K |
| 146. | Bezemer, G. L.; Bitter, J. H.; Kuipers, H.; Oosterbeek, H.; Holewijn, J. E.; Xu, X. D.; Kapteijn, F.; van Dillen, A. J.; de Jong, K. P. J. Am. Chem. Soc. 2006, 128, 3956. doi:10.1021/ja058282w |
| 138. | Enthaler, S.; von Langermann, J.; Schmidt, T. Energy Environ. Sci. 2010, 3, 1207. doi:10.1039/b907569k |
| 140. | Boddien, A.; Gärtner, F.; Federsel, C.; Sponholz, P.; Mellmann, D.; Jackstell, R.; Junge, H.; Beller, M. Angew. Chem., Int. Ed. 2011, 50, 6411. doi:10.1002/anie.201101995 |
| 141. | Enthaler, S. ChemSusChem 2008, 1, 801. doi:10.1002/cssc.200800101 |
| 138. | Enthaler, S.; von Langermann, J.; Schmidt, T. Energy Environ. Sci. 2010, 3, 1207. doi:10.1039/b907569k |
| 139. | Joo, F. ChemSusChem 2008, 1, 805. doi:10.1002/cssc.200800133 |
| 143. | Lipscomb, G. L.; Schut, G. J.; Thorgersen, M. P.; Nixon, W. J.; Kelly, R. M.; Adams, M. W. W. J. Biol. Chem. 2014, 289, 2873. doi:10.1074/jbc.M113.530725 |
| 144. | Hu, H.; Wood, T. K. Biochem. Biophys. Res. Commun. 2010, 391, 1033. doi:10.1016/j.bbrc.2009.12.013 |
| 142. | Sinha, P.; Roy, S.; Das, D. Int. J. Hydrogen Energy 2015, 40, 8806. doi:10.1016/j.ijhydene.2015.05.076 |
| 131. | Kuwabata, S.; Tsuda, R.; Nishida, K.; Yoneyama, H. Chem. Lett. 1993, 22, 1631. doi:10.1246/cl.1993.1631 |
| 132. | Kuwabata, S.; Tsuda, R.; Yoneyama, H. J. Am. Chem. Soc. 1994, 116, 5437. doi:10.1021/ja00091a056 |
| 78. | Long, S. P.; Zhu, X.-G.; Naidu, S. L.; Ort, D. R. Plant, Cell Environ. 2006, 29, 315. doi:10.1111/j.1365-3040.2005.01493.x |
| 137. | Yadav, R. K.; Oh, G. H.; Park, N.-J.; Kumar, A.; Kong, K.-j.; Baeg, J.-O. J. Am. Chem. Soc. 2014, 136, 16728. doi:10.1021/ja509650r |
| 79. | McGrath, J. M.; Long, S. P. Plant Physiol. 2014, 164, 2247. doi:10.1104/pp.113.232611 |
| 133. | Cazelles, R.; Drone, J.; Fajula, F.; Ersen, O.; Moldovan, S.; Galarneau, A. New J. Chem. 2013, 37, 3721. doi:10.1039/c3nj00688c |
| 134. | Luo, J.; Meyer, A. S.; Mateiu, R. V.; Pinelo, M. New Biotechnol. 2015, 32, 319. doi:10.1016/j.nbt.2015.02.006 |
| 135. | Amao, Y.; Watanabe, T. Chem. Lett. 2004, 33, 1544. doi:10.1246/cl.2004.1544 |
| 136. | Amao, Y.; Watanabe, T. Appl. Catal., B 2009, 86, 109. doi:10.1016/j.apcatb.2008.08.008 |
| 77. | Savir, Y.; Noor, E.; Milo, R.; Tlusty, T. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 3475. doi:10.1073/pnas.0911663107 |
| 39. | Lin, M. T.; Occhialini, A.; Andralojc, P. J.; Parry, M. A. J.; Hanson, M. R. Nature 2014, 513, 547. doi:10.1038/nature13776 |
| 74. | Mueller-Cajar, O.; Whitney, S. M. Photosynth. Res. 2008, 98, 667. doi:10.1007/s11120-008-9324-z |
| 76. | Mueller-Cajar, O.; Morell, M.; Whitney, S. M. Biochemistry 2007, 46, 14067. doi:10.1021/bi700820a |
| 39. | Lin, M. T.; Occhialini, A.; Andralojc, P. J.; Parry, M. A. J.; Hanson, M. R. Nature 2014, 513, 547. doi:10.1038/nature13776 |
| 41. | Bonacci, W.; Teng, P. K.; Afonso, B.; Niederholtmeyer, H.; Grob, P.; Silver, P. A.; Savage, D. F. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 478. doi:10.1073/pnas.1108557109 |
| 156. | Siegel, J. B.; Smith, A. L.; Poust, S.; Wargacki, A. J.; Bar-Even, A.; Louw, C.; Shen, B. W.; Eiben, C. B.; Tran, H. M.; Noor, E.; Gallaher, J. L.; Bale, J.; Yoshikuni, Y.; Gelb, M. H.; Keasling, J. D.; Stoddard, B. L.; Lidstrom, M. E.; Baker, D. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 3704. doi:10.1073/pnas.1500545112 |
| 42. | Berg, I. A.; Kockelkorn, D.; Ramos-Vera, W. H.; Say, R. F.; Zarzycki, J.; Hügler, M.; Alber, B. E.; Fuchs, G. Nat. Rev. Microbiol. 2010, 8, 447. doi:10.1038/nrmicro2365 |
| 155. | Mattozzi, M. D.; Ziesack, M.; Voges, M. J.; Silver, P. A.; Way, J. C. Metab. Eng. 2013, 16, 130. doi:10.1016/j.ymben.2013.01.005 |
| 80. | Whitney, S. M.; Birch, R.; Kelso, C.; Beck, J. L.; Kapralov, M. V. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 3564. doi:10.1073/pnas.1420536112 |
| 152. | Gaida, S. M.; Al-Hinai, M. A.; Indurthi, D. C.; Nicolaou, S. A.; Papoutsakis, E. T. Nucleic Acids Res. 2013, 41, 8726. doi:10.1093/nar/gkt651 |
| 153. | Zingaro, K. A.; Nicolaou, S. A.; Papoutsakis, E. T. Trends Biotechnol. 2013, 31, 643. doi:10.1016/j.tibtech.2013.08.005 |
| 26. | Erb, T. J. Appl. Environ. Microbiol. 2011, 77, 8466. doi:10.1128/AEM.05702-11 |
| 154. | Fuchs, G.; Berg, I. A. J. Biotechnol. 2014, 192, 314. doi:10.1016/j.jbiotec.2014.02.015 |
| 18. | Rosgaard, L.; de Porcellinis, A. J.; Jacobsen, J. H.; Frigaard, N.-U.; Sakuragi, Y. J. Biotechnol. 2012, 162, 134. doi:10.1016/j.jbiotec.2012.05.006 |
| 40. | Price, G. D.; Pengelly, J. J. L.; Forster, B.; Du, J.; Whitney, S. M.; von Caemmerer, S.; Badger, M. R.; Howitt, S. M.; Evans, J. R. J. Exp. Bot. 2013, 64, 753. doi:10.1093/jxb/ers257 |
| 48. | Singh, J.; Pandey, P.; James, D.; Chandrasekhar, K.; Achary, V. M. M.; Kaul, T.; Tripathy, B. C.; Reddy, M. K. Plant Biotechnol. J. 2014, 12, 1217. doi:10.1111/pbi.12246 |
| 149. | Hu, Y.; Lee, C. C.; Ribbe, M. W. Science 2011, 333, 753. doi:10.1126/science.1206883 |
| 49. | Studer, R. A.; Christin, P.-A.; Williams, M. A.; Orengo, C. A. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 2223. doi:10.1073/pnas.1310811111 |
| 147. | Woolerton, T. W.; Sheard, S.; Reisner, E.; Pierce, E.; Ragsdale, S. W.; Armstrong, F. A. J. Am. Chem. Soc. 2010, 132, 2132. doi:10.1021/ja910091z |
| 148. | Woolerton, T. W.; Sheard, S.; Pierce, E.; Ragsdale, S. W.; Armstrong, F. A. Energy Environ. Sci. 2011, 4, 2393. doi:10.1039/c0ee00780c |
| 46. | Zhu, X.-G.; Long, S. P.; Ort, D. R. Annu. Rev. Plant Biol. 2010, 61, 235. doi:10.1146/annurev-arplant-042809-112206 |
| 150. | Muschiol, J.; Peters, C.; Oberleitner, N.; Mihovilovic, M. D.; Bornscheuer, U. T.; Rudroff, F. Chem. Commun. 2015, 51, 5798. doi:10.1039/C4CC08752F |
| 151. | Ford, T. J.; Silver, P. A. Curr. Opin. Chem. Biol. 2015, 28, 20. doi:10.1016/j.cbpa.2015.05.012 |
| 47. | Shively, J. M.; van Keulen, G.; Meijer, W. G. Annu. Rev. Microbiol. 1998, 52, 191. doi:10.1146/annurev.micro.52.1.191 |
| 111. | Xia, S.; Zhao, X.; Frigo-Vaz, B.; Zheng, W.; Kim, J.; Wang, P. Bioresour. Technol. 2015, 182, 368. doi:10.1016/j.biortech.2015.01.093 |
| 150. | Muschiol, J.; Peters, C.; Oberleitner, N.; Mihovilovic, M. D.; Bornscheuer, U. T.; Rudroff, F. Chem. Commun. 2015, 51, 5798. doi:10.1039/C4CC08752F |
| 43. | Nisbet, E. G.; Grassineau, N. V.; Howe, C. J.; Abell, P. I.; Regelous, M.; Nisbet, R. E. R. Geobiology 2007, 5, 311. doi:10.1111/j.1472-4669.2007.00127.x |
| 45. | Berg, I. A. Appl. Environ. Microbiol. 2011, 77, 1925. doi:10.1128/AEM.02473-10 |
| 43. | Nisbet, E. G.; Grassineau, N. V.; Howe, C. J.; Abell, P. I.; Regelous, M.; Nisbet, R. E. R. Geobiology 2007, 5, 311. doi:10.1111/j.1472-4669.2007.00127.x |
| 44. | Galmés, J.; Kapralov, M. V.; Andralojc, P. J.; Conesa, M. À.; Keys, A. J.; Parry, M. A. J.; Flexas, J. Plant, Cell Environ. 2014, 37, 1989. doi:10.1111/pce.12335 |
| 48. | Singh, J.; Pandey, P.; James, D.; Chandrasekhar, K.; Achary, V. M. M.; Kaul, T.; Tripathy, B. C.; Reddy, M. K. Plant Biotechnol. J. 2014, 12, 1217. doi:10.1111/pbi.12246 |
| 48. | Singh, J.; Pandey, P.; James, D.; Chandrasekhar, K.; Achary, V. M. M.; Kaul, T.; Tripathy, B. C.; Reddy, M. K. Plant Biotechnol. J. 2014, 12, 1217. doi:10.1111/pbi.12246 |
| 24. | Fuchs, G. Annu. Rev. Microbiol. 2011, 65, 631. doi:10.1146/annurev-micro-090110-102801 |
| 20. | Li, H.; Liao, J. C. Energy Environ. Sci. 2013, 6, 2892. doi:10.1039/c3ee41847b |
| 24. | Fuchs, G. Annu. Rev. Microbiol. 2011, 65, 631. doi:10.1146/annurev-micro-090110-102801 |
| 158. | Michel, H. Angew. Chem., Int. Ed. 2012, 51, 2516. doi:10.1002/anie.201200218 |
| 54. | Ljungdahl, L.; Wood, H. G. Annu. Rev. Microbiol. 1969, 23, 515. doi:10.1146/annurev.mi.23.100169.002503 |
| 55. | Ragsdale, S. W.; Pierce, E. Biochim. Biophys. Acta, Proteins Proteomics 2008, 1784, 1873. doi:10.1016/j.bbapap.2008.08.012 |
| 157. | Cuaresma, M.; Janssen, M.; Vilchez, C.; Wijffels, R. H. Bioresour. Technol. 2011, 102, 5129. doi:10.1016/j.biortech.2011.01.078 |
| 23. | Ducat, D. C.; Silver, P. A. Curr. Opin. Chem. Biol. 2012, 16, 337. doi:10.1016/j.cbpa.2012.05.002 |
| 26. | Erb, T. J. Appl. Environ. Microbiol. 2011, 77, 8466. doi:10.1128/AEM.05702-11 |
| 160. | Guo, K.; Prévoteau, A.; Patil, S. A.; Rabaey, K. Curr. Opin. Biotechnol. 2015, 33, 149. doi:10.1016/j.copbio.2015.02.014 |
| 161. | Freguia, S.; Teh, E. H.; Boon, N.; Leung, K. M.; Keller, J.; Rabaey, K. Bioresour. Technol. 2010, 101, 1233. doi:10.1016/j.biortech.2009.09.054 |
| 162. | Rabaey, K.; Rodríguez, J.; Blackall, L. L.; Keller, J.; Gross, P.; Batstone, D.; Verstraete, W.; Nealson, K. H. ISME J. 2007, 1, 9. doi:10.1038/ismej.2007.4 |
| 26. | Erb, T. J. Appl. Environ. Microbiol. 2011, 77, 8466. doi:10.1128/AEM.05702-11 |
| 20. | Li, H.; Liao, J. C. Energy Environ. Sci. 2013, 6, 2892. doi:10.1039/c3ee41847b |
| 159. | Hawkins, A. S.; Han, Y.; Lian, H.; Loder, A. J.; Menon, A. L.; Iwuchukwu, I. J.; Keller, M.; Leuko, T. T.; Adams, M. W. W.; Kelly, R. M. ACS Catal. 2011, 1, 1043. doi:10.1021/cs2003017 |
| 52. | Buchanan, B. B.; Arnon, D. I. Photosynth. Res. 1990, 24, 47. doi:10.1007/BF00032643 |
| 53. | Evans, M. C. W.; Buchanan, B. B.; Arnon, D. I. Proc. Natl. Acad. Sci. U. S. A. 1966, 55, 928. doi:10.1073/pnas.55.4.928 |
| 50. | Hügler, M.; Sievert, S. M. Annu. Rev. Mar. Sci. 2011, 3, 261. doi:10.1146/annurev-marine-120709-142712 |
| 51. | Hugler, M.; Huber, H.; Stetter, K. O.; Fuchs, G. Arch. Microbiol. 2003, 179, 160. |
| 20. | Li, H.; Liao, J. C. Energy Environ. Sci. 2013, 6, 2892. doi:10.1039/c3ee41847b |
| 50. | Hügler, M.; Sievert, S. M. Annu. Rev. Mar. Sci. 2011, 3, 261. doi:10.1146/annurev-marine-120709-142712 |
| 110. | Lupa, B.; Lyon, D.; Shaw, L. N.; Sieprawska-Lupa, M.; Wiegel, J. Can. J. Microbiol. 2008, 54, 75. doi:10.1139/W07-113 |
| 101. | Yoshida, T.; Fujita, K.; Nagasawa, T. Biosci., Biotechnol., Biochem. 2002, 66, 2388. doi:10.1271/bbb.66.2388 |
| 105. | Wuensch, C.; Pavkov-Keller, T.; Steinkellner, G.; Gross, J.; Fuchs, M.; Hromic, A.; Lyskowski, A.; Fauland, K.; Gruber, K.; Glueck, S. M.; Faber, K. Adv. Synth. Catal. 2015, 357, 1909. doi:10.1002/adsc.201401028 |
| 106. | Pesci, L.; Glueck, S. M.; Gurikov, P.; Smirnova, I.; Faber, K.; Liese, A. FEBS J. 2015, 282, 1334. doi:10.1111/febs.13225 |
| 107. | Wuensch, C.; Gross, J.; Steinkellner, G.; Lyskowski, A.; Gruber, K.; Glueck, S. M.; Faber, K. RSC Adv. 2014, 4, 9673. doi:10.1039/c3ra47719c |
| 108. | Wuensch, C.; Glueck, S. M.; Gross, J.; Koszelewski, D.; Schober, M.; Faber, K. Org. Lett. 2012, 14, 1974. doi:10.1021/ol300385k |
| 109. | Wuensch, C.; Schmidt, N.; Gross, J.; Grischek, B.; Glueck, S. M.; Faber, K. J. Biotechnol. 2013, 168, 264. doi:10.1016/j.jbiotec.2013.07.017 |
| 102. | Yoshida, T.; Inami, Y.; Matsui, T.; Nagasawa, T. Biotechnol. Lett. 2010, 32, 701. doi:10.1007/s10529-010-0210-3 |
| 99. | Yoshida, T.; Nagasawa, T. J. Biosci. Bioeng. 2000, 89, 111. doi:10.1016/S1389-1723(00)88723-X |
| 100. | Wieser, M.; Yoshida, T.; Nagasawa, T. J. Mol. Catal. B: Enzym. 2001, 11, 179. doi:10.1016/S1381-1177(00)00038-2 |
| 101. | Yoshida, T.; Fujita, K.; Nagasawa, T. Biosci., Biotechnol., Biochem. 2002, 66, 2388. doi:10.1271/bbb.66.2388 |
| 102. | Yoshida, T.; Inami, Y.; Matsui, T.; Nagasawa, T. Biotechnol. Lett. 2010, 32, 701. doi:10.1007/s10529-010-0210-3 |
| 103. | Kirimura, K.; Gunji, H.; Wakayama, R.; Hattori, T.; Ishii, Y. Biochem. Biophys. Res. Commun. 2010, 394, 279. doi:10.1016/j.bbrc.2010.02.154 |
| 104. | Kirimura, K.; Yanaso, S.; Kosaka, S.; Koyama, K.; Hattori, T.; Ishii, Y. Chem. Lett. 2011, 40, 206. doi:10.1246/cl.2011.206 |
| 12. | Glueck, S. M.; Gümüs, S.; Fabian, W. M. F.; Faber, K. Chem. Soc. Rev. 2010, 39, 313. doi:10.1039/B807875K |
| 25. | Bar-Even, A.; Noor, E.; Lewis, N. E.; Milo, R. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 8889. doi:10.1073/pnas.0907176107 |
| 26. | Erb, T. J. Appl. Environ. Microbiol. 2011, 77, 8466. doi:10.1128/AEM.05702-11 |
| 27. | Bar-Even, A.; Noor, E.; Milo, R. J. Exp. Bot. 2012, 63, 2325. doi:10.1093/jxb/err417 |
| 28. | Knowles, J. R. Annu. Rev. Biochem. 1989, 58, 195. doi:10.1146/annurev.bi.58.070189.001211 |
| 7. | Barzagli, F.; Mani, F.; Peruzzini, M. Green Chem. 2011, 13, 1267. doi:10.1039/c0gc00674b |
| 23. | Ducat, D. C.; Silver, P. A. Curr. Opin. Chem. Biol. 2012, 16, 337. doi:10.1016/j.cbpa.2012.05.002 |
| 24. | Fuchs, G. Annu. Rev. Microbiol. 2011, 65, 631. doi:10.1146/annurev-micro-090110-102801 |
| 21. | Gong, F.; Liu, G.; Zhai, X.; Zhou, J.; Cai, Z.; Li, Y. Biotechnol. Biofuels 2015, 8, 86. doi:10.1186/s13068-015-0268-1 |
| 112. | Bar-Even, A.; Noor, E.; Flamholz, A.; Milo, R. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 1039. doi:10.1016/j.bbabio.2012.10.013 |
| 12. | Glueck, S. M.; Gümüs, S.; Fabian, W. M. F.; Faber, K. Chem. Soc. Rev. 2010, 39, 313. doi:10.1039/B807875K |
| 19. | Hawkins, A. S.; McTernan, P. M.; Lian, H.; Kelly, R. M.; Adams, M. W. W. Curr. Opin. Biotechnol. 2013, 24, 376. doi:10.1016/j.copbio.2013.02.017 |
| 22. | Matthessen, R.; Fransaer, J.; Binnemans, K.; De Vos, D. E. Beilstein J. Org. Chem. 2014, 10, 2484. doi:10.3762/bjoc.10.260 |
| 100. | Wieser, M.; Yoshida, T.; Nagasawa, T. J. Mol. Catal. B: Enzym. 2001, 11, 179. doi:10.1016/S1381-1177(00)00038-2 |
| 19. | Hawkins, A. S.; McTernan, P. M.; Lian, H.; Kelly, R. M.; Adams, M. W. W. Curr. Opin. Biotechnol. 2013, 24, 376. doi:10.1016/j.copbio.2013.02.017 |
| 20. | Li, H.; Liao, J. C. Energy Environ. Sci. 2013, 6, 2892. doi:10.1039/c3ee41847b |
| 111. | Xia, S.; Zhao, X.; Frigo-Vaz, B.; Zheng, W.; Kim, J.; Wang, P. Bioresour. Technol. 2015, 182, 368. doi:10.1016/j.biortech.2015.01.093 |
| 25. | Bar-Even, A.; Noor, E.; Lewis, N. E.; Milo, R. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 8889. doi:10.1073/pnas.0907176107 |
| 23. | Ducat, D. C.; Silver, P. A. Curr. Opin. Chem. Biol. 2012, 16, 337. doi:10.1016/j.cbpa.2012.05.002 |
| 25. | Bar-Even, A.; Noor, E.; Lewis, N. E.; Milo, R. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 8889. doi:10.1073/pnas.0907176107 |
| 27. | Bar-Even, A.; Noor, E.; Milo, R. J. Exp. Bot. 2012, 63, 2325. doi:10.1093/jxb/err417 |
| 36. | Long, B. M.; Badger, M. R.; Whitney, S. M.; Price, G. D. J. Biol. Chem. 2007, 282, 29323. doi:10.1074/jbc.M703896200 |
| 37. | Chen, A. H.; Robinson-Mosher, A.; Savage, D. F.; Silver, P. A.; Polka, J. K. PLoS One 2013, 8, e76127. doi:10.1371/journal.pone.0076127 |
| 38. | Lin, M. T.; Occhialini, A.; Andralojc, P. J.; Devonshire, J.; Hines, K. M.; Parry, M. A. J.; Hanson, M. R. Plant J. 2014, 79, 1. doi:10.1111/tpj.12536 |
| 39. | Lin, M. T.; Occhialini, A.; Andralojc, P. J.; Parry, M. A. J.; Hanson, M. R. Nature 2014, 513, 547. doi:10.1038/nature13776 |
| 40. | Price, G. D.; Pengelly, J. J. L.; Forster, B.; Du, J.; Whitney, S. M.; von Caemmerer, S.; Badger, M. R.; Howitt, S. M.; Evans, J. R. J. Exp. Bot. 2013, 64, 753. doi:10.1093/jxb/ers257 |
| 26. | Erb, T. J. Appl. Environ. Microbiol. 2011, 77, 8466. doi:10.1128/AEM.05702-11 |
| 28. | Knowles, J. R. Annu. Rev. Biochem. 1989, 58, 195. doi:10.1146/annurev.bi.58.070189.001211 |
| 35. | Chen, A. H.; Robinson-Mosher, A.; Savage, D. F.; Silver, P. A.; Polka, J. K. PLoS One 2013, 8, e76127. doi:10.1371/journal.pone.0076127 |
| 36. | Long, B. M.; Badger, M. R.; Whitney, S. M.; Price, G. D. J. Biol. Chem. 2007, 282, 29323. doi:10.1074/jbc.M703896200 |
| 28. | Knowles, J. R. Annu. Rev. Biochem. 1989, 58, 195. doi:10.1146/annurev.bi.58.070189.001211 |
| 32. | Tong, L. Cell. Mol. Life Sci. 2013, 70, 863. doi:10.1007/s00018-012-1096-0 |
| 17. | Ort, D. R.; Merchant, S. S.; Alric, J.; Barkan, A.; Blankenship, R. E.; Bock, R.; Croce, R.; Hanson, M. R.; Hibberd, J. M.; Long, S. P.; Moore, T. A.; Moroney, J.; Niyogi, K. K.; Parry, M. A. J.; Peralta-Yahya, P. P.; Prince, R. C.; Redding, K. E.; Spalding, M. H.; van Wijk, K. J.; Vermaas, W. F. J.; von Caemmerer, S.; Weber, A. P. M.; Yeates, T. O.; Yuan, J. S.; Zhu, X. G. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 8529. doi:10.1073/pnas.1424031112 |
| 33. | Christin, P.-A.; Arakaki, M.; Osborne, C. P.; Bräutigam, A.; Sage, R. F.; Hibberd, J. M.; Kelly, S.; Covshoff, S.; Wong, G. K.-S.; Hancock, L.; Edwards, E. J. J. Exp. Bot. 2014, 65, 3609. doi:10.1093/jxb/eru087 |
| 34. | Winter, K.; Holtum, J. A. M. J. Exp. Bot. 2014, 65, 3425. doi:10.1093/jxb/eru063 |
| 30. | Moroney, J. V.; Ma, Y.; Frey, W. D.; Fusilier, K. A.; Pham, T. T.; Simms, T. A.; DiMario, R. J.; Yang, J.; Mukherjee, B. Photosynth. Res. 2011, 109, 133. doi:10.1007/s11120-011-9635-3 |
| 31. | Khalifah, R. G. Proc. Natl. Acad. Sci. U. S. A. 1973, 70, 1986. doi:10.1073/pnas.70.7.1986 |
© 2015 Alissandratos and Easton; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)
