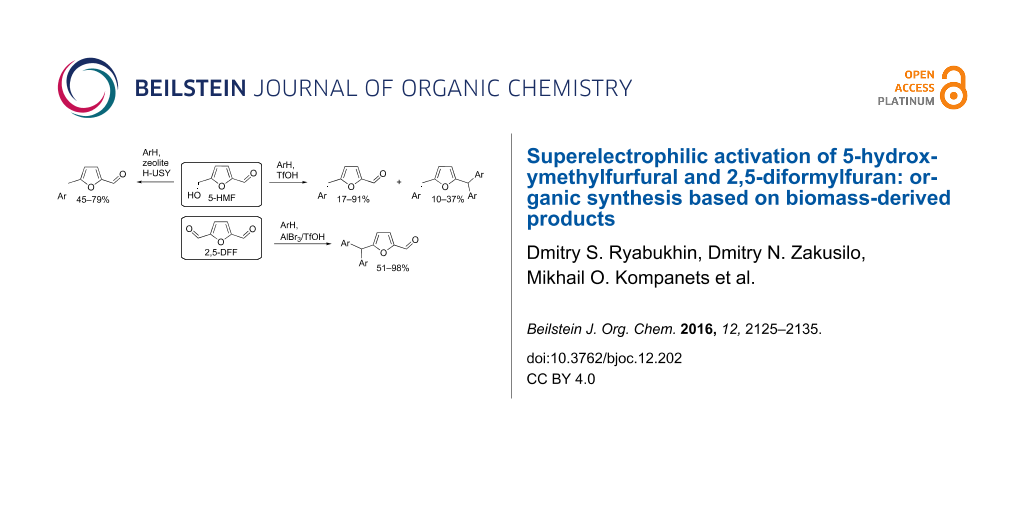
Abstract
The reaction of 5-hydroxymethylfurfural (5-HMF) with arenes in superacidic trifluoromethanesulfonic acid (triflic acid, TfOH) as the solvent at room temperature for 1–24 h gives rise to 5-arylmethylfurfurals (yields of 17–91%) and 2-arylmethyl-5-(diarylmethyl)furans (yields of 10–37%). The formation of these two types of reaction products depends on the nucleophilicity of the arene. The same reactions under the action of acidic zeolites H-USY in high pressure tubes at 130 °C for 1 h result in the formation of only 5-arylmethylfurfurals (yields of 45–79%). 2,5-Diformylfuran (2,5-DFF) in the reaction with arenes under the action of AlBr3 at room temperature for 1 h leads to 5-(diarylmethyl)furfurals (yields of 51–90%). The reactive protonated species of 5-HMF and 2,5-DFF were characterized by NMR spectroscopy in TfOH and studied by DFT calculations. These reactions show possibilities of organic synthesis based on biomass-derived 5-HMF and 2,5-DFF.

Graphical Abstract
Introduction
Nowadays great attention is paid to the use of renewable resources for obtaining fine chemicals and fuels (see numerous reviews [1-16]). The biorefinery of renewable lignin-carbohydrate materials affords various low-molecular weight organic molecules, such as alcohols, carboxylic acids, (hetero)aromatic ketones and aldehydes, phenols, etc. These biomass-derived platform chemicals are considered as an alternative and displacement to petroleum chemistry [17,18]. Among all these compounds, the preparation and reaction of 5-hydroxymethylfurfural (5-HMF) attracts special attention (see fundamental review from 2013 [19] and recent papers [20-25]). The high functionality and reactivity of 5-HMF, due to the presence of oxymethyl and aldehyde substituents, along with the furan moiety, allows many transformations and therefore the production of new useful organic substances [26-30].
Based on our recent study on the synthesis of 5-HMF and its oxidation to 2,5-diformylfuran (2,5-DFF) [31] (Figure 1), this work is focused on developing methods of organic synthesis on the basis of electrophilic activation of these biomass-derived products.
![[1860-5397-12-202-1]](/bjoc/content/figures/1860-5397-12-202-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Formation of 5-HMF from D-glucose or D-fructose followed by oxidation to 2,5-DFF.
Figure 1: Formation of 5-HMF from D-glucose or D-fructose followed by oxidation to 2,5-DFF.
Superelectrophilic activation is the generation of highly reactive di-, tri- (or even higher) cationic species by protonation and protosolvation of organic molecules with low nucleophilic Brønsted superacids, such as CF3SO3H (TfOH) or FSO3H [32]. The same activation may be achieved with strong Lewis acids (AlX3, X = Cl, Br) by their coordination with basic centers of organic compounds, or with acidic zeolites, possessing both Brønsted and Lewis acidity [33].
The main goal of this work was a study of reactions of 5-HMF and 2,5-DFF with arenes under electrophilic activation with Brønsted and Lewis superacids. Previously superelectrophilic activation of aldehyde groups was achieved for heteroaromatic aldehydes [34-37], substituted benzaldehydes and o-phthalic dicarboxaldehyde [38]. Based on these findings, one would expect the activation of an aldehyde group of 5-HMF and 2,5-DFF, and its participation in the hydroxyalkylation of arenes. However, furan carboxaldehydes in such reactions were studied in this work for the first time.
It should be noted, that reactions of arenes with hydroxymethyl and aldehyde groups of 5-HMF and 2,5-DFF may lead to various arylmethyl- and diarylmethyl-substituted furans, which are otherwise hardly available molecules [39-46] and used for the synthesis of bioactive compounds [47]. Thus, the superelectrophilic activation of 5-HMF and 2,5-DFF could be of great value for organic synthesis.
Results and Discussion
The protonation of the carbonyl oxygen and hydroxy group of 5-HMF (1a) in strong acids gives rise to cationic species A, the dehydration of the latter may result in the formation of heteroaromatic benzyl-type dication B (Scheme 1). Protonation of the aldehyde groups in 2,5-DFF (2) leads to cation C and dication D (Scheme 1). All species A, B, C, and D may play a role as reactive intermediates derived from 1a and 2 in superacids. To estimate the electrophilic properties of cations A, B, C, and D we performed quantum chemical calculations by the DFT method (Table 1). HOMO and LUMO energies, global electrophilicity index ω [48,49], charge distribution, and contribution of atomic orbitals into the LUMO were calculated. The dications B and D having high ω values of 9.6 and 8 eV, respectively, should be very reactive electrophiles. The carbon atom of the protonated aldehyde group in species A, B, C, and D bears a large positive charge and shows a great contribution in the LUMO. This indicates that this carbon atom may be an electrophilic reactive center from both charge and orbital point of view. Apart from that, the increase of positive charge on heteroaromatic carbons C1, C4 and the decrease of negative charge on atoms C2, C3, and Ofuran upon protonation of furans 1a and 2 reveal a significant positive charge delocalization into the furan ring in species A, B, C, and D. However, from these calculations no unambiguous answer could be obtained that reveals what cations in pairs A or B, and C or D may take part in electrophilic reactions.
![[1860-5397-12-202-i1]](/bjoc/content/inline/1860-5397-12-202-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Protonation of 5-HMF (1a) and 2,5-DFF (2) leading to cationic species A, B, C, D.
Scheme 1: Protonation of 5-HMF (1a) and 2,5-DFF (2) leading to cationic species A, B, C, D.
Table 1: Selected electronic characteristics (DFT calculations) of starting furans 1a, 2 and cationic species A, B, C, and D.
| Species | EHOMO, eV | ELUMO, eV | ω,a eV | q(CH2),b e | q(C=O),b e | k(CH2)LUMO,с % | k(C=O)LUMO,с % |
|---|---|---|---|---|---|---|---|
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-202-i2.svg?max-width=637&scale=1.0)
1a |
−6.82 | −2.20 | 2.2 | −0.066 | 0.379 | 0.6 | 27.8 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-202-i3.svg?max-width=637&scale=1.0)
A |
−8.93 | −4.59 | 5.3 | −0.060 | 0.428 (COH+) | 6.9 |
26.3
(COH+) |
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-202-i4.svg?max-width=637&scale=1.0)
B |
−10.43 | −6.65 | 9.6 | 0.054 | 0.528 (COH+) | 26.1 |
24.0
(COH+) |
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-202-i5.svg?max-width=637&scale=1.0)
2 |
−7.43 | −3.06 | 3.1 | – | 0.391 | – | 15.4 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-12-202-i6.svg?max-width=637&scale=1.0)
C |
−8.57 | −4.60 | 5.5 | – |
0.400
0.417 (COH+) |
− |
5.7
31.2 (COH+) |
![[Graphic 6]](/bjoc/content/inline/1860-5397-12-202-i7.svg?max-width=637&scale=1.0)
D |
−9.78 | −5.91 | 8.0 | – | 0.488 (COH+) | – |
22.9
(COH+) |
| Species | q(C1),b e | q(C2),b e | q(C3),b e | q(C4),b e | q(Ofuran),b e | ||
![[Graphic 7]](/bjoc/content/inline/1860-5397-12-202-i8.svg?max-width=637&scale=1.0)
1a |
0.152 | −0.192 | −0.302 | 0.352 | −0.464 | ||
![[Graphic 8]](/bjoc/content/inline/1860-5397-12-202-i9.svg?max-width=637&scale=1.0)
A |
0.158 | −0.097 | −0.220 | 0.371 | −0.406 | ||
![[Graphic 9]](/bjoc/content/inline/1860-5397-12-202-i10.svg?max-width=637&scale=1.0)
B |
0.313 | −0.162 | −0.010 | 0.209 | −0.374 | ||
![[Graphic 10]](/bjoc/content/inline/1860-5397-12-202-i11.svg?max-width=637&scale=1.0)
2 |
0.210 | −0.209 | −0.209 | 0.210 | −0.568 | ||
![[Graphic 11]](/bjoc/content/inline/1860-5397-12-202-i12.svg?max-width=637&scale=1.0)
C |
0.161 | −0.106 | −0.217 | 0.330 | −0.404 | ||
![[Graphic 12]](/bjoc/content/inline/1860-5397-12-202-i13.svg?max-width=637&scale=1.0)
D |
0.228 | −0.119 | −0.119 | 0.228 | −0.376 | ||
aGlobal electrophilicity index ω = (EHOMO + ELUMO)2/8(ELUMO − EHOMO). bNatural charges. cContribution of atomic orbitals into the molecular orbital.
To investigate this issue in more detail we studied the protonation of furans 1a and 2 in the superacid TfOH by NMR spectroscopy. Upon dissolving 1a and 2 in TfOH in the NMR tube the formation of deep-red solutions was observed. 1H and 13C NMR data of the generated cationic species are collected in Table 2 (see spectral figures in Supporting Information File 1). The spectral data clearly prove that protonation of 1a in TfOH gives rise to dication A (Scheme 1). Thus, in the 13C NMR spectrum the signal at δ 67.5 ppm belongs to the protonated hydroxymethyl group CH2O+H2. This means that dehydration of this group does not occur in TfOH and cation B is not formed. For comparison, chemical shifts of carbocationic centers in various benzyl cations (R2)ArC+ lie in a much more down-field range of ~182–270 ppm [50-57]. Comparison of 1H and 13C NMR spectra shows that signals of protons and carbons in the furan ring of A are substantially down-field shifted in comparison with the corresponding signals of its neutral precursor 1a. This reveals a significant positive charge delocalization in the furan ring in A that is in good agreement with the DFT calculations (vide supra).
Table 2: 1H and 13C NMR data of furans 1a, 2 in CDCl3 and species A, D in TfOH at room temperature.
| Species | Solvent | NMR data | |
|---|---|---|---|
| 1H | 13C | ||
![[Graphic 13]](/bjoc/content/inline/1860-5397-12-202-i14.svg?max-width=637&scale=1.0)
1a |
CDCl3 | 2.84 (s, 1H, OH), 4.71 (s, 2H, CH2), 6.51 (d,(J = 3.5 Hz, 1H, H2), 7.21 (d, J = 3.5 Hz, 1H, H3), 9.57 (s, 1H, CHO) | 57.6 (CH2), 109.9 (C2), 122.8 (C3), 152.4 (C4), 160.7 (C1), 177.7 (CHO) |
![[Graphic 14]](/bjoc/content/inline/1860-5397-12-202-i15.svg?max-width=637&scale=1.0)
A |
TfOH | 5.85 (s, 2H, CH2), 7.44 (d, J = 4.2 Hz, 1H, H2), 8.79 (d, J = 4.2 Hz, 1H, H3), 9.04 (s, C=OH+) | 67.5 (CH2), 121.0 (C2), 148.5 (C3), 152.8 (C4), 171.9 (C1), 175.9 (C=OH+) |
![[Graphic 15]](/bjoc/content/inline/1860-5397-12-202-i16.svg?max-width=637&scale=1.0)
2 |
CDCl3 | 7.33 (s, 2H, H2), 9.86 (s, CHO) | 119.1 (C2), 154.2 (C1), 179.2 (CHO) |
![[Graphic 16]](/bjoc/content/inline/1860-5397-12-202-i17.svg?max-width=637&scale=1.0)
D |
TfOH | 8.48 (s, 2H), 9.84 (s, C=OH+) | 136.8 (C2), 156.0 (C1), 186.6 (C=OH+) |
According to the NMR data (Table 2 and Supporting Information File 1) protonation of 2 in TfOH leads to the O,O-diprotonated species D. Analogous to dication A, a down-field shift of signals of protons and carbons of the furan ring in 1H and 13C NMR spectra of D compared to the signals for 2 was observed, pointing out a charge delocalization in the furan moiety. Also, the down-field shift of the 13C signal of the protonated aldehyde group in species D indicates that this carbon should be a reactive electrophilic center. Contrary to that, the signal of the carbon of the protonated aldehyde group in A is even slightly up-field shifted compared to the same signal in 1a. This indicates weak electrophilic properties of the protonated aldehyde group in A. Signals of protons bonded to aldehyde and oxymethyl groups in species A and D were not registered in 1H NMR due to the fast proton exchange with the superacidic medium at room temperature.
Thus, the NMR data revealed that protonation of furans 1a and 2 in superacid TfOH resulted in the formation of dications A and D, respectively, although DFT calculations (Table 1) did not manifest that clearly.
Next, 5-HMF (1a), 5-(chloromethyl)furfural (1b) and 5-(bromomethyl)furfural (1c), both of which are also promising biomass-derived products [58,59], were reacted with benzene under the action of various acids (Table 3). In all cases 5-(phenylmethyl)furfural (3a) as Friedel–Crafts reaction product was obtained. Thus, only the hydroxymethyl or halogenomethyl group in furans 1a–c were involved in the reactions. The aldehyde group remained intact despite the DFT calculations predicted a high electrophilicity for the carbon of the protonated aldehyde group (Table 1). The best results (the highest yield of 3a) were achieved with TfOH at room temperature for 1 h (Table 3, entries 1, 7, and 9). Other acids were less efficient, leading to 3a in lower yields or harsher conditions (130 °C) were required for acidic zeolites H-USY, CBV-720 and CBV-500 (Table 3, entries 3, 4, 8). Since the NMR data showed the generation of dication A from 1a (Table 2), this reaction, most probably, proceeds through an SN2 pathway, where “pure” heteroaromatic cation B is not formed. At least the reaction may go through late transition state, in which the C–O bond in the CH2O+H2 group is rather elongated, resulting in a larger positive charge on this carbon than it has been predicted by calculations (see Table 1). It should be noted, that the yields of reaction products in Tables 3–5 are isolated yields after column chromatographic separation. The remaining materials are some oligomeric compounds.
Table 3: Reactions of furans 1a–c with benzene under the action of various acids.
![[Graphic 17]](/bjoc/content/inline/1860-5397-12-202-i18.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Reaction conditions | Yield of 3a,a % | |||
|---|---|---|---|---|---|
| Furan | Acid | T, °C | t, h | ||
| 1 | 1a | TfOH (20 equiv) | rt | 1 | 91 |
| 2 | 1a | TfOH (20 equiv) | rt | 1 | –b |
| 3 | 1a | zeolite CBV-500c | 130 | 3 | 47 |
| 4 | 1a | zeolite CBV-720c | 130 | 1 | 45 |
| 5 | 1a | H2SO4 (50 equiv) | rt | 1 | 19 |
| 6 | 1a | AlCl3 (5 equiv) | rt | 1 | oligomers |
| 7 | 1b | TfOH (20 equiv) | rt | 1 | 75 |
| 8 | 1b | zeolite CBV-500 | 130 | 3 | 25 |
| 9 | 1c | TfOH (20 equiv) | rt | 1 | 68 |
aIsolated yields. bReaction was carried out without benzene, and starting 1a was quantitatively recovered. cThe ratio of 1a–c/zeolite Brønsted acidic sites was around 2:1.
Using these conditions (TfOH, rt, 1 h), we carried out reactions of 5-HMF (1a) with various arenes in TfOH. Additionally we investigated the reactions under the action of zeolite CBV-720, since zeolites are considered as “green” catalysts in organic synthesis [60-66] and the data are collected in Table 4. Similarly to the reaction with benzene (Table 3), 1a with other arenes yielded 5-arylmethylfurfurals 3b–o in TfOH or with zeolite CBV-720. However, the use of activated arenes, such as toluene, xylenes and pseudocumene, afforded additional Friedel–Crafts products, namely furans 4a–e (Table 4, entries 1, 2, 4–8, 11–13, and 15). These compounds were formed by hydroxyalkylation due to the interaction of species A with arenes (see related reactions [34-38]). The polymethylated arenes possess a sufficient π-nucleophilicity for the reaction with the protonated aldehyde group of the intermediates generated from 1a in TfOH. Whereas less nucleophilic arenes, such as benzene (Table 3) and 1,2-dichlorobenzene (Table 4, entries 18 and 19), did not give rise to the corresponding furans 4. Also, the reactions with anisole and veratrole led only to furans 3l, m and 3n, respectively, and no formation of compounds 4 was observed (Table 4, entries 20–23). Under the superacidic conditions the substrates anisole and veratrole may be protonated at the oxygen atoms [33], thus leading to a decreased π-nucleophilicity of these arenes. It should be noted that in the case of zeolites we explored CS2 as low coordinating solvent to avoid blocking zeolite acidic sites by π-donating arenes.
Table 4: Reactions of 5-HMF (1a) with arenes under the action of TfOH or acidic zeolite CBV-720.
![[Graphic 18]](/bjoc/content/inline/1860-5397-12-202-i19.svg?max-width=637&scale=1.0)
|
|||||||||
| Entry | ArH | Reaction conditions | Reaction productsa | Total yielda, % | |||||
|---|---|---|---|---|---|---|---|---|---|
| ArH (equiv) | acid (equiv) | T, °C | t, h | 3 | 4 | ||||
| 1 | toluene | 1.2 | TfOH (20) | rt | 1 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-12-202-i20.svg?max-width=637&scale=1.0)
3b (23%), 3c (23%) |
![[Graphic 20]](/bjoc/content/inline/1860-5397-12-202-i21.svg?max-width=637&scale=1.0)
isomers-4a (10%) |
56 | |
| 2 | toluene | 4 | TfOH (20) | rt | 3 | 3b (44%), 3c (44%) | isomers-4a (10%) | 98 | |
| 3 | toluene | 125 | CBV-720b | 130 | 1 | 3b (24%), 3c (24%) | – | 48 | |
| 4 | o-xylene | 4 | TfOH (20) | rt | 1 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-12-202-i22.svg?max-width=637&scale=1.0)
3d (32%), 3e (22%) |
![[Graphic 22]](/bjoc/content/inline/1860-5397-12-202-i23.svg?max-width=637&scale=1.0)
4b (23%) |
77 | |
| 5 | o-xylene | 4 | TfOH (20) | rt | 24 | 3d (37%), 3e (22%) | 4b (37%) | 96 | |
| 6 | m-xylene | 1.2 | TfOH (20) | rt | 1 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-12-202-i24.svg?max-width=637&scale=1.0)
3f (28%) |
![[Graphic 24]](/bjoc/content/inline/1860-5397-12-202-i25.svg?max-width=637&scale=1.0)
4c (6%) |
34 | |
| 7 | m-xylene | 4 | TfOH (20) | rt | 3 | 3f (75%) | 4c (20%) | 95 | |
| 8 | m-xylene | 4 | TfOH (20) | rt | 24 | 3f (12%) | 4c (32%) | 44 | |
| 9 | m-xylene | 4 | TfOH (20) | rt | 72 | oligomers | – | ||
| 10 | m-xylene | 2.5 | CBV-720b, CS2 | 130 | 1 | 3f (79%) | – | 79 | |
| 11 | p-xylene | 1.2 | TfOH (20) | rt | 1 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-12-202-i26.svg?max-width=637&scale=1.0)
3g (53%) |
![[Graphic 26]](/bjoc/content/inline/1860-5397-12-202-i27.svg?max-width=637&scale=1.0)
4d (18%) |
71 | |
| 12 | p-xylene | 4 | TfOH (20) | rt | 3 | 3g (91%) | 4d (5%) | 96 | |
| 13 | p-xylene | 4 | TfOH (20) | rt | 24 | 3g (21%) | 4d (17%) | 38 | |
| 14 | p-xylene | 4 | CBV-720b, CS2 | 130 | 1 | 3g (78%) | – | 78 | |
| 15 |
pseudo-
cumene |
4 | TfOH (20) | rt | 1 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-12-202-i28.svg?max-width=637&scale=1.0)
3h (28%), 3i (12%) |
![[Graphic 28]](/bjoc/content/inline/1860-5397-12-202-i29.svg?max-width=637&scale=1.0)
4e (32%) |
72 | |
| 16 |
pseudo-
cumene |
10 | TfOH (20) | rt | 24 | oligomers | – | ||
| 17 |
pseudo-
cumene |
4 | CBV-720b, CS2 | 130 | 1 | 3h (53%), 3i (23%) | – | 76 | |
| 18 | 1,2-dichloro-benzene | 4 | TfOH (20) | rt | 1 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-12-202-i30.svg?max-width=637&scale=1.0)
3j (36%), 3k (17%) |
– | 53 | |
| 19 | 1,2-dichloro-benzene | 4 | CBV-720a, CS2 | 130 | 1 | 3j (7%), 3k (4%) | – | 11 | |
| 20 | anisole | 4 | TfOH (20) | rt | 1 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-12-202-i31.svg?max-width=637&scale=1.0)
3l (42%), 3m (9%) |
– | 51 | |
| 21 | anisole | 3 | CBV-720b, CS2 | 130 | 1 | 3l (34%), 3m (18%) | – | 52 | |
| 22 | veratrole | 4 | TfOH (20) | rt | 2 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-12-202-i32.svg?max-width=637&scale=1.0)
3n (62%) |
– | 62 | |
| 23 | veratrole | 4 | CBV-720b, CS2 | 130 | 1 | 3n (21%) | – | 21 | |
| 24 | mesitylene | 5 | TfOH (20) | rt | 2 |
![[Graphic 32]](/bjoc/content/inline/1860-5397-12-202-i33.svg?max-width=637&scale=1.0)
3o (17%) |
– | 17 | |
aIsolated yields. bThe ratio of 1a/zeolite Brønsted acidic sites was around 2:1.
Individually isolated compounds 3f and 3g being dissolved in TfOH at room temperature for 24 h gave rise to mixtures containing furans 4c and 4d along with starting 3f and 3g, respectively. This may prove that compounds 4 are the secondary reaction products. It is interesting to note that compounds 4 were not observed in reactions with zeolite (Table 4, entries 3, 10, 14 and 17), most likely, due to lower acidity of the zeolite compared to TfOH and spatial restrictions in zeolite cages, diminishing the contact between protonated aldehyde groups and the substrate (arene) molecules.
We also varied reaction conditions (time, amount of arene) in TfOH to check yields of compounds 3 and 4. In general, increasing the reaction time up to 3–24 h led to an increase in the yield of furans 4 and a decreased yield of compounds 3 (compare pairs of entries in Table 4: 4 and 5, 7 and 8, 12 and 13). But during long reaction times (24–72 h) compounds 3 and 4 were completely cleaved in TfOH (Table 4, entries 9 and 16), showing complex behavior of these furans under superacidic conditions.
Concerning the reaction mechanism, according to NMR data (see Table 2) the reactive species, derived from 1a in TfOH, is cation A. In zeolite cages the activation of the CH2OH group of 1a takes place due to the coordination of the basic centers of this molecule with Brønsted and Lewis acidic sites of zeolite.
2,5-DFF (2) reacted with arenes under electrophilic activation with formation of 5-diarylmethylfurfurals 5a–g (Table 5, Figure 2). Contrary to compound 1a, which was activated with the Brønsted superacid TfOH to achieve Friedel–Crafts products 3 and 4 (Table 3 and Table 4), compound 2 gave better results with strong Lewis acids AlX3 (X = Cl, Br) (Table 5, entries 4, 5, 9, 11, 13, and 15). The coordination of aluminum halides with the aldehyde group results in the activation of one of the aldehyde groups, catalyzing the reaction subsequently with two arene molecules resulting in the 1,1-diarylated product. The second aldehyde group does not take part in the reaction (compare with data from ref. [38]). This is due to both, an insufficient electrophilic activation by these particular acids and the low nucleophilicity of benzene. Also the reaction of 2 with benzene was promoted by zeolite CBV-500, which is more acidic than CBV-720 (see Supporting Information File 1), and the latter was not be able to give rise to 5a (Table 5, entries 6–8).
Table 5: Reactions of 2,5-DFF 2 with arenes under the action of various acids.
![[Graphic 33]](/bjoc/content/inline/1860-5397-12-202-i34.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | ArH | Reaction conditions | Reaction products, 5 | ||||
|---|---|---|---|---|---|---|---|
| acid (equiv) | T, °C | t, h | substituents R in Ar | yielda (%) | |||
| 1 | benzene | TfOH (20) | rt | 2 | H | 5a (90%) | |
| 2 | benzene | H2SO4 (50) | rt | 2 | H | 5a (74%) | |
| 3 | benzene | FSO3H (20), SO2 | −45 | 2 | H | 5a (84%) | |
| 4 | benzene | AlCl3 (5) | rt | 1 | H | 5a (92%) | |
| 5 | benzene | AlBr3 (5) | rt | 1 | H | 5a (98%) | |
| 6 | benzene | CBV-500b | 130 | 1 | H | 5a (13%)c | |
| 7 | benzene | CBV-500b | 130 | 10 | H | 5a (46%) | |
| 8 | benzene | CBV-720b | 130 | 10 | H | –d | |
| 9 | o-xylene | AlBr3 (5) | rt | 1 | 3,4-Me2 | 5b (72%) | |
| 10 | o-xylene | TfOH (20) | rt | 1 | oligomers | ||
| 11 | m-xylene | AlBr3 (5) | rt | 1 | 3,5-Me2 | 5c (36%) | |
| 2,4-Me2 | 5d (15%) | ||||||
| 12 | m-xylene | TfOH (20) | rt | 1 | oligomers | ||
| 13 | p-xylene | AlBr3 (5) | rt | 1 | 2,5-Me2 | 5e (78%) | |
| 14 | p-xylene | TfOH (20) | rt | 1 | 2,5-Me2 | 5e (87%) | |
| 15 | 1,2-dichlorobenzene | TfOH (20) | rt | 1 | 3,4-Cl2 | 5f (82%) | |
| 2,3-Cl2 | 5g (16%) | ||||||
| 16 | 1,2-dichlorobenzene | AlBr3 (5) | rt | 1 | –d | ||
aIsolated yields. bThe ratio of 2/zeolite Brønsted acidic sites was around 2:1. cIncomplete conversion; 55% of starting 2 was recovered. dNo reaction and quantitative recovery of starting 2.
Electron-donating xylenes in the reaction with 2 showed a more complex behavior. The different isomeric xylenes led to compounds 5b–e in the presence of AlBr3 (Table 5, entries 9, 11, and 13). On the other hand, in the Brønsted superacid TfOH o- and m-xylenes gave rise to complex oligomeric mixtures (Table 5, entries 10 and 12) having masses up to 1400–1500 Da according to MALDI–MS (see Supporting Information File 1). These oligomers may have formed due to the participation of both aldehyde groups of species D (see Scheme 1 and Table 2) in the reaction with these electron-rich arenes. Under the action of AlBr3 2,5-DFF did not react with 1,2-dichlorobenzene, due to a too low nucleophilicity of the latter. However, this reaction took place in TfOH (Table 5, entries 15 and 16).
![[1860-5397-12-202-2]](/bjoc/content/figures/1860-5397-12-202-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray crystal structure of compounds 5a (a), and 5c (b) (ORTEP diagrams, ellipsoid contour of probability levels is 50%, CCDC reference numbers 5a: 1483523, 5c: 1483524).
Figure 2: X-ray crystal structure of compounds 5a (a), and 5c (b) (ORTEP diagrams, ellipsoid contour of proba...
Conclusion
We have developed a simple and effective synthesis of various arylmethyl and diarylmethyl-substituted furans by reactions of 5-HMF and 2,5-DFF with arenes under electrophilic activation by Brønsted/Lewis (super)acids or acidic zeolites H-USY.
In these reactions 5-HMF in TfOH gives rise to 5-(arylmethyl)furfurals and 2-(arylmethyl)-5-(diarylmethyl)furans. The latter compounds are formed in reactions with donating arenes. Reactions of 5-HMF with arenes under the action of acidic zeolites H-USY result in the selective formation of only 5-(arylmethyl)furfurals. 2,5-DFF in reactions with arenes under the action of AlBr3 leads solely to 5-(diarylmethyl)furfurals.
The electrophilic intermediates derived from the protonation of 5-HMF and 2,5-DFF were investigated by means of DFT calculations and NMR in the superacid TfOH. These reactions are a contribution to organic syntheses based on biomass-derived products 5-HMF and 2,5-DFF.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization of compounds, 1H, 13C, 19F NMR spectra, and data on DFT calculations. | ||
| Format: PDF | Size: 4.4 MB | Download |
References
-
Barta, K.; Ford, P. C. Acc. Chem. Res. 2014, 47, 1503–1512. doi:10.1021/ar4002894
Return to citation in text: [1] -
Colmenares, J. C.; Luque, R. Chem. Soc. Rev. 2014, 43, 765–778. doi:10.1039/c3cs60262a
Return to citation in text: [1] -
Straathof, A. J. J. Chem. Rev. 2014, 114, 1871–1908. doi:10.1021/cr400309c
Return to citation in text: [1] -
Besson, M.; Gallezot, P.; Pinel, C. Chem. Rev. 2014, 114, 1827–1870. doi:10.1021/cr4002269
Return to citation in text: [1] -
Moiseev, I. I. Russ. Chem. Rev. 2013, 82, 616–623. doi:10.1070/RC2013v082n07ABEH004393
Return to citation in text: [1] -
Alonso, D. M.; Wettstein, S. G.; Dumesic, J. A. Chem. Soc. Rev. 2012, 41, 8075–8098. doi:10.1039/c2cs35188a
Return to citation in text: [1] -
Gallezot, P. Chem. Soc. Rev. 2012, 41, 1538–1558. doi:10.1039/C1CS15147A
Return to citation in text: [1] -
Ponomarev, A. V.; Ershov, B. G. Russ. Chem. Rev. 2012, 81, 918–935. doi:10.1070/RC2012v081n10ABEH004266
Return to citation in text: [1] -
Sun, N.; Rodriguez, H.; Rahman, M.; Rogers, R. D. Chem. Commun. 2011, 47, 1405–1421. doi:10.1039/C0CC03990J
Return to citation in text: [1] -
Varfolomeev, S. D.; Efremenko, E. N.; Krylova, L. P. Russ. Chem. Rev. 2010, 79, 491–510. doi:10.1070/RC2010v079n06ABEH004138
Return to citation in text: [1] -
Marshall, A.-L.; Alaimo, P. Chem. – Eur. J. 2010, 16, 4970–4980. doi:10.1002/chem.200903028
Return to citation in text: [1] -
Stöcker, M. Angew. Chem., Int. Ed. 2008, 47, 9200–9211. doi:10.1002/anie.200801476
Return to citation in text: [1] -
Chheda, J. N.; Huber, G. W.; Dumesic, J. A. Angew. Chem., Int. Ed. 2007, 46, 7164–7183. doi:10.1002/anie.200604274
Return to citation in text: [1] -
Huber, G. W.; Corma, A. Angew. Chem., Int. Ed. 2007, 46, 7184–7201. doi:10.1002/anie.200604504
Return to citation in text: [1] -
Corma, A.; Iborra, S.; Velty, A. Chem. Rev. 2007, 107, 2411–2502. doi:10.1021/cr050989d
Return to citation in text: [1] -
Huber, G. W.; Iborra, S.; Corma, A. Chem. Rev. 2006, 106, 4044–4098. doi:10.1021/cr068360d
Return to citation in text: [1] -
Bozell, J. J.; Petersen, G. R. Green Chem. 2010, 12, 539–554. doi:10.1039/B922014C
Return to citation in text: [1] -
Kamm, B. Angew. Chem., Int. Ed. 2007, 46, 5056–5058. doi:10.1002/anie.200604514
Return to citation in text: [1] -
van Putten, R.-J.; van der Waal, J. C.; de Jong, E.; Rasrendra, C. B.; Heeres, H. J.; de Vries, J. G. Chem. Rev. 2013, 113, 1499–1597. doi:10.1021/cr300182k
Return to citation in text: [1] -
Kashin, A. S.; Galkin, K. I.; Khokhlova, E. A.; Ananikov, V. P. Angew. Chem., Int. Ed. 2016, 55, 2161–2166. doi:10.1002/anie.201510090
Return to citation in text: [1] -
Galkin, K. I.; Krivodaeva, E. A.; Romashov, L. V.; Zalesskiy, S. S.; Kachala, V. V.; Burykina, J. V.; Ananikov, V. P. Angew. Chem., Int. Ed. 2016, 55, 8338–8342. doi:10.1002/anie.201602883
Return to citation in text: [1] -
Li, W.; Xu, Z.; Zhang, T.; Li, G.; Jameel, H.; Chang, H.; Ma, L. BioResources 2016, 11, 5839–5853. doi:10.15376/biores.11.3.5839-5853
Return to citation in text: [1] -
Choudhary, V.; Mushrif, S. H.; Ho, C.; Anderko, A.; Nikolakis, V.; Marinkovic, N. S.; Frenkel, A. I.; Sandler, S. I.; Vlachos, D. G. J. Am. Chem. Soc. 2013, 135, 3997–4006. doi:10.1021/ja3122763
Return to citation in text: [1] -
Hu, L.; Zhao, C.; Tang, X.; Wu, Z.; Xu, J.; Lin, L.; Liu, S. Bioresour. Technol. 2013, 148, 501–507. doi:10.1016/j.biortech.2013.09.016
Return to citation in text: [1] -
Yang, J.; De OliveiraVigier, K.; Gu, Y.; Jérôme, F. ChemSusChem 2015, 8, 269–274. doi:10.1002/cssc.201402761
Return to citation in text: [1] -
Iesce, M. R.; Sferruzza, R.; Cermola, F.; DellaGreca, M. Helv. Chim. Acta 2016, 99, 296–301. doi:10.1002/hlca.201500196
Return to citation in text: [1] -
Palframan, M. J.; Pattenden, G. Chem. Commun. 2014, 50, 7223–7242. doi:10.1039/c4cc01196a
Return to citation in text: [1] -
Dijkman, W. P.; Groothuis, D. E.; Fraaije, M. W. Angew. Chem., Int. Ed. 2014, 53, 6515–6518. doi:10.1002/anie.201402904
Return to citation in text: [1] -
Ghezali, W.; De Oliveira Vigier, K.; Kessas, R.; Jérôme, F. Green Chem. 2015, 17, 4459–4464. doi:10.1039/C5GC01336D
Return to citation in text: [1] -
Liu, F.; Audemar, M.; De Oliveira Vigier, K.; Clacens, J.-M.; De Campo, F.; Jérôme, F. ChemSusChem 2014, 7, 2089–2093. doi:10.1002/cssc.201402221
Return to citation in text: [1] -
Kompanets, M. O.; Kushch, O. V.; Litvinov, Yu. E.; Pliekhov, O. L.; Novikova, K. V.; Novokhatko, A. O.; Shendrick, A. N.; Vasilyev, A. V.; Opeida, I. O. Catal. Commun. 2014, 57, 60–63. doi:10.1016/j.catcom.2014.08.005
Return to citation in text: [1] -
Olah, G. A.; Klumpp, D. A. Superelectrophiles and Their Chemistry; Wiley: New York, 2008.
Return to citation in text: [1] -
Olah, G. A.; Prakash, G. K. S.; Molnar, A.; Sommer, J. Superacid Chemistry, 2nd ed.; Wiley: New Jersey, 2009.
Return to citation in text: [1] [2] -
Prakash, G. K. S.; Paknia, F.; Chacko, S.; Mathew, T.; Olah, G. A. Heterocycles 2008, 76, 783–799. doi:10.3987/COM-08-S(N)90
Return to citation in text: [1] [2] -
Klumpp, D. A.; Jones, A.; Lau, S.; de Leon, S.; Garza, M. Synthesis 2000, 1117–1120. doi:10.1055/s-2000-6322
Return to citation in text: [1] [2] -
Klumpp, D. A.; Kindelin, P. J.; Li, A. Tetrahedron Lett. 2005, 46, 2931–2935. doi:10.1016/j.tetlet.2005.02.152
Return to citation in text: [1] [2] -
Sheets, M. R.; Li, A.; Bower, E. A.; Weigel, A. R.; Abbot, M. P.; Gallo, R. M.; Mitton, A. M.; Klumpp, D. A. J. Org. Chem. 2009, 74, 2502–2507. doi:10.1021/jo802798x
Return to citation in text: [1] [2] -
Prakash, G. K. S.; Panja, C.; Shakhmin, A.; Shah, E.; Mathew, T.; Olah, G. A. J. Org. Chem. 2009, 74, 8659–8668. doi:10.1021/jo901668j
Return to citation in text: [1] [2] [3] -
Cabares, J.; Mavoungou-Gomes, L. Bull. Soc. Chim. Fr. 1986, 401–412.
Return to citation in text: [1] -
Skrzyńska, A.; Przydacz, A.; Albrecht, Ł. Org. Lett. 2015, 17, 5682–5685. doi:10.1021/acs.orglett.5b02979
Return to citation in text: [1] -
Onorato, A.; Pavlik, C.; Invernale, M. A.; Berghorn, I. D.; Sotzing, G. A.; Morton, M. D.; Smith, M. B. Carbohydr. Res. 2011, 346, 1662–1670. doi:10.1016/j.carres.2011.04.017
Return to citation in text: [1] -
Ager, D. J. Tetrahedron Lett. 1983, 24, 5441–5444. doi:10.1016/S0040-4039(00)94107-8
Return to citation in text: [1] -
Watanabe, N.; Matsugi, A.; Nakano, K.; Ichikawa, Y.; Kotsuki, H. Synlett 2014, 25, 438–442. doi:10.1055/s-0033-1340343
Return to citation in text: [1] -
Gomes, R. F. A.; Coelho, J. A. S.; Frade, R. F. M.; Trindade, A. F.; Afonso, C. A. M. J. Org. Chem. 2015, 80, 10404–10411. doi:10.1021/acs.joc.5b01875
Return to citation in text: [1] -
Pérez, M.; Mahdi, T.; Hounjet, L. J.; Stephan, D. W. Chem. Commun. 2015, 51, 11301–11304. doi:10.1039/C5CC03572D
Return to citation in text: [1] -
Miao, M.; Luo, Y.; Li, H.; Xu, X.; Chen, Z.; Xu, J.; Ren, H. J. Org. Chem. 2016, 81, 5228–5235. doi:10.1021/acs.joc.6b00734
Return to citation in text: [1] -
Villain-Guillot, P.; Gualtieri, M.; Bastide, L.; Roquet, F.; Martinez, J.; Amblard, M.; Pugniere, M.; Leonetti, J.-P. J. Med. Chem. 2007, 50, 4195–4204. doi:10.1021/jm0703183
Return to citation in text: [1] -
Parr, R. G.; van Szentpály, L.; Liu, S. J. Am. Chem. Soc. 1999, 121, 1922–1924. doi:10.1021/ja983494x
Return to citation in text: [1] -
Chattaraj, P. K.; Giri, S.; Duley, S. Chem. Rev. 2011, 111, PR43–PR75. doi:10.1021/cr100149p
Return to citation in text: [1] -
Alkhafaji, H. M. H.; Ryabukhin, D. S.; Muzalevskiy, V. M.; Vasilyev, A. V.; Fukin, G. K.; Shastin, A. V.; Nenajdenko, V. G. Eur. J. Org. Chem. 2013, 1132–1143. doi:10.1002/ejoc.201201375
Return to citation in text: [1] -
Alkhafaji, H. M. H.; Ryabukhin, D. S.; Muzalevskiy, V. M.; Osetrova, L. V.; Vasilyev, A. V.; Nenajdenko, V. G. Russ. J. Org. Chem. 2013, 49, 327–341. doi:10.1134/S1070428013030032
Return to citation in text: [1] -
Koltunov, K. Y.; Shakirov, M. M.; Repinskaya, I. B.; Koptug, V. A. Zh. Org. Khim. 1991, 27, 2622–2623.
Return to citation in text: [1] -
Koltunov, K. Y.; Repinskaya, I. B. Zh. Org. Khim. 1993, 30, 90–93.
Return to citation in text: [1] -
Walspurger, S.; Vasilyev, A. V.; Sommer, J.; Pale, P. Tetrahedron 2005, 61, 3559–3564. doi:10.1016/j.tet.2005.01.110
Return to citation in text: [1] -
Rendy, R.; Zhang, Y.; McElrea, A.; Gomeez, A.; Klumpp, D. A. J. Org. Chem. 2004, 69, 2340–2347. doi:10.1021/jo030327t
Return to citation in text: [1] -
Mühlthau, F.; Stadler, D.; Goeppert, A.; Olah, G. A.; Prakash, G. K. S.; Bach, T. J. Am. Chem. Soc. 2006, 128, 9668–9675. doi:10.1021/ja062102g
Return to citation in text: [1] -
Olah, G. A.; Spear, R. J.; Forsyth, D. A. J. Am. Chem. Soc. 1977, 99, 2615–2621. doi:10.1021/ja00450a035
Return to citation in text: [1] -
Mascal, M.; Nikitin, E. B. Angew. Chem., Int. Ed. 2008, 47, 7924–7926. doi:10.1002/anie.200801594
Return to citation in text: [1] -
Kumari, N.; Olesen, J. K.; Pedersen, C. M.; Bols, M. Eur. J. Org. Chem. 2011, 1266–1270. doi:10.1002/ejoc.201001539
Return to citation in text: [1] -
Ennaert, T.; Van Aelst, J.; Dijkmans, J.; De Clercq, R.; Schutyser, W.; Dusselier, M.; Verboekend, D.; Sels, B. F. Chem. Soc. Rev. 2016, 45, 584–611. doi:10.1039/C5CS00859J
Return to citation in text: [1] -
Sineva, L. V.; Asalieva, E. Yu.; Mordkovich, V. Z. Russ. Chem. Rev. 2015, 84, 1176–1189. doi:10.1070/RCR4464
Return to citation in text: [1] -
Shi, J.; Wang, Y.; Yang, W.; Tang, Y.; Xie, Z. Chem. Soc. Rev. 2015, 44, 8877–8903. doi:10.1039/C5CS00626K
Return to citation in text: [1] -
Dapsens, P. Y.; Mondelli, C.; Pérez-Ramírez, J. Chem. Soc. Rev. 2015, 44, 7025–7043. doi:10.1039/C5CS00028A
Return to citation in text: [1] -
Van Speybroeck, V.; Hemelsoet, K.; Joos, L.; Waroquier, M.; Bell, R. G.; Catlow, C. R. A. Chem. Soc. Rev. 2015, 44, 7044–7111. doi:10.1039/C5CS00029G
Return to citation in text: [1] -
Primo, A.; Garcia, H. Chem. Soc. Rev. 2014, 43, 7548–7561. doi:10.1039/C3CS60394F
Return to citation in text: [1] -
Li, Y.; Yu, J. Chem. Rev. 2014, 114, 7268–7316. doi:10.1021/cr500010r
Return to citation in text: [1]
| 38. | Prakash, G. K. S.; Panja, C.; Shakhmin, A.; Shah, E.; Mathew, T.; Olah, G. A. J. Org. Chem. 2009, 74, 8659–8668. doi:10.1021/jo901668j |
| 34. | Prakash, G. K. S.; Paknia, F.; Chacko, S.; Mathew, T.; Olah, G. A. Heterocycles 2008, 76, 783–799. doi:10.3987/COM-08-S(N)90 |
| 35. | Klumpp, D. A.; Jones, A.; Lau, S.; de Leon, S.; Garza, M. Synthesis 2000, 1117–1120. doi:10.1055/s-2000-6322 |
| 36. | Klumpp, D. A.; Kindelin, P. J.; Li, A. Tetrahedron Lett. 2005, 46, 2931–2935. doi:10.1016/j.tetlet.2005.02.152 |
| 37. | Sheets, M. R.; Li, A.; Bower, E. A.; Weigel, A. R.; Abbot, M. P.; Gallo, R. M.; Mitton, A. M.; Klumpp, D. A. J. Org. Chem. 2009, 74, 2502–2507. doi:10.1021/jo802798x |
| 38. | Prakash, G. K. S.; Panja, C.; Shakhmin, A.; Shah, E.; Mathew, T.; Olah, G. A. J. Org. Chem. 2009, 74, 8659–8668. doi:10.1021/jo901668j |
| 33. | Olah, G. A.; Prakash, G. K. S.; Molnar, A.; Sommer, J. Superacid Chemistry, 2nd ed.; Wiley: New Jersey, 2009. |
| 1. | Barta, K.; Ford, P. C. Acc. Chem. Res. 2014, 47, 1503–1512. doi:10.1021/ar4002894 |
| 2. | Colmenares, J. C.; Luque, R. Chem. Soc. Rev. 2014, 43, 765–778. doi:10.1039/c3cs60262a |
| 3. | Straathof, A. J. J. Chem. Rev. 2014, 114, 1871–1908. doi:10.1021/cr400309c |
| 4. | Besson, M.; Gallezot, P.; Pinel, C. Chem. Rev. 2014, 114, 1827–1870. doi:10.1021/cr4002269 |
| 5. | Moiseev, I. I. Russ. Chem. Rev. 2013, 82, 616–623. doi:10.1070/RC2013v082n07ABEH004393 |
| 6. | Alonso, D. M.; Wettstein, S. G.; Dumesic, J. A. Chem. Soc. Rev. 2012, 41, 8075–8098. doi:10.1039/c2cs35188a |
| 7. | Gallezot, P. Chem. Soc. Rev. 2012, 41, 1538–1558. doi:10.1039/C1CS15147A |
| 8. | Ponomarev, A. V.; Ershov, B. G. Russ. Chem. Rev. 2012, 81, 918–935. doi:10.1070/RC2012v081n10ABEH004266 |
| 9. | Sun, N.; Rodriguez, H.; Rahman, M.; Rogers, R. D. Chem. Commun. 2011, 47, 1405–1421. doi:10.1039/C0CC03990J |
| 10. | Varfolomeev, S. D.; Efremenko, E. N.; Krylova, L. P. Russ. Chem. Rev. 2010, 79, 491–510. doi:10.1070/RC2010v079n06ABEH004138 |
| 11. | Marshall, A.-L.; Alaimo, P. Chem. – Eur. J. 2010, 16, 4970–4980. doi:10.1002/chem.200903028 |
| 12. | Stöcker, M. Angew. Chem., Int. Ed. 2008, 47, 9200–9211. doi:10.1002/anie.200801476 |
| 13. | Chheda, J. N.; Huber, G. W.; Dumesic, J. A. Angew. Chem., Int. Ed. 2007, 46, 7164–7183. doi:10.1002/anie.200604274 |
| 14. | Huber, G. W.; Corma, A. Angew. Chem., Int. Ed. 2007, 46, 7184–7201. doi:10.1002/anie.200604504 |
| 15. | Corma, A.; Iborra, S.; Velty, A. Chem. Rev. 2007, 107, 2411–2502. doi:10.1021/cr050989d |
| 16. | Huber, G. W.; Iborra, S.; Corma, A. Chem. Rev. 2006, 106, 4044–4098. doi:10.1021/cr068360d |
| 26. | Iesce, M. R.; Sferruzza, R.; Cermola, F.; DellaGreca, M. Helv. Chim. Acta 2016, 99, 296–301. doi:10.1002/hlca.201500196 |
| 27. | Palframan, M. J.; Pattenden, G. Chem. Commun. 2014, 50, 7223–7242. doi:10.1039/c4cc01196a |
| 28. | Dijkman, W. P.; Groothuis, D. E.; Fraaije, M. W. Angew. Chem., Int. Ed. 2014, 53, 6515–6518. doi:10.1002/anie.201402904 |
| 29. | Ghezali, W.; De Oliveira Vigier, K.; Kessas, R.; Jérôme, F. Green Chem. 2015, 17, 4459–4464. doi:10.1039/C5GC01336D |
| 30. | Liu, F.; Audemar, M.; De Oliveira Vigier, K.; Clacens, J.-M.; De Campo, F.; Jérôme, F. ChemSusChem 2014, 7, 2089–2093. doi:10.1002/cssc.201402221 |
| 58. | Mascal, M.; Nikitin, E. B. Angew. Chem., Int. Ed. 2008, 47, 7924–7926. doi:10.1002/anie.200801594 |
| 59. | Kumari, N.; Olesen, J. K.; Pedersen, C. M.; Bols, M. Eur. J. Org. Chem. 2011, 1266–1270. doi:10.1002/ejoc.201001539 |
| 20. | Kashin, A. S.; Galkin, K. I.; Khokhlova, E. A.; Ananikov, V. P. Angew. Chem., Int. Ed. 2016, 55, 2161–2166. doi:10.1002/anie.201510090 |
| 21. | Galkin, K. I.; Krivodaeva, E. A.; Romashov, L. V.; Zalesskiy, S. S.; Kachala, V. V.; Burykina, J. V.; Ananikov, V. P. Angew. Chem., Int. Ed. 2016, 55, 8338–8342. doi:10.1002/anie.201602883 |
| 22. | Li, W.; Xu, Z.; Zhang, T.; Li, G.; Jameel, H.; Chang, H.; Ma, L. BioResources 2016, 11, 5839–5853. doi:10.15376/biores.11.3.5839-5853 |
| 23. | Choudhary, V.; Mushrif, S. H.; Ho, C.; Anderko, A.; Nikolakis, V.; Marinkovic, N. S.; Frenkel, A. I.; Sandler, S. I.; Vlachos, D. G. J. Am. Chem. Soc. 2013, 135, 3997–4006. doi:10.1021/ja3122763 |
| 24. | Hu, L.; Zhao, C.; Tang, X.; Wu, Z.; Xu, J.; Lin, L.; Liu, S. Bioresour. Technol. 2013, 148, 501–507. doi:10.1016/j.biortech.2013.09.016 |
| 25. | Yang, J.; De OliveiraVigier, K.; Gu, Y.; Jérôme, F. ChemSusChem 2015, 8, 269–274. doi:10.1002/cssc.201402761 |
| 60. | Ennaert, T.; Van Aelst, J.; Dijkmans, J.; De Clercq, R.; Schutyser, W.; Dusselier, M.; Verboekend, D.; Sels, B. F. Chem. Soc. Rev. 2016, 45, 584–611. doi:10.1039/C5CS00859J |
| 61. | Sineva, L. V.; Asalieva, E. Yu.; Mordkovich, V. Z. Russ. Chem. Rev. 2015, 84, 1176–1189. doi:10.1070/RCR4464 |
| 62. | Shi, J.; Wang, Y.; Yang, W.; Tang, Y.; Xie, Z. Chem. Soc. Rev. 2015, 44, 8877–8903. doi:10.1039/C5CS00626K |
| 63. | Dapsens, P. Y.; Mondelli, C.; Pérez-Ramírez, J. Chem. Soc. Rev. 2015, 44, 7025–7043. doi:10.1039/C5CS00028A |
| 64. | Van Speybroeck, V.; Hemelsoet, K.; Joos, L.; Waroquier, M.; Bell, R. G.; Catlow, C. R. A. Chem. Soc. Rev. 2015, 44, 7044–7111. doi:10.1039/C5CS00029G |
| 65. | Primo, A.; Garcia, H. Chem. Soc. Rev. 2014, 43, 7548–7561. doi:10.1039/C3CS60394F |
| 66. | Li, Y.; Yu, J. Chem. Rev. 2014, 114, 7268–7316. doi:10.1021/cr500010r |
| 19. | van Putten, R.-J.; van der Waal, J. C.; de Jong, E.; Rasrendra, C. B.; Heeres, H. J.; de Vries, J. G. Chem. Rev. 2013, 113, 1499–1597. doi:10.1021/cr300182k |
| 48. | Parr, R. G.; van Szentpály, L.; Liu, S. J. Am. Chem. Soc. 1999, 121, 1922–1924. doi:10.1021/ja983494x |
| 49. | Chattaraj, P. K.; Giri, S.; Duley, S. Chem. Rev. 2011, 111, PR43–PR75. doi:10.1021/cr100149p |
| 17. | Bozell, J. J.; Petersen, G. R. Green Chem. 2010, 12, 539–554. doi:10.1039/B922014C |
| 18. | Kamm, B. Angew. Chem., Int. Ed. 2007, 46, 5056–5058. doi:10.1002/anie.200604514 |
| 50. | Alkhafaji, H. M. H.; Ryabukhin, D. S.; Muzalevskiy, V. M.; Vasilyev, A. V.; Fukin, G. K.; Shastin, A. V.; Nenajdenko, V. G. Eur. J. Org. Chem. 2013, 1132–1143. doi:10.1002/ejoc.201201375 |
| 51. | Alkhafaji, H. M. H.; Ryabukhin, D. S.; Muzalevskiy, V. M.; Osetrova, L. V.; Vasilyev, A. V.; Nenajdenko, V. G. Russ. J. Org. Chem. 2013, 49, 327–341. doi:10.1134/S1070428013030032 |
| 52. | Koltunov, K. Y.; Shakirov, M. M.; Repinskaya, I. B.; Koptug, V. A. Zh. Org. Khim. 1991, 27, 2622–2623. |
| 53. | Koltunov, K. Y.; Repinskaya, I. B. Zh. Org. Khim. 1993, 30, 90–93. |
| 54. | Walspurger, S.; Vasilyev, A. V.; Sommer, J.; Pale, P. Tetrahedron 2005, 61, 3559–3564. doi:10.1016/j.tet.2005.01.110 |
| 55. | Rendy, R.; Zhang, Y.; McElrea, A.; Gomeez, A.; Klumpp, D. A. J. Org. Chem. 2004, 69, 2340–2347. doi:10.1021/jo030327t |
| 56. | Mühlthau, F.; Stadler, D.; Goeppert, A.; Olah, G. A.; Prakash, G. K. S.; Bach, T. J. Am. Chem. Soc. 2006, 128, 9668–9675. doi:10.1021/ja062102g |
| 57. | Olah, G. A.; Spear, R. J.; Forsyth, D. A. J. Am. Chem. Soc. 1977, 99, 2615–2621. doi:10.1021/ja00450a035 |
| 34. | Prakash, G. K. S.; Paknia, F.; Chacko, S.; Mathew, T.; Olah, G. A. Heterocycles 2008, 76, 783–799. doi:10.3987/COM-08-S(N)90 |
| 35. | Klumpp, D. A.; Jones, A.; Lau, S.; de Leon, S.; Garza, M. Synthesis 2000, 1117–1120. doi:10.1055/s-2000-6322 |
| 36. | Klumpp, D. A.; Kindelin, P. J.; Li, A. Tetrahedron Lett. 2005, 46, 2931–2935. doi:10.1016/j.tetlet.2005.02.152 |
| 37. | Sheets, M. R.; Li, A.; Bower, E. A.; Weigel, A. R.; Abbot, M. P.; Gallo, R. M.; Mitton, A. M.; Klumpp, D. A. J. Org. Chem. 2009, 74, 2502–2507. doi:10.1021/jo802798x |
| 39. | Cabares, J.; Mavoungou-Gomes, L. Bull. Soc. Chim. Fr. 1986, 401–412. |
| 40. | Skrzyńska, A.; Przydacz, A.; Albrecht, Ł. Org. Lett. 2015, 17, 5682–5685. doi:10.1021/acs.orglett.5b02979 |
| 41. | Onorato, A.; Pavlik, C.; Invernale, M. A.; Berghorn, I. D.; Sotzing, G. A.; Morton, M. D.; Smith, M. B. Carbohydr. Res. 2011, 346, 1662–1670. doi:10.1016/j.carres.2011.04.017 |
| 42. | Ager, D. J. Tetrahedron Lett. 1983, 24, 5441–5444. doi:10.1016/S0040-4039(00)94107-8 |
| 43. | Watanabe, N.; Matsugi, A.; Nakano, K.; Ichikawa, Y.; Kotsuki, H. Synlett 2014, 25, 438–442. doi:10.1055/s-0033-1340343 |
| 44. | Gomes, R. F. A.; Coelho, J. A. S.; Frade, R. F. M.; Trindade, A. F.; Afonso, C. A. M. J. Org. Chem. 2015, 80, 10404–10411. doi:10.1021/acs.joc.5b01875 |
| 45. | Pérez, M.; Mahdi, T.; Hounjet, L. J.; Stephan, D. W. Chem. Commun. 2015, 51, 11301–11304. doi:10.1039/C5CC03572D |
| 46. | Miao, M.; Luo, Y.; Li, H.; Xu, X.; Chen, Z.; Xu, J.; Ren, H. J. Org. Chem. 2016, 81, 5228–5235. doi:10.1021/acs.joc.6b00734 |
| 33. | Olah, G. A.; Prakash, G. K. S.; Molnar, A.; Sommer, J. Superacid Chemistry, 2nd ed.; Wiley: New Jersey, 2009. |
| 47. | Villain-Guillot, P.; Gualtieri, M.; Bastide, L.; Roquet, F.; Martinez, J.; Amblard, M.; Pugniere, M.; Leonetti, J.-P. J. Med. Chem. 2007, 50, 4195–4204. doi:10.1021/jm0703183 |
| 32. | Olah, G. A.; Klumpp, D. A. Superelectrophiles and Their Chemistry; Wiley: New York, 2008. |
| 31. | Kompanets, M. O.; Kushch, O. V.; Litvinov, Yu. E.; Pliekhov, O. L.; Novikova, K. V.; Novokhatko, A. O.; Shendrick, A. N.; Vasilyev, A. V.; Opeida, I. O. Catal. Commun. 2014, 57, 60–63. doi:10.1016/j.catcom.2014.08.005 |
| 38. | Prakash, G. K. S.; Panja, C.; Shakhmin, A.; Shah, E.; Mathew, T.; Olah, G. A. J. Org. Chem. 2009, 74, 8659–8668. doi:10.1021/jo901668j |
© 2016 Ryabukhin et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)