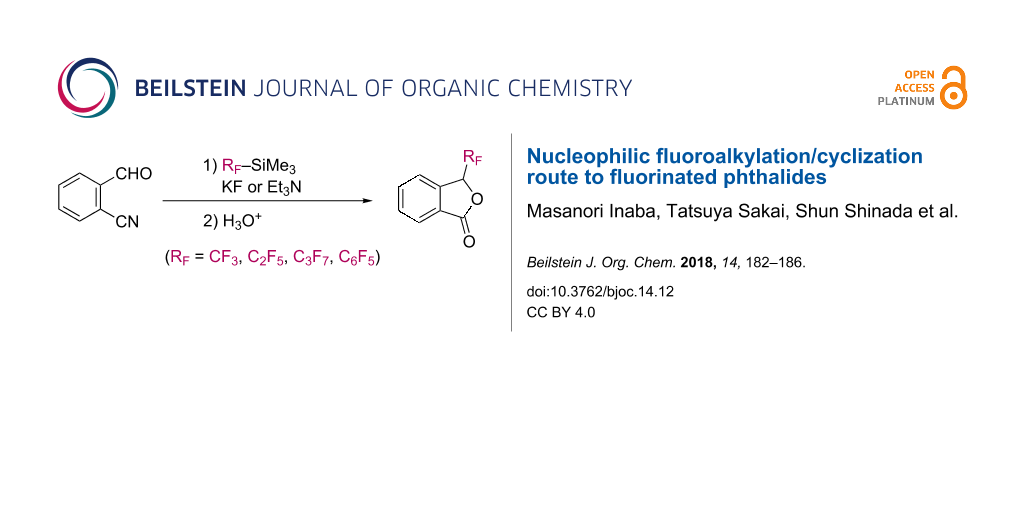
Abstract
Discribed in this article is a versatile and practical method for the synthesis of C3-perfluoroalkyl-substituted phthalides in a one-pot manner. Upon treatment of KF or triethylamine, 2-cyanobenzaldehyde reacted with (perfluoroalkyl)trimethylsilanes via nucleophilic addition and subsequent intramolecular cyclization to give perfluoroalkylphthalides in good yields.

Graphical Abstract
Introduction
Phthalides (1(3H)-isobenzofuranones) are frequently found in natural products and exhibit a range of bioactivity (Scheme 1) [1,2]. Substituted phthalides have been used as building blocks for the synthesis of useful bioactive compounds. There is a growing interest in the usefulness of phthalides and their derivatives. Organofluorine compounds often show attractive physical, chemical, and biological properties and are widely used in many fields, such as pharmaceuticals, agrochemicals, and materials [3-10]. Selective incorporation of fluorine or a fluoroalkyl group into a molecule is a topic of significant interest in organic chemistry. Fluorinated phthalides are considered to be one of the most fascinating organofluorine compounds. However, to our best knowledge, there have been few reports on the preparation of fluoroalkyl phthalides [11-19]. The first synthesis of 3-(trifluoromethyl)phthalides was accomplished by Reinecke and Chen in 1993. They studied ortho-lithiation of phenyloxazolines and the subsequent reactions with pentafluoroacetone and hexafluoroacetone to give 3-(trifluoromethyl)phthalide derivatives [11]. In 2006, Pedrosa et al. reported the nucleophilic trifluoromethylation of protected ortho-phthalaldehyde, followed by deprotection and oxidation to afford 3-(trifluoromethyl)phthalide [12]. Pohmakotr et al. demonstrated the nucleophilic trifluoromethylation of acid anhydrides to produce γ-hydroxy-γ-trifluoromethyl-γ-butyrolactones, which acted as good precursors in the synthesis of γ-trifluoromethyl-γ-butyrolactones with organometallic reagents [16]. All these protocols involve multiple steps to obtain trifluoromethylphthalides. An operational simple procedure for a short-step synthesis of 3-(trifluoromethyl)phthalide is required due to their great potential in a variety of applications. Herein, we wish to report a general and convenient synthesis of 3-(perfluoroalkyl)phthalides 1 by nucleophilic perfluoroalkylation of 2-cyanobenzaldehyde (2) and subsequent intramolecular cyclization.
![[1860-5397-14-12-i1]](/bjoc/content/inline/1860-5397-14-12-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Phthalide and fluorinated phthalides (1).
Scheme 1: Phthalide and fluorinated phthalides (1).
Results and Discussion
The reaction procedure is very simple. A mixture of 2-cyanobenzaldehyde (2), CF3–SiMe3 (so-called Ruppert–Prakash reagent) [20,21], and a catalytic amount of KF in anhydrous DMF was stirred at room temperature for 1 h. After work-up under acidic conditions, 3-(trifluoromethyl)-1(3H)-isobenzofuranone (1a) was obtained in 99% NMR yield (95% isolated yield) (Table 1, entry 1). As a good alternative activator of CF3–SiMe3, the use of a Lewis base such as triethylamine [22,23] worked well for the synthesis of 3-(trifluoromethyl)phthalide (1a, Table 1, entries 2–4). When aldehyde 2 was treated with CF3–SiMe3 in the presence of Et3N at 50 °C, the cascade trifluoromethylation/cyclization proceeded smoothly to afford phthalide 1a in 70% isolated yield (Table 1, entry 4).
Table 1: Trifluoromethylation/cyclization of 2-cyanobenzaldehyde.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-12-i6.svg?max-width=637&scale=1.0)
|
|||
| Entry | Base (equiv) | Conditions | Yield of 1a (%)a,b |
|---|---|---|---|
| 1 | KF (0.1) | rt, 1 h | 99 (95) |
| 2 | Et3N (0.5) | rt, 1 h | 80 |
| 3 | Et3N (0.5) | 50 °C, 1 h | 86 |
| 4 | Et3N (1.0) | 50 °C, 1 h | 91 (70) |
aYields were determined by 19F NMR analysis using 1,3-bis(trifluoromethyl)benzene as an internal standard. bThe values in parentheses indicate the isolated yield of 1a.
The formation of phthalide 1a can be explained by assuming the pathway shown in Scheme 2. The formyl group in aldehyde 2 undergoes nucleophilic trifluoromethylation triggered by a catalytic amount of KF to give the ortho-cyanobenzyl silyl ether 3. Upon treatment with aq HCl, the subsequent lactonization of 4 takes place to afford trifluoromethylphthalide 1a.
![[1860-5397-14-12-i2]](/bjoc/content/inline/1860-5397-14-12-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Plausible reaction mechanism for the formation of phthalide 1a.
Scheme 2: Plausible reaction mechanism for the formation of phthalide 1a.
Other examples of the one-pot synthesis of fluorinated phthalides 1 are given in Scheme 3. The reactions of 2-cyanobenzaldehyde (2) with organosilicon compounds (RF–SiMe3) proceeded cleanly by the use of KF (conditions A) or Et3N (conditions B). As a consequence, pentafluoroethyl, heptafluoropropyl, and pentafluorophenyl [24,25] groups were successfully installed at the C3-position of phthalides 1b–d.
![[1860-5397-14-12-i3]](/bjoc/content/inline/1860-5397-14-12-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of fluorinated phthalides 1.
Scheme 3: Synthesis of fluorinated phthalides 1.
Thus, the selective formation of fluorinated phthalides 1 represents a synthetic usefulness for further applications. In particular, the asymmetric synthesis of C3-substituted phthalides is of considerable importance in chemistry [26-32]. Enantioselective fluoroalkylation/lactonization reactions are worth investigating since a new stereogenic carbon center next to the fluoroalkyl groups is generated in products 1. To the best of our knowledge, only one successful example of an asymmetric synthesis of 3-(trifluoromethyl)phthalide (1a) using a chiral auxiliary was published to date. In 2006, Pedrosa and co-workers discribed the diastereoselective nucleophilic trifluoromethylation of aldehyde 5, which was prepared by condensation of ortho-phthalaldehyde with (−)-8-benzylaminomenthol (Scheme 4) [12]. Only diastereoisomer 6 was detected in the NMR analysis. Acid-promoted deprotection of hemiaminal 6 and subsequent oxidation of acetal 7 gave the enantiopure phthalide 1a in good yield.
![[1860-5397-14-12-i4]](/bjoc/content/inline/1860-5397-14-12-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Asymmetric synthesis of 1a using a chiral auxiliary.
Scheme 4: Asymmetric synthesis of 1a using a chiral auxiliary.
Although a high control of diastereoselectivity (using stoichiometric auxiliary strategy) was achieved for the asymmetric synthesis of trifluoromethylphthalide 1a, it is desirable to reduce the amount of the chiral sources. Next, we undertook the development of a catalytic asymmetric synthesis of 1 in a one-pot manner. The results of our trial are summarized in Table 2.
Table 2: Trifluoromethylation/cyclization of 2-cyanobenzaldehyde (2) in the presence of chiral catalysts.
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-12-i7.svg?max-width=637&scale=1.18182)
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Conditions | Yield of 1a (%)a | % eeb |
| 1 | 8 | DMF | rt, 1 h | 79 | 0 |
| 2 | 8 | DMF | 0 °C, 1 h | 62 | 0 |
| 3 | 9a | toluene/CH2Cl2 (2:1) | −60 °C, 24 h | 61 | 0 |
| 4 | 9b | toluene/CH2Cl2 (2:1) | −60 °C, 24 h | 51 | 12 |
| 5 | 9c | toluene/CH2Cl2 (2:1) | −60 °C, 24 h | 76 | 6 |
aIsolated yield of 1a. bEach enantiomeric excess (ee) was determined by HPLC analyses.
For the catalytic asymmetric synthesis of 1, we carried out the nucleophilic trifluoromethylation of 2 employing a small amount of chiral tertiary amines. However, the use of (1R,2R)-(N,N,N’,N’-tetramethyl)-1,2-diaminocyclohexane (8) as a chiral catalyst resulted in the formation of a racemic mixture of 1a (Table 2, entries 1 and 2).
Previously, Shibata et al. reported a cinchona alkaloid/Me4NF-catalyzed nucleophilic enantioselective trifluoromethylation of carbonyl compounds [33-35]. Initially, we tried to conduct the reaction of 2-cyanobenzaldehyde (2) with CF3–SiMe3 in the presence of cinchona alkaloids 9/TMAF combination (Table 2, entries 3–5). By employing catalyst 9b, the reaction proceeded at −60 °C to give phthalide 1a in 51% yield with 12% ee (Table 2, entry 4).
To improve the enantioselectivity of the present nucleophilic trifluorometylation/lactonization, we surveyed suitable conditions for the catalytic asymmetric transformation. After many experiments, we found that the use of ethyl 2-formylbenzoate (10) [36] instead of nitrile 2 resulted in the formation of 1a with 27% ee upon exposure to organocatalyst 9b (0.1 equiv) and tetramethylammonium fluoride (0.5 equiv, Scheme 5).
![[1860-5397-14-12-i5]](/bjoc/content/inline/1860-5397-14-12-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Catalytic asymmetric synthesis of 1a.
Scheme 5: Catalytic asymmetric synthesis of 1a.
Conclusion
In summary, we have demonstrated a convenient route to fluorinated phthalides from 2-cyanobenzaldehyde or 2-formylbenzoates in a one-pot manner. In the transformations, installation of fluorinated substituents at the C3-position of phthalides has been achieved. The issue of the low stereoselectivity of the catalytic asymmetric fluoroalkylation should be solved in the future. Further examples for the promising utilization of fluorinated phthalides as building blocks can be found in [37].
Supporting Information
| Supporting Information File 1: General methods, synthetic procedures, 1H and 19F NMR spectra for known compound 1a and full characterization of all new compounds. | ||
| Format: PDF | Size: 1.2 MB | Download |
Acknowledgements
This article is dedicated to the memory of Professor Yoshihiko Ito (1937-2006) on the occasion of the 10th anniversary of his sudden death. The financial support of JSPS KAKENHI Grant No. JP 16H04143 (Grant-in-Aid for Scientific Research (B)), JP16H01003 in Precisely Designed Catalysts with Customized Scaffolding, JP16H01129 in Middle Molecular Strategy, and Japan Science and Technology Agency (JST) (ACT-C: Creation of Advanced Catalytic Transformation for the Sustainable Manufacturing at Low Energy, Low Environmental Load) is acknowledged. We would like to thank Prof. Hiroshi Sano (Gunma University) for his useful suggestions.
References
-
Lin, G.; Chan, S.-K. S.; Chung, H.-S.; Li, S.-L. Chemistry and biological activities of naturally occurring phthalides. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, 2005; Vol. 32, pp 611–670.
Return to citation in text: [1] -
Knepper, K.; Ziegert, R. E.; Bräse, S. Tetrahedron 2004, 60, 8591–8603. doi:10.1016/j.tet.2004.05.111
Return to citation in text: [1] -
Hiyama, T.; Kanie, K.; Kusumoto, T.; Morizawa, Y.; Shimizu, M. In Organofluorine Compounds: Chemistry and Applications; Yamamoto, H., Ed.; Springer-Verlag: Berlin, 2000. doi:10.1007/978-3-662-04164-2
Return to citation in text: [1] -
Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/352760393X
Return to citation in text: [1] -
Chambers, R. D. Fluorine in Organic Chemistry; Blackwell: Oxford, 2004.
Return to citation in text: [1] -
Uneyama, K. Organofluorine Chemistry; Blackwell: Oxford, 2006.
Return to citation in text: [1] -
Bégué, J.-P.; Bonnet-Delpon, D. Bioorganic and Medicinal Chemistry of Fluorine; John Wiley & Sons, Inc.: Hoboken, NJ, 2008. doi:10.1002/9780470281895
Return to citation in text: [1] -
Ojima, I., Ed. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Chichester, West Sussex, 2009. doi:10.1002/9781444312096
Return to citation in text: [1] -
Fluorine in Pharmaceutical and Medicinal Chemistry: From Biophysical Aspects to Clinical Applications; Gouverneur, V.; Müller, K., Eds.; Molecular Medicine and Medicinal Chemistry, Vol. 6; Imperial College Press: London, 2012. doi:10.1142/p746
Return to citation in text: [1] -
Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879
Return to citation in text: [1] -
Reinecke, M. G.; Chen, L.-J. Acta Chem. Scand. 1993, 47, 318–322. doi:10.3891/acta.chem.scand.47-0318
Return to citation in text: [1] [2] -
Pedrosa, R.; Sayalero, S.; Vicente, M. Tetrahedron 2006, 62, 10400–10407. doi:10.1016/j.tet.2006.08.058
Return to citation in text: [1] [2] [3] -
Zonov, Y. V.; Karpov, V. M.; Platonov, V. E.; Rybalova, T. V.; Gatilov, Y. V. J. Fluorine Chem. 2006, 127, 1574–1583. doi:10.1016/j.jfluchem.2006.08.006
Return to citation in text: [1] -
Kinoshita, K.; Yamada, S.; Iwama, S.; Kawasaki-Takasuka, T.; Yamazaki, T. 20th International Symposium on Fluorine Chemistry, Kyoto, Japan, July 22–27, 2012; Abstr., No. P-41.
Return to citation in text: [1] -
Shi, X.; Li, C.-J. Adv. Synth. Catal. 2012, 354, 2933–2938. doi:10.1002/adsc.201200690
Return to citation in text: [1] -
Masusai, C.; Soorukram, D.; Kuhakarn, C.; Tuchinda, P.; Reutrakul, V.; Pohmakotr, M. J. Fluorine Chem. 2013, 154, 37–42. doi:10.1016/j.jfluchem.2013.06.006
Return to citation in text: [1] [2] -
Xu, L.; Jiang, H.; Hao, J.; Zhao, G. Tetrahedron 2014, 70, 4373–4378. doi:10.1016/j.tet.2014.04.072
Return to citation in text: [1] -
Egami, H.; Asada, J.; Sato, K.; Hashizume, D.; Kawato, Y.; Hamashima, Y. J. Am. Chem. Soc. 2015, 137, 10132–10135. doi:10.1021/jacs.5b06546
Return to citation in text: [1] -
Fustero, S.; Moscardó, J.; Sánchez-Rosselló, M.; Rodriguez, E.; Barrio, P. Org. Lett. 2010, 12, 5494–5497. doi:10.1021/ol102341n
Return to citation in text: [1] -
Prakash, G. K. S.; Krishnamurti, R.; Olah, G. A. J. Am. Chem. Soc. 1989, 111, 393–395. doi:10.1021/ja00183a073
Return to citation in text: [1] -
Prakash, G. K. S.; Yudin, A. K. Chem. Rev. 1997, 97, 757–786. doi:10.1021/cr9408991
Return to citation in text: [1] -
Hagiwara, T.; Kobayashi, T.; Fuchikami, T. Nippon Kagaku Kaishi 1997, 869–875. doi:10.1246/nikkashi.1997.869
Return to citation in text: [1] -
Hagiwara, T.; Kobayashi, T.; Fuchikami, T. Main Group Chem. 1997, 2, 13–15. doi:10.1080/13583149712331338829
Return to citation in text: [1] -
Brogan, S.; Carter, N. B.; Lam, H. W. Synlett 2010, 615–617. doi:10.1055/s-0029-1219159
Return to citation in text: [1] -
Du, G.-F.; Xing, F.; Gu, C.-Z.; Dai, B.; He, L. RSC Adv. 2015, 5, 35513–35517. doi:10.1039/C5RA05487G
Return to citation in text: [1] -
Kitamura, M.; Okhuma, T.; Inoue, S.; Sayo, N.; Kumobayshi, H.; Akutagawa, S.; Ohta, T.; Takaya, H.; Noyori, R. J. Am. Chem. Soc. 1988, 110, 629–631. doi:10.1021/ja00210a070
Return to citation in text: [1] -
Ramachandran, P. V.; Chen, G.-M.; Brown, H. C. Tetrahedron Lett. 1996, 37, 2205–2208. doi:10.1016/0040-4039(96)00260-2
Return to citation in text: [1] -
Witulski, B.; Zimmermann, A. Synlett 2002, 1855–1859. doi:10.1055/s-2002-34883
Return to citation in text: [1] -
Kosaka, M.; Sekiguchi, S.; Naito, J.; Uemura, M.; Kuwahara, S.; Watanabe, M.; Harada, N.; Hiroi, K. Chirality 2005, 17, 218–232. doi:10.1002/chir.20156
Return to citation in text: [1] -
Chang, H.-T.; Jeganmohan, M.; Cheng, C.-H. Chem. – Eur. J. 2007, 13, 4356–4363. doi:10.1002/chem.200601880
Return to citation in text: [1] -
Phan, D. H. T.; Kim, B.; Dong, V. M. J. Am. Chem. Soc. 2009, 131, 15608–15609. doi:10.1021/ja907711a
Return to citation in text: [1] -
Yasukawa, T.; Kobayashi, S. Chem. Lett. 2017, 46, 98–100. doi:10.1246/cl.160862
Return to citation in text: [1] -
Mizuta, S.; Shibata, N.; Akiti, S.; Fujimoto, H.; Nakamura, S.; Toru, T. Org. Lett. 2007, 9, 3707–3710. doi:10.1021/ol701791r
Return to citation in text: [1] -
Kawai, H.; Kusuda, A.; Nakamura, S.; Shiro, M.; Shibata, N. Angew. Chem., Int. Ed. 2009, 48, 6324–6327. doi:10.1002/anie.200902457
Return to citation in text: [1] -
Kawai, H.; Mizuta, S.; Tokunaga, E.; Shibata, N. J. Fluorine Chem. 2013, 152, 46–50. doi:10.1016/j.jfluchem.2013.01.032
Return to citation in text: [1] -
Cai, C.; Chen, T. Preparation method of 3-trifluoromethyl phthalide. Chin. Pat. Appl CN 2016-11198471, Dec 22, 2016.
While this article was in preparation, on May 17, 2017, a related observation using 2-formylbenzoates was reported in the above stated patent.
Return to citation in text: [1] -
Hamura, T.; Nakayama, R. Chem. Lett. 2013, 42, 1013–1015. doi:10.1246/cl.130398
Return to citation in text: [1]
| 1. | Lin, G.; Chan, S.-K. S.; Chung, H.-S.; Li, S.-L. Chemistry and biological activities of naturally occurring phthalides. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, 2005; Vol. 32, pp 611–670. |
| 2. | Knepper, K.; Ziegert, R. E.; Bräse, S. Tetrahedron 2004, 60, 8591–8603. doi:10.1016/j.tet.2004.05.111 |
| 12. | Pedrosa, R.; Sayalero, S.; Vicente, M. Tetrahedron 2006, 62, 10400–10407. doi:10.1016/j.tet.2006.08.058 |
| 11. | Reinecke, M. G.; Chen, L.-J. Acta Chem. Scand. 1993, 47, 318–322. doi:10.3891/acta.chem.scand.47-0318 |
| 11. | Reinecke, M. G.; Chen, L.-J. Acta Chem. Scand. 1993, 47, 318–322. doi:10.3891/acta.chem.scand.47-0318 |
| 12. | Pedrosa, R.; Sayalero, S.; Vicente, M. Tetrahedron 2006, 62, 10400–10407. doi:10.1016/j.tet.2006.08.058 |
| 13. | Zonov, Y. V.; Karpov, V. M.; Platonov, V. E.; Rybalova, T. V.; Gatilov, Y. V. J. Fluorine Chem. 2006, 127, 1574–1583. doi:10.1016/j.jfluchem.2006.08.006 |
| 14. | Kinoshita, K.; Yamada, S.; Iwama, S.; Kawasaki-Takasuka, T.; Yamazaki, T. 20th International Symposium on Fluorine Chemistry, Kyoto, Japan, July 22–27, 2012; Abstr., No. P-41. |
| 15. | Shi, X.; Li, C.-J. Adv. Synth. Catal. 2012, 354, 2933–2938. doi:10.1002/adsc.201200690 |
| 16. | Masusai, C.; Soorukram, D.; Kuhakarn, C.; Tuchinda, P.; Reutrakul, V.; Pohmakotr, M. J. Fluorine Chem. 2013, 154, 37–42. doi:10.1016/j.jfluchem.2013.06.006 |
| 17. | Xu, L.; Jiang, H.; Hao, J.; Zhao, G. Tetrahedron 2014, 70, 4373–4378. doi:10.1016/j.tet.2014.04.072 |
| 18. | Egami, H.; Asada, J.; Sato, K.; Hashizume, D.; Kawato, Y.; Hamashima, Y. J. Am. Chem. Soc. 2015, 137, 10132–10135. doi:10.1021/jacs.5b06546 |
| 19. | Fustero, S.; Moscardó, J.; Sánchez-Rosselló, M.; Rodriguez, E.; Barrio, P. Org. Lett. 2010, 12, 5494–5497. doi:10.1021/ol102341n |
| 36. |
Cai, C.; Chen, T. Preparation method of 3-trifluoromethyl phthalide. Chin. Pat. Appl CN 2016-11198471, Dec 22, 2016.
While this article was in preparation, on May 17, 2017, a related observation using 2-formylbenzoates was reported in the above stated patent. |
| 3. | Hiyama, T.; Kanie, K.; Kusumoto, T.; Morizawa, Y.; Shimizu, M. In Organofluorine Compounds: Chemistry and Applications; Yamamoto, H., Ed.; Springer-Verlag: Berlin, 2000. doi:10.1007/978-3-662-04164-2 |
| 4. | Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/352760393X |
| 5. | Chambers, R. D. Fluorine in Organic Chemistry; Blackwell: Oxford, 2004. |
| 6. | Uneyama, K. Organofluorine Chemistry; Blackwell: Oxford, 2006. |
| 7. | Bégué, J.-P.; Bonnet-Delpon, D. Bioorganic and Medicinal Chemistry of Fluorine; John Wiley & Sons, Inc.: Hoboken, NJ, 2008. doi:10.1002/9780470281895 |
| 8. | Ojima, I., Ed. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Chichester, West Sussex, 2009. doi:10.1002/9781444312096 |
| 9. | Fluorine in Pharmaceutical and Medicinal Chemistry: From Biophysical Aspects to Clinical Applications; Gouverneur, V.; Müller, K., Eds.; Molecular Medicine and Medicinal Chemistry, Vol. 6; Imperial College Press: London, 2012. doi:10.1142/p746 |
| 10. | Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879 |
| 37. | Hamura, T.; Nakayama, R. Chem. Lett. 2013, 42, 1013–1015. doi:10.1246/cl.130398 |
| 24. | Brogan, S.; Carter, N. B.; Lam, H. W. Synlett 2010, 615–617. doi:10.1055/s-0029-1219159 |
| 25. | Du, G.-F.; Xing, F.; Gu, C.-Z.; Dai, B.; He, L. RSC Adv. 2015, 5, 35513–35517. doi:10.1039/C5RA05487G |
| 12. | Pedrosa, R.; Sayalero, S.; Vicente, M. Tetrahedron 2006, 62, 10400–10407. doi:10.1016/j.tet.2006.08.058 |
| 22. | Hagiwara, T.; Kobayashi, T.; Fuchikami, T. Nippon Kagaku Kaishi 1997, 869–875. doi:10.1246/nikkashi.1997.869 |
| 23. | Hagiwara, T.; Kobayashi, T.; Fuchikami, T. Main Group Chem. 1997, 2, 13–15. doi:10.1080/13583149712331338829 |
| 33. | Mizuta, S.; Shibata, N.; Akiti, S.; Fujimoto, H.; Nakamura, S.; Toru, T. Org. Lett. 2007, 9, 3707–3710. doi:10.1021/ol701791r |
| 34. | Kawai, H.; Kusuda, A.; Nakamura, S.; Shiro, M.; Shibata, N. Angew. Chem., Int. Ed. 2009, 48, 6324–6327. doi:10.1002/anie.200902457 |
| 35. | Kawai, H.; Mizuta, S.; Tokunaga, E.; Shibata, N. J. Fluorine Chem. 2013, 152, 46–50. doi:10.1016/j.jfluchem.2013.01.032 |
| 20. | Prakash, G. K. S.; Krishnamurti, R.; Olah, G. A. J. Am. Chem. Soc. 1989, 111, 393–395. doi:10.1021/ja00183a073 |
| 21. | Prakash, G. K. S.; Yudin, A. K. Chem. Rev. 1997, 97, 757–786. doi:10.1021/cr9408991 |
| 16. | Masusai, C.; Soorukram, D.; Kuhakarn, C.; Tuchinda, P.; Reutrakul, V.; Pohmakotr, M. J. Fluorine Chem. 2013, 154, 37–42. doi:10.1016/j.jfluchem.2013.06.006 |
| 26. | Kitamura, M.; Okhuma, T.; Inoue, S.; Sayo, N.; Kumobayshi, H.; Akutagawa, S.; Ohta, T.; Takaya, H.; Noyori, R. J. Am. Chem. Soc. 1988, 110, 629–631. doi:10.1021/ja00210a070 |
| 27. | Ramachandran, P. V.; Chen, G.-M.; Brown, H. C. Tetrahedron Lett. 1996, 37, 2205–2208. doi:10.1016/0040-4039(96)00260-2 |
| 28. | Witulski, B.; Zimmermann, A. Synlett 2002, 1855–1859. doi:10.1055/s-2002-34883 |
| 29. | Kosaka, M.; Sekiguchi, S.; Naito, J.; Uemura, M.; Kuwahara, S.; Watanabe, M.; Harada, N.; Hiroi, K. Chirality 2005, 17, 218–232. doi:10.1002/chir.20156 |
| 30. | Chang, H.-T.; Jeganmohan, M.; Cheng, C.-H. Chem. – Eur. J. 2007, 13, 4356–4363. doi:10.1002/chem.200601880 |
| 31. | Phan, D. H. T.; Kim, B.; Dong, V. M. J. Am. Chem. Soc. 2009, 131, 15608–15609. doi:10.1021/ja907711a |
| 32. | Yasukawa, T.; Kobayashi, S. Chem. Lett. 2017, 46, 98–100. doi:10.1246/cl.160862 |
© 2018 Inaba et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)