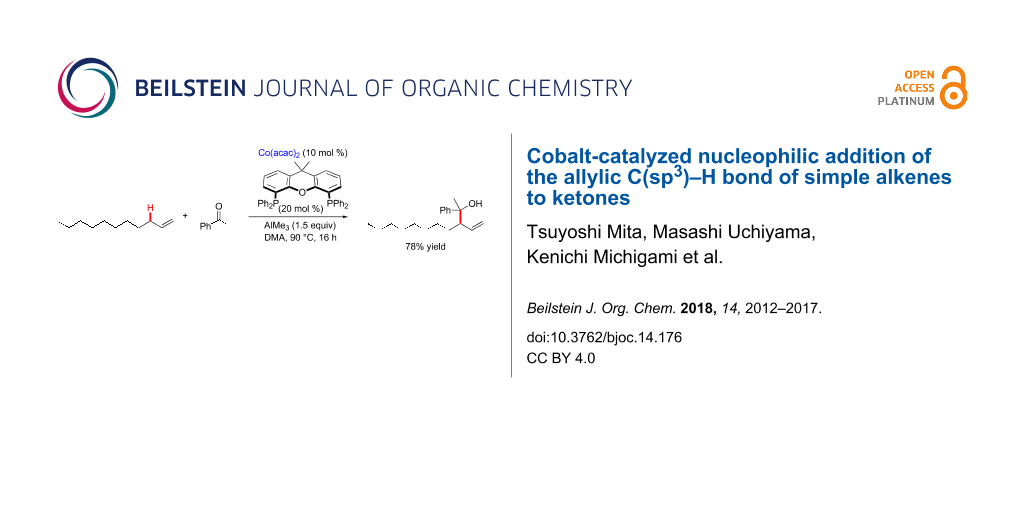
Abstract
We herein describe a cobalt/Xantphos-catalyzed regioselective addition of simple alkenes to acetophenone derivatives, affording branched homoallylic alcohols in high yields with perfect branch selectivities. The intermediate of the reaction would be a nucleophilic allylcobalt(I) species generated via cleavage of the low reactive allylic C(sp3)–H bond of simple terminal alkenes.

Graphical Abstract
Introduction
The cleavage of C–H bonds of unreactive hydrocarbon followed by functionalization should be an ideal method for constructing complex molecules without introduction of reactive functionality in advance [1-9]. Since terminal alkenes including α-olefins (CxH2x) are abundantly present in nature or are readily accessible, they should be appropriate starting materials for C–C bond forming reactions to create organic frameworks of value-added compounds such as natural products, drugs, and fine chemicals. There have been tremendous synthetic methods involving catalytic C–C bond construction with the double bond of terminal alkenes (e.g., Heck reaction, hydrometalation followed by functionalization, carbometalation, and olefin metathesis) [10-13]. However, direct C–C bond formation of the allylic C(sp3)–H bond adjacent to double bonds has remained underdeveloped even though C–O bond formation of allylic C(sp3)–H bonds was firmly established by using SeO2 [14] or CrO3/3,5-dimethylpyrazole [15] (ene-type allylic oxidation). Although the most prominent work on catalytic allylic functionalization studied thus far is considered to be a palladium-catalyzed C–C bond formation using a stoichiometric amount of an oxidant [16-22], the π-allylpalladium intermediate [23-25] is an electrophilic species that exclusively reacts with nucleophiles. Therefore, it would be a formidable challenge for the generation of a nucleophilic π-allylmetal complex that reacts with electrophiles, triggered by allylic C(sp3)–H activation. To this end, Schneider [26], Kanai [27], and we [28,29] reported in 2017 catalytic allylic C(sp3)–H activation of alkenes to react carbonyl electrophiles such as imines, ketones, and CO2 via nucleophilic allylmetal species (Figure 1). However, the substrates employed have been restricted to allylarenes and 1,4-enyne, and 1,4-diene derivatives and α-olefins were totally unexplored. Therefore, the next challenge would be to use less reactive α-olefins (pKa value of 1-propene = 43). In this paper, we describe an allylic C(sp3)–H addition of α-olefins, mainly 1-undecene and their analogues, to ketone electrophiles.
![[1860-5397-14-176-1]](/bjoc/content/figures/1860-5397-14-176-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Precedent examples of catalytic allylic C(sp3)–H additions to carbonyl electrophiles.
Figure 1: Precedent examples of catalytic allylic C(sp3)–H additions to carbonyl electrophiles.
Results and Discussion
We initially conducted screening of conditions using 1 equiv of 1-undecene (1a) and 3 equiv of acetophenone (2a) as starting materials (Table 1). When the reaction was conducted at 60 °C in DMA according to our previously established catalytic conditions (Co(acac)2 (10 mol %), Xantphos (20 mol %), and AlMe3 (1.0 equiv)) [29], branched homoallylic alcohol 3aa was obtained in only 23% yield with 1.6:1 diastereoselectivity (Table 1, entry 1). In constant to the C(sp3)–H addition of allylarene to acetophenone that exhibited high linear selectivity [29], perfect branch selectivity was observed using 1a as a substrate. When the reaction temperature was raised, the yield of 3aa was improved to 45% yield at 90 °C (Table 1, entries 2 and 3). An increase in the amount of AlMe3 to 1.5 equiv further improved the yield of 3aa to 54% yield (Table 1, entry 4). The moderate yield was attributed to the generation of the olefin isomerization product derived from 1a (vide infra). We then changed the equivalents of reagents 1a and 2a. Although the yield of 3aa was decreased when the reaction was conducted using a 1:1 ratio of 1a and 2a (Table 1, entry 5), the use of an excess amount of 1a (3 equiv) greatly improved the yield to 70% (Table 1, entry 6: optimized conditions). When 1-octadecene (1b) was subjected to the optimized reaction conditions without adding 2a, internal olefins were exclusively obtained (mixture of positional and geometric isomers) in 97% yield. It was shown by 1H NMR analysis that the mixture contained about 60% of 2-octadecene (E/Z mixture).
Table 1: Screening of reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-176-i1.svg?max-width=637&scale=1.0)
|
||||
| Entry | 1a:2a | Temp (°C) | AlMe3 (x equiv) | 3aa (dr) (%)a |
| 1 | 1:3 | 60 | 1.0 | 23 (1.6:1) |
| 2 | 1:3 | 80 | 1.0 | 44 (1.3:1) |
| 3 | 1:3 | 90 | 1.0 | 45 (1.3:1) |
| 4b | 1:3 | 90 | 1.5 | 54 (1.3:1) |
| 5 | 1:1 | 90 | 1.5 | 46 (1.2:1) |
| 6 | 3:1 | 90 | 1.5 | 70c (1.3:1) |
aYields were determined by 1H NMR analysis using 1,1,2,2-tetrachloroethane as an internal standard. The diastereoselectivity (dr) was determined by 1H NMR analysis. bThe olefin isomerization product was obtained in 32% yield. cIsolated yield.
Having established the reaction conditions, we then screened the scope and limitation of substituted acetophenone derivatives using an excess amount of 1-undecene (1a, 3 equiv, Figure 2). Electron-neutral and electron-donating substituents such as H (2a), Me (2b), and OMe (2c) at the para-position efficiently promoted the allylic C(sp3)–H addition, in which the reaction of 2a could be scaled-up (1 mmol) to afford 3aa in a slightly higher yield (78%). Electron-withdrawing substituents such as F (2d) and CO2Me (2e) also promoted the reaction with similar levels without damaging the ester functionality. Furthermore, 2-naphtophenone (2f) and propiophenone (2g) were tolerated well, affording branched products selectively in over 60% yield. However, p-CF3-acetophenone (18%), acetone (14%), cyclohexanone (29%), and benzophenone (25%) were not suitable substrates for C(sp3)–H addition of 1a (figures not shown).
![[1860-5397-14-176-2]](/bjoc/content/figures/1860-5397-14-176-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Substrate scope for acetophenone derivatives. aPreparative scale synthesis using 1 mmol of 2a.
Figure 2: Substrate scope for acetophenone derivatives. aPreparative scale synthesis using 1 mmol of 2a.
We next examined several α-olefins 1 (3 equiv) for allylic C(sp3)–H addition to acetophenone (2a). Not only 1-undecene (1a) but also 1-octadecene (1b) and 6-phenyl-1-hexene (1c) were tolerable to afford the corresponding products in around 70% yield with perfect branch selectivity (Figure 3). Although the allylic C(sp3)–H bond of α-olefins is weakly acidic (pKa value of 1-propene = 43), it is noteworthy that thermal cleavage of allylic C(sp3)–H bonds is possible without using highly basic organolithium or organomagnesium reagents (Grignard reagents) that react with ketones rather than deprotonating the allylic C(sp3)–H bonds.
![[1860-5397-14-176-3]](/bjoc/content/figures/1860-5397-14-176-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Based on the observed perfect branch selectivity, we propose the catalytic cycle of the C(sp3)–H addition of 1-undecene (1a) to acetophenone (2a, Figure 4). First, methylcobalt(I) I should be generated from Co(acac)2, Xantphos, and AlMe3 [28,29]. Oxidative addition of the allylic C(sp3)–H bond to I would proceed to afford η3-allylcobalt(III) intermediate II, which is tautomerized to η1-allylcobalt(III) III by the assistance of the oxygen atom in the Xantphos ligand [30]. When using α-olefin as a substrate, the cobalt atom should be located at the terminal position due to the avoidance of steric repulsion between the bulky Xantphos ligand and an alkyl substitution (similar to the case of nucleophilic η1-allylpalladium species [31-39]), whereas the cobalt atom preferred to reside at the internal position when allylarenes and 1,4-dienes were employed in our previous studies [28,29]. Subsequently, reductive elimination of methane from III would lead to a low-valent allylcobalt(I) species, and then C–C bond formation of IV with 2a would proceed at the γ-position to produce cobalt alkoxide(I) V [28,29,31-39]. Transmetalation between V and AlMe3 would furnish branched aluminium alkoxide VI along with the regeneration of I. Alkoxide VI is converted to homoallylic alcohol 3aa by usual work-up.
![[1860-5397-14-176-4]](/bjoc/content/figures/1860-5397-14-176-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, we have successfully developed a cobalt-catalyzed nucleophilic addition of the C(sp3)–H bond of simple alkenes to ketones. This novel transformation could realize perfect branch selectivity for all substrates. Much effort toward the development of an asymmetric variant is ongoing. We are also conducting computational analysis to explain the observed perfect regioselectivity. These results will be reported in due course.
Experimental
Representative procedure: To an oven-dried test tube was placed Co(acac)2 (5.2 mg, 20 μmol, 10 mol %) and Xantphos (23.1 mg, 40 μmol, 20 mol %) in DMA (2 mL). The resulting mixture was stirred at room temperature until the materials had been completely dissolved. After the solution had been cooled to 0 ºC, it was stirred for 1 minute, and then AlMe3 (2 M in toluene, 0.15 mL, 0.3 mmol, 1.5 equiv) was added. The dark green solution was stirred for another 1 minute, and then alkene 1 (0.6 mmol, 3.0 equiv) was added followed by the addition of ketone 2 (0.2 mmol, 1.0 equiv). The resulting mixture was stirred at 90 ºC for 16 h. After cooling the mixture to 0 ºC, the reaction was quenched by 1 M HCl aq and extracted with ethyl acetate (3 times). The combined organic layer was washed with brine and dried over Na2SO4. After the solids had been filtered off, the solvent was removed under reduced pressure and the residue was dried under vacuum to afford the crude mixture. The approximate yield of 3 was determined at this stage using 1,1,2,2-tetrachloroethane (δ = 6.1 ppm in CDCl3, 2H) as an internal standard. If the ketone remained, NaBH4 was added to convert it into the corresponding alcohol, which could be easily separated from 3 by silica-gel column chromatography. It was then purified by silica-gel column chromatography to afford the products 3.
Supporting Information
| Supporting Information File 1: Experimental details and characterization data. | ||
| Format: PDF | Size: 2.3 MB | Download |
Acknowledgements
This work was financially supported by Grant-in-Aid for Scientific Research (C) (No. 18K05096), Grant-in-Aid for Scientific Research (B) (No. 26293001) from JSPS, and also by JST ACT-C (No. JPMJCR12YM). T.M. thanks the Naito Foundation for financial support. K.M. thanks JSPS for a fellowship (No. 14J08052).
References
-
Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147–1169. doi:10.1021/cr900184e
Return to citation in text: [1] -
Jazzar, R.; Hitce, J.; Renaudat, A.; Sofack-Kreutzer, J.; Baudoin, O. Chem. – Eur. J. 2010, 16, 2654–2672. doi:10.1002/chem.200902374
Return to citation in text: [1] -
Wendlandt, A. E.; Suess, A. M.; Stahl, S. S. Angew. Chem., Int. Ed. 2011, 50, 11062–11087. doi:10.1002/anie.201103945
Return to citation in text: [1] -
Dastbaravardeh, N.; Christakakou, M.; Haider, M.; Schnürch, M. Synthesis 2014, 46, 1421–1439. doi:10.1055/s-0033-1338625
Return to citation in text: [1] -
Guo, X.-X.; Gu, D.-W.; Wu, Z.; Zhang, W. Chem. Rev. 2015, 115, 1622–1651. doi:10.1021/cr500410y
Return to citation in text: [1] -
Hartwig, J. F.; Larsen, M. A. ACS Cent. Sci. 2016, 2, 281–292. doi:10.1021/acscentsci.6b00032
Return to citation in text: [1] -
Gensch, T.; Hopkinson, M. N.; Glorius, F.; Wencel-Delord, J. Chem. Soc. Rev. 2016, 45, 2900–2936. doi:10.1039/C6CS00075D
Return to citation in text: [1] -
Yoshino, T.; Matsunaga, S. Adv. Synth. Catal. 2017, 359, 1245–1262. doi:10.1002/adsc.201700042
Return to citation in text: [1] -
He, J.; Wasa, M.; Chan, K. S. L.; Shao, Q.; Yu, J.-Q. Chem. Rev. 2017, 117, 8754–8786. doi:10.1021/acs.chemrev.6b00622
Return to citation in text: [1] -
Dounay, A. B.; Overman, L. E. Chem. Rev. 2003, 103, 2945–2964. doi:10.1021/cr020039h
Return to citation in text: [1] -
Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746–1787. doi:10.1021/cr9002424
Return to citation in text: [1] -
Crossley, S. W. M.; Obradors, C.; Martinez, R. M.; Shenvi, R. A. Chem. Rev. 2016, 116, 8912–9000. doi:10.1021/acs.chemrev.6b00334
Return to citation in text: [1] -
Shigehisa, H. Chem. Pharm. Bull. 2018, 66, 339–346. doi:10.1248/cpb.c17-01006
Return to citation in text: [1] -
Młochowski, J.; Wójtowicz-Młochowska, H. Molecules 2015, 20, 10205–10243. doi:10.3390/molecules200610205
Return to citation in text: [1] -
Salmond, W. G.; Barta, M. A.; Havens, J. L. J. Org. Chem. 1978, 43, 2057–2059. doi:10.1021/jo00404a049
Return to citation in text: [1] -
Chen, M. S.; White, M. C. J. Am. Chem. Soc. 2004, 126, 1346–1347. doi:10.1021/ja039107n
Return to citation in text: [1] -
Young, A. J.; White, M. C. J. Am. Chem. Soc. 2008, 130, 14090–14091. doi:10.1021/ja806867p
Return to citation in text: [1] -
Kondo, H.; Yu, F.; Yamaguchi, J.; Liu, G.; Itami, K. Org. Lett. 2014, 16, 4212–4215. doi:10.1021/ol5019135
Return to citation in text: [1] -
Ammann, S. E.; Liu, W.; White, M. C. Angew. Chem., Int. Ed. 2016, 55, 9571–9575. doi:10.1002/anie.201603576
Return to citation in text: [1] -
Cochet, T.; Bellosta, V.; Roche, D.; Ortholand, J.-Y.; Greiner, A.; Cossy, J. Chem. Commun. 2012, 48, 10745–10747. doi:10.1039/c2cc36067e
Return to citation in text: [1] -
Shibata, Y.; Kudo, E.; Sugiyama, H.; Uekusa, H.; Tanaka, K. Organometallics 2016, 35, 1547–1552. doi:10.1021/acs.organomet.6b00143
Return to citation in text: [1] -
Burman, J. S.; Blakey, S. B. Angew. Chem., Int. Ed. 2017, 56, 13666–13669. doi:10.1002/anie.201707021
Return to citation in text: [1] -
Hüttel, R.; Kratzer, J.; Bechter, M. Chem. Ber. 1961, 94, 766–780. doi:10.1002/cber.19610940329
Return to citation in text: [1] -
Hüttel, R.; Christ, H. Chem. Ber. 1963, 96, 3101–3104. doi:10.1002/cber.19630961202
Return to citation in text: [1] -
Trost, B. M.; Fullerton, T. J. J. Am. Chem. Soc. 1973, 95, 292–294. doi:10.1021/ja00782a080
Return to citation in text: [1] -
Bao, W.; Kossen, H.; Schneider, U. J. Am. Chem. Soc. 2017, 139, 4362–4365. doi:10.1021/jacs.7b01542
Return to citation in text: [1] -
Wei, X.-F.; Xie, X.-W.; Shimizu, Y.; Kanai, M. J. Am. Chem. Soc. 2017, 139, 4647–4650. doi:10.1021/jacs.7b01254
Return to citation in text: [1] -
Michigami, K.; Mita, T.; Sato, Y. J. Am. Chem. Soc. 2017, 139, 6094–6097. doi:10.1021/jacs.7b02775
Return to citation in text: [1] [2] [3] [4] -
Mita, T.; Hanagata, S.; Michigami, K.; Sato, Y. Org. Lett. 2017, 19, 5876–5879. doi:10.1021/acs.orglett.7b02871
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Ren, P.; Pike, S. D.; Pernik, I.; Weller, A. S.; Willis, M. C. Organometallics 2015, 34, 711–723. doi:10.1021/om500984y
Return to citation in text: [1] -
Tamaru, Y. J. Organomet. Chem. 1999, 576, 215–231. doi:10.1016/S0022-328X(98)01060-2
Return to citation in text: [1] [2] -
Marshall, J. A. Chem. Rev. 2000, 100, 3163–3186. doi:10.1021/cr000003u
Return to citation in text: [1] [2] -
Szabó, K. J. Chem. – Eur. J. 2004, 10, 5268–5275. doi:10.1002/chem.200400261
Return to citation in text: [1] [2] -
Szabó, K. J. Synlett 2006, 811–824. doi:10.1055/s-2006-933137
Return to citation in text: [1] [2] -
Zanoni, G.; Pontiroli, A.; Marchetti, A.; Vidari, G. Eur. J. Org. Chem. 2007, 3599–3611. doi:10.1002/ejoc.200700054
Return to citation in text: [1] [2] -
Spielmann, K.; Niel, G.; de Figueiredo, R. M.; Campagne, J.-M. Chem. Soc. Rev. 2018, 47, 1159–1173. doi:10.1039/C7CS00449D
Return to citation in text: [1] [2] -
Mita, T.; Higuchi, Y.; Sato, Y. Chem. – Eur. J. 2015, 21, 16391–16394. doi:10.1002/chem.201503359
Return to citation in text: [1] [2] -
Mita, T.; Tanaka, H.; Higuchi, Y.; Sato, Y. Org. Lett. 2016, 18, 2754–2757. doi:10.1021/acs.orglett.6b01231
Return to citation in text: [1] [2] -
Higuchi, Y.; Mita, T.; Sato, Y. Org. Lett. 2017, 19, 2710–2713. doi:10.1021/acs.orglett.7b01055
Return to citation in text: [1] [2]
| 1. | Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147–1169. doi:10.1021/cr900184e |
| 2. | Jazzar, R.; Hitce, J.; Renaudat, A.; Sofack-Kreutzer, J.; Baudoin, O. Chem. – Eur. J. 2010, 16, 2654–2672. doi:10.1002/chem.200902374 |
| 3. | Wendlandt, A. E.; Suess, A. M.; Stahl, S. S. Angew. Chem., Int. Ed. 2011, 50, 11062–11087. doi:10.1002/anie.201103945 |
| 4. | Dastbaravardeh, N.; Christakakou, M.; Haider, M.; Schnürch, M. Synthesis 2014, 46, 1421–1439. doi:10.1055/s-0033-1338625 |
| 5. | Guo, X.-X.; Gu, D.-W.; Wu, Z.; Zhang, W. Chem. Rev. 2015, 115, 1622–1651. doi:10.1021/cr500410y |
| 6. | Hartwig, J. F.; Larsen, M. A. ACS Cent. Sci. 2016, 2, 281–292. doi:10.1021/acscentsci.6b00032 |
| 7. | Gensch, T.; Hopkinson, M. N.; Glorius, F.; Wencel-Delord, J. Chem. Soc. Rev. 2016, 45, 2900–2936. doi:10.1039/C6CS00075D |
| 8. | Yoshino, T.; Matsunaga, S. Adv. Synth. Catal. 2017, 359, 1245–1262. doi:10.1002/adsc.201700042 |
| 9. | He, J.; Wasa, M.; Chan, K. S. L.; Shao, Q.; Yu, J.-Q. Chem. Rev. 2017, 117, 8754–8786. doi:10.1021/acs.chemrev.6b00622 |
| 16. | Chen, M. S.; White, M. C. J. Am. Chem. Soc. 2004, 126, 1346–1347. doi:10.1021/ja039107n |
| 17. | Young, A. J.; White, M. C. J. Am. Chem. Soc. 2008, 130, 14090–14091. doi:10.1021/ja806867p |
| 18. | Kondo, H.; Yu, F.; Yamaguchi, J.; Liu, G.; Itami, K. Org. Lett. 2014, 16, 4212–4215. doi:10.1021/ol5019135 |
| 19. | Ammann, S. E.; Liu, W.; White, M. C. Angew. Chem., Int. Ed. 2016, 55, 9571–9575. doi:10.1002/anie.201603576 |
| 20. | Cochet, T.; Bellosta, V.; Roche, D.; Ortholand, J.-Y.; Greiner, A.; Cossy, J. Chem. Commun. 2012, 48, 10745–10747. doi:10.1039/c2cc36067e |
| 21. | Shibata, Y.; Kudo, E.; Sugiyama, H.; Uekusa, H.; Tanaka, K. Organometallics 2016, 35, 1547–1552. doi:10.1021/acs.organomet.6b00143 |
| 22. | Burman, J. S.; Blakey, S. B. Angew. Chem., Int. Ed. 2017, 56, 13666–13669. doi:10.1002/anie.201707021 |
| 28. | Michigami, K.; Mita, T.; Sato, Y. J. Am. Chem. Soc. 2017, 139, 6094–6097. doi:10.1021/jacs.7b02775 |
| 29. | Mita, T.; Hanagata, S.; Michigami, K.; Sato, Y. Org. Lett. 2017, 19, 5876–5879. doi:10.1021/acs.orglett.7b02871 |
| 15. | Salmond, W. G.; Barta, M. A.; Havens, J. L. J. Org. Chem. 1978, 43, 2057–2059. doi:10.1021/jo00404a049 |
| 28. | Michigami, K.; Mita, T.; Sato, Y. J. Am. Chem. Soc. 2017, 139, 6094–6097. doi:10.1021/jacs.7b02775 |
| 29. | Mita, T.; Hanagata, S.; Michigami, K.; Sato, Y. Org. Lett. 2017, 19, 5876–5879. doi:10.1021/acs.orglett.7b02871 |
| 31. | Tamaru, Y. J. Organomet. Chem. 1999, 576, 215–231. doi:10.1016/S0022-328X(98)01060-2 |
| 32. | Marshall, J. A. Chem. Rev. 2000, 100, 3163–3186. doi:10.1021/cr000003u |
| 33. | Szabó, K. J. Chem. – Eur. J. 2004, 10, 5268–5275. doi:10.1002/chem.200400261 |
| 34. | Szabó, K. J. Synlett 2006, 811–824. doi:10.1055/s-2006-933137 |
| 35. | Zanoni, G.; Pontiroli, A.; Marchetti, A.; Vidari, G. Eur. J. Org. Chem. 2007, 3599–3611. doi:10.1002/ejoc.200700054 |
| 36. | Spielmann, K.; Niel, G.; de Figueiredo, R. M.; Campagne, J.-M. Chem. Soc. Rev. 2018, 47, 1159–1173. doi:10.1039/C7CS00449D |
| 37. | Mita, T.; Higuchi, Y.; Sato, Y. Chem. – Eur. J. 2015, 21, 16391–16394. doi:10.1002/chem.201503359 |
| 38. | Mita, T.; Tanaka, H.; Higuchi, Y.; Sato, Y. Org. Lett. 2016, 18, 2754–2757. doi:10.1021/acs.orglett.6b01231 |
| 39. | Higuchi, Y.; Mita, T.; Sato, Y. Org. Lett. 2017, 19, 2710–2713. doi:10.1021/acs.orglett.7b01055 |
| 14. | Młochowski, J.; Wójtowicz-Młochowska, H. Molecules 2015, 20, 10205–10243. doi:10.3390/molecules200610205 |
| 30. | Ren, P.; Pike, S. D.; Pernik, I.; Weller, A. S.; Willis, M. C. Organometallics 2015, 34, 711–723. doi:10.1021/om500984y |
| 10. | Dounay, A. B.; Overman, L. E. Chem. Rev. 2003, 103, 2945–2964. doi:10.1021/cr020039h |
| 11. | Vougioukalakis, G. C.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746–1787. doi:10.1021/cr9002424 |
| 12. | Crossley, S. W. M.; Obradors, C.; Martinez, R. M.; Shenvi, R. A. Chem. Rev. 2016, 116, 8912–9000. doi:10.1021/acs.chemrev.6b00334 |
| 13. | Shigehisa, H. Chem. Pharm. Bull. 2018, 66, 339–346. doi:10.1248/cpb.c17-01006 |
| 31. | Tamaru, Y. J. Organomet. Chem. 1999, 576, 215–231. doi:10.1016/S0022-328X(98)01060-2 |
| 32. | Marshall, J. A. Chem. Rev. 2000, 100, 3163–3186. doi:10.1021/cr000003u |
| 33. | Szabó, K. J. Chem. – Eur. J. 2004, 10, 5268–5275. doi:10.1002/chem.200400261 |
| 34. | Szabó, K. J. Synlett 2006, 811–824. doi:10.1055/s-2006-933137 |
| 35. | Zanoni, G.; Pontiroli, A.; Marchetti, A.; Vidari, G. Eur. J. Org. Chem. 2007, 3599–3611. doi:10.1002/ejoc.200700054 |
| 36. | Spielmann, K.; Niel, G.; de Figueiredo, R. M.; Campagne, J.-M. Chem. Soc. Rev. 2018, 47, 1159–1173. doi:10.1039/C7CS00449D |
| 37. | Mita, T.; Higuchi, Y.; Sato, Y. Chem. – Eur. J. 2015, 21, 16391–16394. doi:10.1002/chem.201503359 |
| 38. | Mita, T.; Tanaka, H.; Higuchi, Y.; Sato, Y. Org. Lett. 2016, 18, 2754–2757. doi:10.1021/acs.orglett.6b01231 |
| 39. | Higuchi, Y.; Mita, T.; Sato, Y. Org. Lett. 2017, 19, 2710–2713. doi:10.1021/acs.orglett.7b01055 |
| 28. | Michigami, K.; Mita, T.; Sato, Y. J. Am. Chem. Soc. 2017, 139, 6094–6097. doi:10.1021/jacs.7b02775 |
| 29. | Mita, T.; Hanagata, S.; Michigami, K.; Sato, Y. Org. Lett. 2017, 19, 5876–5879. doi:10.1021/acs.orglett.7b02871 |
| 29. | Mita, T.; Hanagata, S.; Michigami, K.; Sato, Y. Org. Lett. 2017, 19, 5876–5879. doi:10.1021/acs.orglett.7b02871 |
| 27. | Wei, X.-F.; Xie, X.-W.; Shimizu, Y.; Kanai, M. J. Am. Chem. Soc. 2017, 139, 4647–4650. doi:10.1021/jacs.7b01254 |
| 28. | Michigami, K.; Mita, T.; Sato, Y. J. Am. Chem. Soc. 2017, 139, 6094–6097. doi:10.1021/jacs.7b02775 |
| 29. | Mita, T.; Hanagata, S.; Michigami, K.; Sato, Y. Org. Lett. 2017, 19, 5876–5879. doi:10.1021/acs.orglett.7b02871 |
| 26. | Bao, W.; Kossen, H.; Schneider, U. J. Am. Chem. Soc. 2017, 139, 4362–4365. doi:10.1021/jacs.7b01542 |
| 23. | Hüttel, R.; Kratzer, J.; Bechter, M. Chem. Ber. 1961, 94, 766–780. doi:10.1002/cber.19610940329 |
| 24. | Hüttel, R.; Christ, H. Chem. Ber. 1963, 96, 3101–3104. doi:10.1002/cber.19630961202 |
| 25. | Trost, B. M.; Fullerton, T. J. J. Am. Chem. Soc. 1973, 95, 292–294. doi:10.1021/ja00782a080 |
| 29. | Mita, T.; Hanagata, S.; Michigami, K.; Sato, Y. Org. Lett. 2017, 19, 5876–5879. doi:10.1021/acs.orglett.7b02871 |
© 2018 Mita et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)