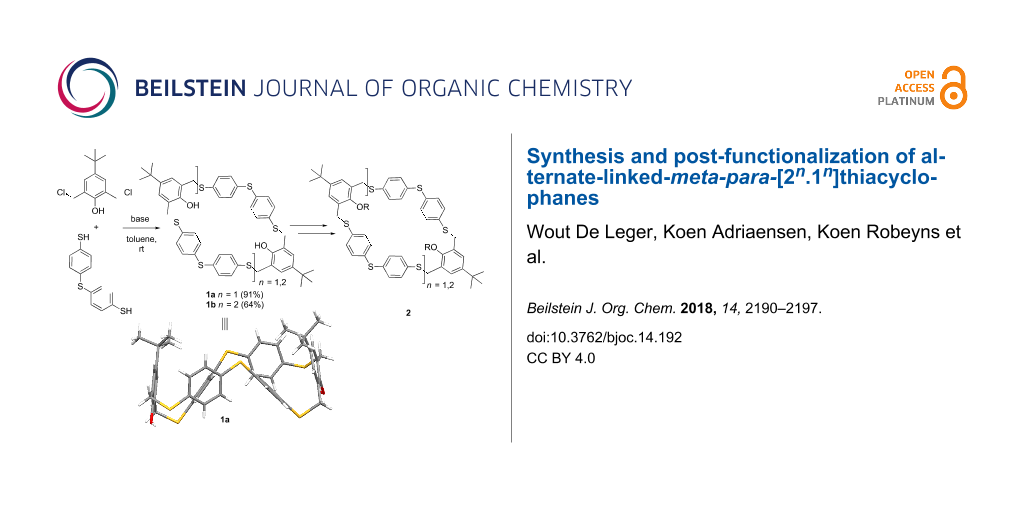
Abstract
In recent decades, considerable research attention has been devoted to new synthetic procedures for thiacyclophanes. Thiacyclophanes are widely used as host molecules for the molecular recognition of organic compounds as well as metals. Herein, we report the selective and high-yielding synthesis of novel alternate-linked-meta-para-thiacyclophanes. These novel thiacyclophanes are selectively synthesized in high-yielding procedures. Furthermore, post-functionalization of the phenolic moieties was successfully performed. The 3D structure of the alternate-linked-meta-para-[22.12]thiacyclophane was further elucidated via X-ray crystallographic analysis.

Graphical Abstract
Introduction
The ability of cyclophanes to form three-dimensional cavities is interesting for various potential applications, e.g., as supramolecular hosts. Synthetic procedures towards novel cyclophanes have attracted much interest in the scientific community [1-4]. An interesting subclass of cyclophanes is formed by thiacyclophanes, in which the thioether linkages impose less conformational strain and which have an increased cavity size compared to other (oxa/aza)cyclophanes. Thiacalix[n]arenes are among the most widely known thiacyclophanes with significant ability for molecular recognition [5-9]. Our group has experience in the synthesis of homothiacalix[n]arenes, a subclass of the thiacalix[n]arenes that has so far received little attention compared to other homoheteracalix[n]arenes [10,11]. Homothiacalix[4]arenes were successfully synthesized via nucleophilic substitution and homodithiacalix[4]arenes by dynamic covalent chemistry [12-14]. Functionalization of homothiacalix[4]arenes was made possible by changing the precursors before macrocyclization (Scheme 1) [12].
![[1860-5397-14-192-i1]](/bjoc/content/inline/1860-5397-14-192-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Macrocyclization towards homothiacalixarenes 3a and 3b [12].
Scheme 1: Macrocyclization towards homothiacalixarenes 3a and 3b [12].
Recently, pillar[n]arenes have attracted much interest as new supramolecular receptors due to their pillar-shaped structure [15-17]. Wang et al. were the first to report a one-pot procedure towards the synthesis of various thiapillararenes [18-20]. Our group reported a disulfide-bridged [2n] pillararene-like molecule in a two-step procedure [21]. In contrast to meta-para-bridged azacyclophanes [22-25], less synthetic work has been performed on the synthesis of sulfur-linked cyclophanes with an alternating meta-para-bridge [26]. Herein we report a one-pot macrocyclization of meta-para-bridged thiacyclophanes by means of a substitution reaction between the biselectrophile 2,6-bis(chloromethyl)-4-tert-butylphenol (4) and the bisnucleophile 4,4’-thiobisbenzenethiol (5). Either the [2 + 2] adduct 6 or the [3 + 3] adduct 7 were selectively obtained by varying the reaction conditions. Post-functionalization of the phenolic moieties was successfully performed. Alternate-linked-meta-para-[22.12]thiacyclophane 6 was further analysed by X-ray diffraction.
Results and Discussion
Macrocyclization
Macrocyclization and post-functionalization of cyclophanes is of high interest for the development of various applications in molecular recognition. Based on our previous one-pot procedure towards homothiacalixarenes [12], we now report the development of an alternate meta-para-thiacyclophane which could be post-functionalized.
The precursors 2,6-bis(chloromethyl)-4-tert-butylphenol (4) and commercially available 4,4’-thiobisbenzenethiol (5) were chosen as the biselectrophile and bisnucleophile, respectively [27]. Cyclocondensation of 4 and 5 under highly diluted conditions (employing a syringe pump) resulted in the formation of a mixture of the [2 + 2] adduct 6 and the [3 + 3] adduct 7 (Scheme 2 and Table 1, entry 1).
![[1860-5397-14-192-i2]](/bjoc/content/inline/1860-5397-14-192-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Cyclocondensation reaction of 4 and 5 towards [2 + 2] and [3 + 3] adducts.
Scheme 2: Cyclocondensation reaction of 4 and 5 towards [2 + 2] and [3 + 3] adducts.
Table 1: Optimisation towards alternate-linked-meta-para-thiacyclophane 6 and 7.
| entry | basea | solvent | concentration (mM) | temperature (°C) | time (h) | NMR yield of 6b | NMR yield of 7b |
|---|---|---|---|---|---|---|---|
| 1 | K2CO3 | THF | 20c | rt | 18 | 26 | 61 |
| 2 | K2CO3 | THF | 40 | rt | 1 | 14 | 62 |
| 3 | KOH | THF | 40 | rt | 1 | 3 | 82 |
| 4 | K2CO3 | toluene | 40 | rt | 1 | 21 | 67 |
| 5 | K2CO3 | toluene | 4d | rt | 1 | 68 | 17 |
| 6 | K2CO3 | toluene | 4 | rt | 72 | 79 | 21 |
| 7 | K2CO3 | toluene | 4 | rt | 168 | –e (91%)f | – |
| 8 | t-BuOK (2 equiv) | toluene | 4 | rt | 1.5 | 2 | 92 (64%)f |
a1.2 Equivalents used unless otherwise stated. bConversions in all reactions were measured by 1H NMR spectroscopy in CDCl3 at 25 °C. The conversion was calculated using the signals from the aromatic peaks of the phenol moiety (6.88 ppm for [3 + 3], 6.86–6.83 ppm for oligomers and 6.67 ppm for [2 + 2] adduct). cBiselectrophile and bisnucleophile added with syringe pump over 6 h. dO2 free conditions by flushing with argon. eReaction product precipitated from the reaction mixture. fIsolated yield.
The optimization study was further conducted under diluted conditions and shorter reaction time (Table 1, entries 2–5). As summarized in Table 1, the reaction is strongly affected by time, base and solvent. At higher concentrations, a decreased yield of 6 is observed. A more significant effect on the selectivity towards 7 was observed by screening different bases (Supporting Information File 1, Table S1, entries 2, 3, 9, and 10). Interestingly, stronger bases let to the almost exclusive formation of the [3 + 3] adduct 7 (Table 1, entry 3) over [2 + 2] adduct 6. A higher proportion of macrocycle 6 was obtained using K2CO3 as the base and toluene as the solvent, the yield rose from 14% (Table 1, entry 2) to 21% (Table 1, entry 4). Working at low concentrations (4 mM) and under O2 free conditions gave rise to a significant increase in the formation of 6 (Table 1, entry 5). The [3 + 3] adduct 7 is significantly disfavoured under these conditions. On the basis of these data it could be hypothesized that the [2 + 2] adduct 6 is the more thermodynamically favoured product and the [3 + 3] adduct 7 is the kinetic product. When the reaction was carried out at higher temperatures, an increase in the formation of open chain oligomers was observed (Supporting Information File 1, Table S1, entries 13 and 14). A longer reaction time at low concentration resulted in a higher yield of the [2 + 2] adduct 6. After 6 days a white precipitate was observed in the reaction mixture (Table 1, entry 7). Filtration of the precipitate followed by successively washing with water and methanol resulted in an isolated yield of 91%. The 3D structure of macrocycle 6 was confirmed by single crystal X-ray diffraction and shows approximate twofold rotational symmetry (point group C2, Figure 1). The dihedral angles between the aromatic rings are given in Table S2 (Supporting Information File 1, ring numbering as shown in Figure 1). The conformation in the solid state is stabilised by intramolecular O–H···S hydrogen bonds (O···S distances 3.2081 (16) and 3.4179 (17) Å) and shows no central void.
![[1860-5397-14-192-1]](/bjoc/content/figures/1860-5397-14-192-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray crystal structure of alternate-linked-meta-para-thiacyclophane 6: (a) ball-and-stick representation, with O–H···S hydrogen bonds shown as green dashed lines, (b) space-filling representation viewed along the pseudo twofold axis.
Figure 1: X-ray crystal structure of alternate-linked-meta-para-thiacyclophane 6: (a) ball-and-stick represen...
The formation of the [3 + 3] product is favoured by stronger bases and shorter reaction times. Therefore, in entry 8 of Table 1, we report our method of choice to selectively synthesize 7. Purification of [3 + 3] adduct 7 was successful via precipitation (CHCl3/MeOH) in good yield (64%).
Proposed mechanism and stability
In Scheme 3 a reaction mechanism for the formation of the macrocycles is proposed. It is believed that after deprotonation of the OH group and subsequent chloride loss of 4 an o-quinoid structure 9 is formed in situ, that quickly reacts with the deprotonated thiol 10 via Michael addition (Scheme 3, route A) [28-30]. However, as aromatic thiols at the benzylic position are good leaving groups, conversely β-elimination (route B) can easily occur, leading to reversal of the addition reactions.
![[1860-5397-14-192-i3]](/bjoc/content/inline/1860-5397-14-192-i3.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed reaction mechanism towards alternate-linked-meta-para-thiacyclophanes.
Scheme 3: Proposed reaction mechanism towards alternate-linked-meta-para-thiacyclophanes.
This was observed in a stability experiment where pure [3 + 3] macrocycle 7 in CDCl3 (without adding base) over two weeks’ time converted into a mixture of [3 + 3] macrocycle 7 (9%), acyclic oligomers (50%) and the more thermodynamically stable [2 + 2] macrocycle 6 (41%, see Figure S13, Supporting Information File 1). Due to the instability of the macrocycles, purification of the reaction mixtures via column chromatography (silica/alumina) or selective crystallization was difficult.
Changing the biselectrophile precursor to anisole derivative 13, for which the ortho-quinoid formation is not possible, did not led to the formation of macrocycle 14 or 15 under the optimized conditions, supporting the proposed mechanism (Scheme 4). Mainly starting material was observed in the reaction mixture.
![[1860-5397-14-192-i4]](/bjoc/content/inline/1860-5397-14-192-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Attempted cyclocondensation with anisole derivative 13, products 14 and 15 were not formed.
Scheme 4: Attempted cyclocondensation with anisole derivative 13, products 14 and 15 were not formed.
The syntheses of various phenolic thiamacrocycles are reported under basic conditions [31-34]. To the best of our knowledge, reactions of thiamacrocycles under acidic conditions have not been reported in contrast to homooxacalix[n]arenes, for example [35,36].
A reaction under acidic conditions with precursor 16 was investigated based on similar conditions as reported by Cragg et al. [35]. The reaction in the presence of p-toluenesulfonic acid (0.05 equiv) led mainly to oligomerization, although in the 1H NMR spectrum traces of macrocycles 6 and 7 were observed (Scheme 5).
![[1860-5397-14-192-i5]](/bjoc/content/inline/1860-5397-14-192-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Macrocyclization under acidic conditions, with only traces of 6 and 7 observed.
Scheme 5: Macrocyclization under acidic conditions, with only traces of 6 and 7 observed.
Post-functionalization
As the macrocycles are not stable in solution or in basic medium, most of the initial attempts to post-functionalization with ethyl bromoacetate (17) resulted in complex reaction mixtures. Reactions at room temperature or higher temperature (25–60 °C) mainly resulted in the transformation of the [2 + 2] adduct 6 to the functionalized [3 + 3] adduct 19. Traces of unidentified oligomers and functionalized [2 + 2] macrocycle were also observed in the reaction mixture. It can be argued that, due to steric hindrance, the alkylation of macrocycle 6 is slow. The less sterically hindered cyclic trimer 7 and the linear oligomers are therefore faster alkylated and removed from the equilibrium between macrocycle 6 and 7. Macrocyclization under the optimal conditions (Table 1), followed by in situ post-functionalization (DBU 2.2 equiv, ethyl bromoacetate 3 equiv) in a one-pot procedure also led to a shift towards the functionalized [3 + 3] adduct 19. Purification of these complex reaction mixtures was not successful. Therefore, in order to prevent β-elimination, lower temperatures were applied. Cooling of the reaction mixtures to −20 °C had a positive effect on the stability of the macrocycles. Despite this, no full conversion was obtained, even with a strong base (NaH). Further exploration of the reaction conditions towards the functionalized macrocycles 18 and 19 indicated that the reaction proceeds best at 0 °C, using NaH as a base combined with a large excess of the appropriate electrophile 17 (15 equivalents per hydroxy moiety, Scheme 6). Functionalized macrocycles 18 and 19 were obtained in good yields 93% and 77%, respectively.
![[1860-5397-14-192-i6]](/bjoc/content/inline/1860-5397-14-192-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Post-functionalization of thiacyclophanes 6 and 7 with ethyl bromoacetate (17).
Scheme 6: Post-functionalization of thiacyclophanes 6 and 7 with ethyl bromoacetate (17).
Further modification of the functionalized [2 + 2] macrocycle 18 towards the amide derivative 20 and the acid derivative 21 were successfully performed in good yields (Scheme 7). Functionalization of the phenolic moieties afforded a stable macrocycle under various conditions (basic medium, heat). This also indicates that ring opening occurs via an o-quinoid structure like 9 (Scheme 3).
![[1860-5397-14-192-i7]](/bjoc/content/inline/1860-5397-14-192-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Modification of the functionalized [2 + 2] adduct 18 towards an amide derivative 20 and acid derivative 21.
Scheme 7: Modification of the functionalized [2 + 2] adduct 18 towards an amide derivative 20 and acid deriva...
Conclusion
In conclusion, a selective procedure was developed towards alternate-linked-meta-para-thiacyclophanes. Starting from readily available materials, the [2 + 2] adduct 6 was synthesized in a high-yielding protocol. A major benefit of the procedure is the simple work-up as the product precipitates from the reaction mixture. Furthermore, it was also possible to selectively synthesize the [3 + 3] adduct 7 in good yield, while avoiding chromatography. The unfunctionalized macrocycles are labile in neutral solution or basic medium. However, post-functionalization of the macrocycles was successfully realized at low temperatures and with a large excess of the electrophile. Functionalization of the [2 + 2] macrocycle 6 towards an amide derivative 20 and acid derivative 21 was performed with good overall yields (three steps), 80% and 54%. In the near future, the binding properties of these interesting alternate-linked-meta-para-thiacyclophanes will be investigated.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 1.0 MB | Download |
References
-
Gutsche, C. D. Calixarenes Revisited; Royal Society of Chemistry: Cambridge, 1998.
Return to citation in text: [1] -
Liu, Z.; Nalluri, S. K. M.; Stoddart, J. F. Chem. Soc. Rev. 2017, 46, 2459–2478. doi:10.1039/C7CS00185A
Return to citation in text: [1] -
Böhmer, V. Angew. Chem., Int. Ed. Engl. 1995, 34, 713–745. doi:10.1002/anie.199507131
Return to citation in text: [1] -
Gutsche, C. D. Calixarenes; Royal Society of Chemistry: Cambridge, 1989.
Return to citation in text: [1] -
Lhoták, P. Eur. J. Org. Chem. 2004, 1675–1692. doi:10.1002/ejoc.200300492
Return to citation in text: [1] -
Morohashi, N.; Narumi, F.; Iki, N.; Hattori, T.; Miyano, S. Chem. Rev. 2006, 106, 5291–5316. doi:10.1021/cr050565j
Return to citation in text: [1] -
König, B.; Fonseca, M. H. Eur. J. Inorg. Chem. 2000, 2303–2310. doi:10.1002/1099-0682(200011)2000:11<2303::AID-EJIC2303>3.0.CO;2-Y
Return to citation in text: [1] -
Kumar, R.; Lee, Y. O.; Bhalla, V.; Kumar, M.; Kim, J. S. Chem. Soc. Rev. 2014, 43, 4824. doi:10.1039/c4cs00068d
Return to citation in text: [1] -
Wang, M.-X. Chem. Commun. 2008, 4541–4551. doi:10.1039/b809287g
Return to citation in text: [1] -
Cottet, K.; Marcos, P. M.; Cragg, P. J. Beilstein J. Org. Chem. 2012, 8, 201–226. doi:10.3762/bjoc.8.22
Return to citation in text: [1] -
Thomas, J.; Gusak, A. S.; Dehaen, W. Homoheteracalix[n]arenes (X = S, Se, N). In Calixarenes and Beyond; Neri, P.; Sessler, J. L.; Wang, M.-X., Eds.; Springer International Publishing: Cham, 2016.
Return to citation in text: [1] -
Thomas, J.; Van Hecke, K.; Robeyns, K.; Van Rossom, W.; Sonawane, M. P.; Van Meervelt, L.; Smet, M.; Maes, W.; Dehaen, W. Chem. – Eur. J. 2011, 17, 10339–10349. doi:10.1002/chem.201101690
Return to citation in text: [1] [2] [3] [4] -
Sonawane, M. P.; Van Hecke, K.; Jacobs, J.; Thomas, J.; Van Meervelt, L.; Dehaen, W.; Van Rossom, W. J. Org. Chem. 2012, 77, 8444–8450. doi:10.1021/jo301321w
Return to citation in text: [1] -
Thomas, J.; Reekmans, G.; Adriaensens, P.; Van Meervelt, L.; Smet, M.; Maes, W.; Dehaen, W.; Dobrzańska, L. Angew. Chem., Int. Ed. 2013, 52, 10237–10240. doi:10.1002/anie.201302822
Return to citation in text: [1] -
Ogoshi, T.; Yamagishi, T.-a.; Nakamoto, Y. Chem. Rev. 2016, 116, 7937–8002. doi:10.1021/acs.chemrev.5b00765
Return to citation in text: [1] -
Xue, M.; Yang, Y.; Chi, X.; Zhang, Z.; Huang, F. Acc. Chem. Res. 2012, 45, 1294–1308. doi:10.1021/ar2003418
Return to citation in text: [1] -
Yang, K.; Pei, Y.; Wen, J.; Pei, Z. Chem. Commun. 2016, 52, 9316–9326. doi:10.1039/C6CC03641D
Return to citation in text: [1] -
Fu, Z.-D.; Guo, Q.-H.; Zhao, L.; Wang, D.-X.; Wang, M.-X. Org. Lett. 2016, 18, 2668–2671. doi:10.1021/acs.orglett.6b01112
Return to citation in text: [1] -
Guo, Q.-H.; Zhao, L.; Wang, M.-X. Chem. – Eur. J. 2016, 22, 6947–6955. doi:10.1002/chem.201600462
Return to citation in text: [1] -
Guo, Q.-H.; Zhao, L.; Wang, M.-X. Angew. Chem., Int. Ed. 2015, 54, 8386–8389. doi:10.1002/anie.201503179
Return to citation in text: [1] -
Sonawane, M. P.; Jacobs, J.; Thomas, J.; Van Meervelt, L.; Dehaen, W. Chem. Commun. 2013, 49, 6310–6312. doi:10.1039/c3cc42984a
Return to citation in text: [1] -
Ito, A.; Ono, Y.; Tanaka, K. Angew. Chem., Int. Ed. 2000, 39, 1072–1075. doi:10.1002/(SICI)1521-3773(20000317)39:6<1072::AID-ANIE1072>3.0.CO;2-1
Return to citation in text: [1] -
Kurata, R.; Sakamaki, D.; Ito, A. Org. Lett. 2017, 19, 3115–3118. doi:10.1021/acs.orglett.7b01229
Return to citation in text: [1] -
Ito, A.; Uebe, M.; Kurata, R.; Yano, S.; Fueno, H.; Matsumoto, T. Chem. – Asian J. 2018, 13, 754–760. doi:10.1002/asia.201701717
Return to citation in text: [1] -
Sakamaki, D.; Ito, A.; Furukawa, K.; Kato, T.; Tanaka, K. Chem. Commun. 2009, 4524–4526. doi:10.1039/b907792h
Return to citation in text: [1] -
Seppälä, T.; Wegelius, E.; Rissanen, K. New J. Chem. 1998, 22, 789–791. doi:10.1039/a803490g
Return to citation in text: [1] -
Paine, R. T.; Tan, Y.-C.; Gan, X.-M. Inorg. Chem. 2001, 40, 7009–7013. doi:10.1021/ic0107633
Return to citation in text: [1] -
Walden, D. M.; Jaworski, A. A.; Johnston, R. C.; Hovey, M. T.; Baker, H. V.; Meyer, M. P.; Scheidt, K. A.; Cheong, P. H.-Y. J. Org. Chem. 2017, 82, 7183–7189. doi:10.1021/acs.joc.7b00697
Return to citation in text: [1] -
Lewis, R. S.; Garza, C. J.; Dang, A. T.; Pedro, T. K. A.; Chain, W. J. Org. Lett. 2015, 17, 2278–2281. doi:10.1021/acs.orglett.5b00972
Return to citation in text: [1] -
Merijan, A.; Shoulders, B. A.; Gardner, P. D. J. Org. Chem. 1963, 28, 2148–2149. doi:10.1021/jo01043a507
Return to citation in text: [1] -
Ashram, M. J. Inclusion Phenom. Macrocyclic Chem. 2006, 54, 253–259. doi:10.1007/s10847-005-8648-y
Return to citation in text: [1] -
Ito, K.; Yamamori, Y.; Ohba, Y.; Sone, T. Synth. Commun. 2000, 30, 1167–1177. doi:10.1080/00397910008087137
Return to citation in text: [1] -
Ito, K.; Nagai, T.; Ohba, Y.; Sone, T. J. Heterocycl. Chem. 2000, 37, 1121–1128. doi:10.1002/jhet.5570370516
Return to citation in text: [1] -
Sonawane, M. P.; Boodts, S.; Thomas, J.; Dehaen, W. Supramol. Chem. 2014, 26, 547–551. doi:10.1080/10610278.2013.868898
Return to citation in text: [1] -
Miah, M.; Romanov, N. N.; Cragg, P. J. J. Org. Chem. 2002, 67, 3124–3126. doi:10.1021/jo025597a
Return to citation in text: [1] [2] -
Hampton, P. D.; Bencze, Z.; Tong, W.; Daitch, C. E. J. Org. Chem. 1994, 59, 4838–4843. doi:10.1021/jo00096a026
Return to citation in text: [1]
| 35. | Miah, M.; Romanov, N. N.; Cragg, P. J. J. Org. Chem. 2002, 67, 3124–3126. doi:10.1021/jo025597a |
| 1. | Gutsche, C. D. Calixarenes Revisited; Royal Society of Chemistry: Cambridge, 1998. |
| 2. | Liu, Z.; Nalluri, S. K. M.; Stoddart, J. F. Chem. Soc. Rev. 2017, 46, 2459–2478. doi:10.1039/C7CS00185A |
| 3. | Böhmer, V. Angew. Chem., Int. Ed. Engl. 1995, 34, 713–745. doi:10.1002/anie.199507131 |
| 4. | Gutsche, C. D. Calixarenes; Royal Society of Chemistry: Cambridge, 1989. |
| 12. | Thomas, J.; Van Hecke, K.; Robeyns, K.; Van Rossom, W.; Sonawane, M. P.; Van Meervelt, L.; Smet, M.; Maes, W.; Dehaen, W. Chem. – Eur. J. 2011, 17, 10339–10349. doi:10.1002/chem.201101690 |
| 31. | Ashram, M. J. Inclusion Phenom. Macrocyclic Chem. 2006, 54, 253–259. doi:10.1007/s10847-005-8648-y |
| 32. | Ito, K.; Yamamori, Y.; Ohba, Y.; Sone, T. Synth. Commun. 2000, 30, 1167–1177. doi:10.1080/00397910008087137 |
| 33. | Ito, K.; Nagai, T.; Ohba, Y.; Sone, T. J. Heterocycl. Chem. 2000, 37, 1121–1128. doi:10.1002/jhet.5570370516 |
| 34. | Sonawane, M. P.; Boodts, S.; Thomas, J.; Dehaen, W. Supramol. Chem. 2014, 26, 547–551. doi:10.1080/10610278.2013.868898 |
| 12. | Thomas, J.; Van Hecke, K.; Robeyns, K.; Van Rossom, W.; Sonawane, M. P.; Van Meervelt, L.; Smet, M.; Maes, W.; Dehaen, W. Chem. – Eur. J. 2011, 17, 10339–10349. doi:10.1002/chem.201101690 |
| 13. | Sonawane, M. P.; Van Hecke, K.; Jacobs, J.; Thomas, J.; Van Meervelt, L.; Dehaen, W.; Van Rossom, W. J. Org. Chem. 2012, 77, 8444–8450. doi:10.1021/jo301321w |
| 14. | Thomas, J.; Reekmans, G.; Adriaensens, P.; Van Meervelt, L.; Smet, M.; Maes, W.; Dehaen, W.; Dobrzańska, L. Angew. Chem., Int. Ed. 2013, 52, 10237–10240. doi:10.1002/anie.201302822 |
| 35. | Miah, M.; Romanov, N. N.; Cragg, P. J. J. Org. Chem. 2002, 67, 3124–3126. doi:10.1021/jo025597a |
| 36. | Hampton, P. D.; Bencze, Z.; Tong, W.; Daitch, C. E. J. Org. Chem. 1994, 59, 4838–4843. doi:10.1021/jo00096a026 |
| 10. | Cottet, K.; Marcos, P. M.; Cragg, P. J. Beilstein J. Org. Chem. 2012, 8, 201–226. doi:10.3762/bjoc.8.22 |
| 11. | Thomas, J.; Gusak, A. S.; Dehaen, W. Homoheteracalix[n]arenes (X = S, Se, N). In Calixarenes and Beyond; Neri, P.; Sessler, J. L.; Wang, M.-X., Eds.; Springer International Publishing: Cham, 2016. |
| 27. | Paine, R. T.; Tan, Y.-C.; Gan, X.-M. Inorg. Chem. 2001, 40, 7009–7013. doi:10.1021/ic0107633 |
| 5. | Lhoták, P. Eur. J. Org. Chem. 2004, 1675–1692. doi:10.1002/ejoc.200300492 |
| 6. | Morohashi, N.; Narumi, F.; Iki, N.; Hattori, T.; Miyano, S. Chem. Rev. 2006, 106, 5291–5316. doi:10.1021/cr050565j |
| 7. | König, B.; Fonseca, M. H. Eur. J. Inorg. Chem. 2000, 2303–2310. doi:10.1002/1099-0682(200011)2000:11<2303::AID-EJIC2303>3.0.CO;2-Y |
| 8. | Kumar, R.; Lee, Y. O.; Bhalla, V.; Kumar, M.; Kim, J. S. Chem. Soc. Rev. 2014, 43, 4824. doi:10.1039/c4cs00068d |
| 9. | Wang, M.-X. Chem. Commun. 2008, 4541–4551. doi:10.1039/b809287g |
| 28. | Walden, D. M.; Jaworski, A. A.; Johnston, R. C.; Hovey, M. T.; Baker, H. V.; Meyer, M. P.; Scheidt, K. A.; Cheong, P. H.-Y. J. Org. Chem. 2017, 82, 7183–7189. doi:10.1021/acs.joc.7b00697 |
| 29. | Lewis, R. S.; Garza, C. J.; Dang, A. T.; Pedro, T. K. A.; Chain, W. J. Org. Lett. 2015, 17, 2278–2281. doi:10.1021/acs.orglett.5b00972 |
| 30. | Merijan, A.; Shoulders, B. A.; Gardner, P. D. J. Org. Chem. 1963, 28, 2148–2149. doi:10.1021/jo01043a507 |
| 21. | Sonawane, M. P.; Jacobs, J.; Thomas, J.; Van Meervelt, L.; Dehaen, W. Chem. Commun. 2013, 49, 6310–6312. doi:10.1039/c3cc42984a |
| 26. | Seppälä, T.; Wegelius, E.; Rissanen, K. New J. Chem. 1998, 22, 789–791. doi:10.1039/a803490g |
| 18. | Fu, Z.-D.; Guo, Q.-H.; Zhao, L.; Wang, D.-X.; Wang, M.-X. Org. Lett. 2016, 18, 2668–2671. doi:10.1021/acs.orglett.6b01112 |
| 19. | Guo, Q.-H.; Zhao, L.; Wang, M.-X. Chem. – Eur. J. 2016, 22, 6947–6955. doi:10.1002/chem.201600462 |
| 20. | Guo, Q.-H.; Zhao, L.; Wang, M.-X. Angew. Chem., Int. Ed. 2015, 54, 8386–8389. doi:10.1002/anie.201503179 |
| 12. | Thomas, J.; Van Hecke, K.; Robeyns, K.; Van Rossom, W.; Sonawane, M. P.; Van Meervelt, L.; Smet, M.; Maes, W.; Dehaen, W. Chem. – Eur. J. 2011, 17, 10339–10349. doi:10.1002/chem.201101690 |
| 15. | Ogoshi, T.; Yamagishi, T.-a.; Nakamoto, Y. Chem. Rev. 2016, 116, 7937–8002. doi:10.1021/acs.chemrev.5b00765 |
| 16. | Xue, M.; Yang, Y.; Chi, X.; Zhang, Z.; Huang, F. Acc. Chem. Res. 2012, 45, 1294–1308. doi:10.1021/ar2003418 |
| 17. | Yang, K.; Pei, Y.; Wen, J.; Pei, Z. Chem. Commun. 2016, 52, 9316–9326. doi:10.1039/C6CC03641D |
| 12. | Thomas, J.; Van Hecke, K.; Robeyns, K.; Van Rossom, W.; Sonawane, M. P.; Van Meervelt, L.; Smet, M.; Maes, W.; Dehaen, W. Chem. – Eur. J. 2011, 17, 10339–10349. doi:10.1002/chem.201101690 |
| 22. | Ito, A.; Ono, Y.; Tanaka, K. Angew. Chem., Int. Ed. 2000, 39, 1072–1075. doi:10.1002/(SICI)1521-3773(20000317)39:6<1072::AID-ANIE1072>3.0.CO;2-1 |
| 23. | Kurata, R.; Sakamaki, D.; Ito, A. Org. Lett. 2017, 19, 3115–3118. doi:10.1021/acs.orglett.7b01229 |
| 24. | Ito, A.; Uebe, M.; Kurata, R.; Yano, S.; Fueno, H.; Matsumoto, T. Chem. – Asian J. 2018, 13, 754–760. doi:10.1002/asia.201701717 |
| 25. | Sakamaki, D.; Ito, A.; Furukawa, K.; Kato, T.; Tanaka, K. Chem. Commun. 2009, 4524–4526. doi:10.1039/b907792h |
© 2018 De Leger et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)