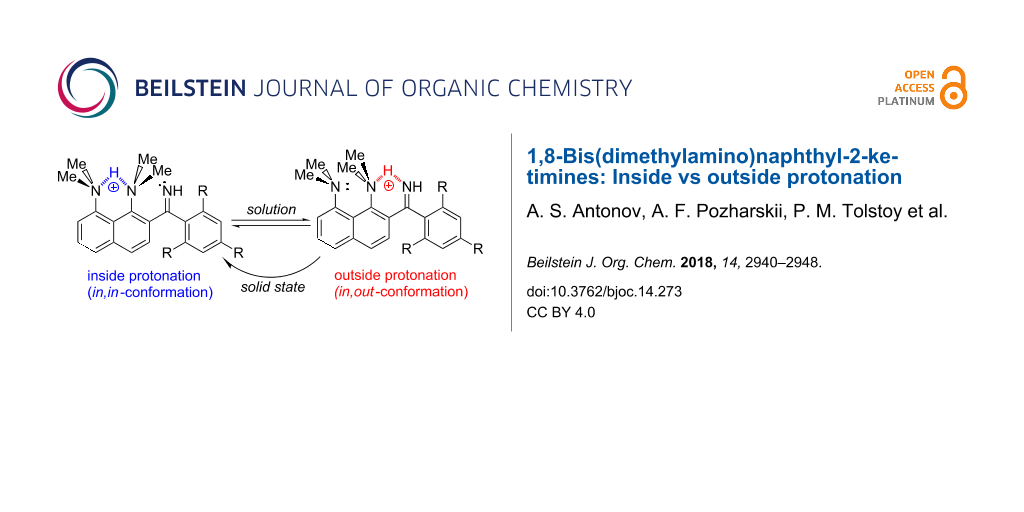
Abstract
The structure and protonation behaviour of four ortho-arylketimines of 1,8-bis(dimethylamonio)naphthalene with a different number of methoxy groups in an aromatic substituent were investigated in solution by NMR (acetone, DMSO, MeCN), in solid state by X-ray analysis and in the gas phase by DFT calculations. Both mono- and diprotonated species were considered. It has been shown that E-isomers of neutral imines can be stabilised by an intramolecular C=N−H···OMe hydrogen bond with a neighbouring methoxy group. Electron-donating OMe groups dramatically increase the basicity of the imino nitrogen, forcing the latter to abstract a proton from the proton sponge moiety in monoprotonated forms. The participation of the out-inverted and protonated 1-NMe2 group in the Me2N−H···NH=C hydrogen bond is experimentally demonstrated. It was shown that the number and position of OMe groups in the aromatic substituents strongly affects the rate of the internal hindered rotation of the NH2+ fragment in dications.

Graphical Abstract
Introduction
It is well known that the extremely high basicity of 1,8-bis(dimethylamino)naphthalene (1, DMAN; pKa = 12.1 in H2O [1,2], 18.62 in MeCN [3], 7.5 in DMSO [4]) and its derivatives originates from the electrostatic repulsion between the unshared electron pairs of nitrogen atoms, which strongly destabilises the neutral base. This repulsion is additionally fortified by the steric inhibition of resonance (both NMe2 groups cannot be conjugated to the aromatic system at the same time) preventing charge delocalisation. The protonation results in the formation of the N–H···N bonded cation 1H+, the removal of the electrostatic and steric strain and thus a considerable free energy gain (Scheme 1) [5]. This is also a reason why the vast majority of DMAN derivatives are protonated to the internitrogen space, even if other centres of basicity are present in the molecule. The only exceptions are compounds 2 and 3 which are protonated to aza- and carbonyl groups, respectively (Scheme 2) [6]. This unusual protonation site originates from the conjugation between the C=N (C=O) and the NMe2 groups, leading to the formation of stabilised cations.
![[1860-5397-14-273-i1]](/bjoc/content/inline/1860-5397-14-273-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-14-273-i2]](/bjoc/content/inline/1860-5397-14-273-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Protonation equilibria for 2 and 3.
Scheme 2: Protonation equilibria for 2 and 3.
In our recent work, we observed for the first time that the ortho-ketimino group in 4a can compete with the proton sponge moiety for the proton, which results in the formation of equilibria between the forms 4b (protonated to the proton sponge fragment) and 4b’ (protonated to the imino group) in the DMSO solution (Scheme 3) [7]. The aim of the present work is to investigate the major factors influencing the effective basicity of the imino functionality. As primary objects for this study, we consider a series of imines 4a–7a (Scheme 4), which differ by the number and position of an OMe group in an aryl substituent. We hope that increasing the number of strong electron-donating groups will result in a significant transfer of basicity to the imino function. Selected series of neutral molecules 4a–7a, as well as their mono- (4b–7b) and diprotonated (4c–7c) species were investigated by 1H NMR spectroscopy in DMSO-d6, CD3CN and acetone-d6 in a wide range of temperatures. Results obtained in solutions were compared to single crystal X-ray structures and calculated equilibrium geometries of isolated molecules in a vacuum.
![[1860-5397-14-273-i3]](/bjoc/content/inline/1860-5397-14-273-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Protonation of imine 4a with perchloric acid.
Scheme 3: Protonation of imine 4a with perchloric acid.
![[1860-5397-14-273-i4]](/bjoc/content/inline/1860-5397-14-273-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of investigated substrates (yields obtained by paths a and b are denoted by a corresponding superscript).
Scheme 4: Synthesis of investigated substrates (yields obtained by paths a and b are denoted by a correspondi...
Results and Discussion
Synthesis of DMAN-ortho-ketimines and their cations
Target compounds 4a–7a were synthesised by techniques previously developed in our laboratory (Scheme 4) [7,8]. Compounds 4a–6a were obtained by the treatment of the ortho-lithium derivative 8 with the corresponding nitrile (path a). Unfortunately, the strong election-donating effect of two and more OMe groups makes this approach not effective for the synthesis of compounds 6a and 7a. To overcome this difficulty, we used a reversed method based on the treatment of ortho-cyanide 9 with the corresponding aryllithiums, which produces the compounds 5a–7a with good to moderate yields (path b). The monocations 4b–7b and dications 4c–7c were prepared by the treatment of the corresponding imines with one or two equivalents of HBF4 in Et2O (Scheme 5). The so obtained tetrafluoroborates were used for NMR measurements after recrystallisation from EtOH.
![[1860-5397-14-273-i5]](/bjoc/content/inline/1860-5397-14-273-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Preparation of protonated forms of imines 4–7.
Scheme 5: Preparation of protonated forms of imines 4–7.
Structural investigations
The structural investigations started with a 1H NMR analysis of the imines 4a–7a in acetone-d6, CD3CN and DMSO-d6 in a wide range of temperatures. The main attention was paid to the chemical shift and the shape of the C=NH and NMe2 groups’ signals. Upon temperature decrease, the NH signal splits into two peaks of unequal intensity, which is best seen in the case of the acetone-d6 solution (Figure 1). For example, for compound 4a, the C=NH proton signals are at δ = 9.9 ppm and 10.7 ppm at −80 °C, with the major signal being in the higher field. In contrast, in the spectra of 5a–7a, bearing OMe groups in ortho-position to the imino group, the major NH signal is located in the lower field (Figures S2–S4 in Supporting Information File 1). We assigned these signals to the NHa and NHb protons of Z- and E-isomers, as shown in Scheme 6. We believe that in solution the Z-form is more stable for 4a, while the E-form is more stable for 5a–7a. Indeed, the X-ray analysis shows that compound 6a in the solid state exists in the E-form stabilised by the N−H···O intramolecular hydrogen bond (IHB) with an ortho-OMe group (Figure 2a), while compound 4a, as it was revealed in our previous work [7], exists exclusively as a Z-isomer (Figure 2b). Moreover, gas phase quantum chemical calculations show that the E-form is preferable for 5a (ΔE = EZ − EE = 1.66 kcal/mol), 6a (ΔE = 1.19 kcal/mol), and 7a (ΔE = 1.90 kcal/mol), while the Z-form dominates for 4a (ΔE = −2.62 kcal/mol, Figure 3). The contribution of the N−H···O IHB to the stabilisation of the E-forms of 5a–7a appears to be small: in the 1H NMR spectra, the NHa signal is shifted to the low field by less than 1 ppm in comparison with Z-4a (Figure 1) [9]; in a DFT calculation, the mutual orientation of C=NH and the OMe groups is not optimal for IHB formation.
![[1860-5397-14-273-1]](/bjoc/content/figures/1860-5397-14-273-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: C=NH Signal regions of temperature-depending 1H NMR spectra for compounds 4a–7a, acetone-d6.
Figure 1: C=NH Signal regions of temperature-depending 1H NMR spectra for compounds 4a–7a, acetone-d6.
![[1860-5397-14-273-i6]](/bjoc/content/inline/1860-5397-14-273-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: E/Z-Isomerisation of ketimines 4a–7a.
Scheme 6: E/Z-Isomerisation of ketimines 4a–7a.
![[1860-5397-14-273-2]](/bjoc/content/figures/1860-5397-14-273-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular structure of imines 6a (a) and 4a (b). The H···O distance for 6a is shown.
Figure 2: Molecular structure of imines 6a (a) and 4a (b). The H···O distance for 6a is shown.
![[1860-5397-14-273-3]](/bjoc/content/figures/1860-5397-14-273-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Optimised geometry and energy difference ΔE = EE – EZ (kcal/mol) for imines 4a–7a (B3LYP/6-311+G(d,p)).
Figure 3: Optimised geometry and energy difference ΔE = EE – EZ (kcal/mol) for imines 4a–7a (B3LYP/6-311+G(d,...
The rate and equilibrium constants of this uncatalysed isomerisation [10] depend on the solvent and temperature. By comparing the results obtained in different solvents at equal temperatures, one can see that the Ha ![[Graphic 1]](/bjoc/content/inline/1860-5397-14-273-i10.svg?max-width=637&scale=0.59091) Hb exchange rate is noticeably lower in DMSO-d6 (Figures S9–S12 in Supporting Information File 1) than in acetone-d6 and CD3CN (Figures S5–S8 in Supporting Information File 1), while the chemical shift differences between the Ha and Hb signals remain similar in all cases (ca. 1 ppm). The increased energy barrier for the isomerisation could be attributed to the stabilisation of the Z-form through formation of a stronger hydrogen bond between the DMSO molecule and the NHb proton [11], which is sterically unhindered and accessible for the solvent. Indeed, X-ray data and quantum chemical calculations show that phenyl and naphthalene rings are almost perpendicular (Figure 2 and Figure 3).
Hb exchange rate is noticeably lower in DMSO-d6 (Figures S9–S12 in Supporting Information File 1) than in acetone-d6 and CD3CN (Figures S5–S8 in Supporting Information File 1), while the chemical shift differences between the Ha and Hb signals remain similar in all cases (ca. 1 ppm). The increased energy barrier for the isomerisation could be attributed to the stabilisation of the Z-form through formation of a stronger hydrogen bond between the DMSO molecule and the NHb proton [11], which is sterically unhindered and accessible for the solvent. Indeed, X-ray data and quantum chemical calculations show that phenyl and naphthalene rings are almost perpendicular (Figure 2 and Figure 3).
After the first protonation of imines 4a–7a with HBF4, the 1H NMR spectra display two sets of signals (Figure 4). One of the sets corresponds to the monocations 4b–7b protonated at the proton sponge site: the NHN bridging proton resonates at ≈19 ppm and the positions of the CH signals of the dimethylaminonaphthalene fragment are typical and characteristic for protonated proton sponges [5]. In the other set of signals, the chemical shifts of the NMe2 groups are practically the same as for the neutral bases 4a–7a. This means that protonation occurs at another basic site, which can only be imine nitrogen, i.e., the structure corresponds to monocations 4b’–7b’ (Scheme 7). Indeed, around 10 ppm, a new signal appears which corresponds to two NH protons. To our knowledge, this is the first example of an imine to be basic enough to successfully compete for a proton with the proton sponge moiety: for example, the basicity of benzophenone imine (pKa = 6.82) and even that of cyclohexanone imine (pKa = 9.15) [12] is much lower than that of DMAN (pKa = 12.1) [1,2]. Moreover, as it was already mentioned in the introduction, this is the third known example of a DMAN derivative with a substituent that is more basic than the peri-dimethylaminonaphthalene core. Such an imine basicity boost can originate from the fact that upon protonation, NMe2 groups lose conjugation with the ring whereas the C=N moiety does not. As a result, the imino nitrogen saves the electron density supply from both OMe and NMe2 groups, which increases the s-character of nitrogen hybridisation thus fortifying the basicity [13].
![[1860-5397-14-273-4]](/bjoc/content/figures/1860-5397-14-273-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: 1H NMR spectra of imines 4b–7b (4b’–7b’) in DMSO-d6, 25 °C (the signals of the forms protonated at the imino function are marked with an asterisk).
Figure 4: 1H NMR spectra of imines 4b–7b (4b’–7b’) in DMSO-d6, 25 °C (the signals of the forms protonated at ...
![[1860-5397-14-273-i7]](/bjoc/content/inline/1860-5397-14-273-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Switching of protonation sites in imines 4b–7b.
Scheme 7: Switching of protonation sites in imines 4b–7b.
The ratio of two monocations, estimated from 1H NMR spectra at room temperature, depends on the position and number of the OMe groups and the solvent (Table 1; the values are practically temperature independent). One could expect that strong electron-donating OMe groups being placed in positions 2, 4 or 6 of a phenylimino substituent should increase the basicity of imine nitrogen. Thus, in CD3CN, both 4b and 5b, containing only one OMe group, show no proton transfer towards the imino function (Figure S13 in Supporting Information File 1). The addition of a second OMe group in the case of 6 leads to the formation of 25% 6b’. Upon the insertion of a third OMe group in 7, the amount of 7b’ expectedly increases to 33%. Switching of the solvent to acetone-d6 facilitates the proton transfer away from the proton sponge moiety (Figure S14, Supporting Information File 1). For example, the relative amount of 7b’ increases from 33% to 66%. Using DMSO-d6 leads to the dramatic shift of the equilibrium, resulting in the formation of 90% 7b’ (Figure 4). We believe that the forms protonated at the imino function are additionally stabilised by hydrogen bonds with solvent molecules and thus the amount of b’ forms correlates with the proton accepting ability of the medium [11]. The gas phase calculations show that without any additional interaction with the medium, the forms protonated at the imino group are lowest in energy for the imines 4, 6, 7 (Figure 5). In the solid state, no proton transfer to the imino group was observed: compounds 4a·HClO4 and 6b·EtOH crystallise in forms protonated at the proton sponge moiety (Figure 6).
![[1860-5397-14-273-5]](/bjoc/content/figures/1860-5397-14-273-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Optimised geometries and energy differences ΔE = Eb’ – Eb (kcal/mol) for different sites of protonation of the studied imines (B3LYP/6-311+G(d,p)).
Figure 5: Optimised geometries and energy differences ΔE = Eb’ – Eb (kcal/mol) for different sites of protona...
![[1860-5397-14-273-6]](/bjoc/content/figures/1860-5397-14-273-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Molecular structure of protonated imines 4a·HClO4 (a) and 6b·EtOH (b).
Figure 6: Molecular structure of protonated imines 4a·HClO4 (a) and 6b·EtOH (b).
We expected that the proton transfer to the C=N group will force the nearest proton sponge moiety to occupy the in,out-conformation [14] with the 1-NMe2 group participating in NHN intramolecular hydrogen bonding with the C=NH2+ group (Scheme 8). Indeed, in the case of 7b’, which contains three OMe groups, we have found direct evidence of this process: the 1-NMe2 group signal shifts from 2.8 to 3.3 ppm – the characteristic region for protonated dimethylamino groups in proton sponges [5] – while the 8-NMe2 group signals stay at 2.8 ppm. Thus, only the 1-NMe2 group is protonated, which is only possible if it is out-inverted. Similar chemical shifts were observed for the cation 10 (Scheme 8, right), which is out-protonated due to steric reasons [15]. In refs. [15-17] it was shown that hydrogen bonding to an out-inverted NMe2 group without the proton transfer does not noticeably change the chemical shift of methyl protons. Thus, in the case of 7b’, the chemical shift at 3.3 ppm proves the proton transfer to the amino nitrogen, as shown in Scheme 8 for (in,out)-7b’. In other words, in the case of 7b’, steric pressure of the trimethoxyphenyl substituent facilitates the 1-NMe2 group’s out-inversion which is stabilised by an IHB, forming N(sp3)/N(sp2) type of proton sponges, which were recently reported in our paper [18]. It was previously shown that the in,out-conformation can be stabilised by placing the following functionalities in position 2(7): tertiary alcoholic groups which are able to form the O–H···N IHB [16,17,19], bulky substituents rendering a steric pressure onto the 1-NMe2 group [7,20], or a metal atom coordinating the NMe2 group as a ligand causing its inversion [21] (Scheme 9). A possibility of out-protonation of 1,8-bis(dimethylamino)naphthalene was discussed in our recent paper [15], while it is experimentally demonstrated here for the first time.
![[1860-5397-14-273-i8]](/bjoc/content/inline/1860-5397-14-273-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Protonation of the out-inverted dialkylamino group.
Scheme 8: Protonation of the out-inverted dialkylamino group.
![[1860-5397-14-273-i9]](/bjoc/content/inline/1860-5397-14-273-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Examples of proton sponges with stabilised in,out-conformation.
Scheme 9: Examples of proton sponges with stabilised in,out-conformation.
We also prepared dications 4c–7c (Scheme 5) and investigated their structure in solution in CD3CN, DMSO-d6 and acetone-d6. It was found that the exchange rate between protons in the C=NH2+ group strongly depends on the position and number of OMe groups and the temperature. In the case of 4c, signals are coalescent even at −80 °C (Figure 7). The presence of an OMe group in the ortho-position to C=N groups in 5c slows down the exchange and results in NH2+ signals splitting at −40 °C. This can be attributed to the formation of the weak NHO intramolecular hydrogen bond, which becomes stronger upon inserting an additional OMe group. Such a NHO bond is clearly visible in the X-ray structure of 6c (Figure 8). As a result, in solutions of 6c and 7c, NH2+ proton signals are observed separately up to 40 °C (for spectra in CD3CN see also Figures S20–S23 in Supporting Information File 1).
![[1860-5397-14-273-7]](/bjoc/content/figures/1860-5397-14-273-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: C=NH2+ Signal regions of temperature-depending spectra for compounds 4c–7c, acetone-d6.
Figure 7: C=NH2+ Signal regions of temperature-depending spectra for compounds 4c–7c, acetone-d6.
![[1860-5397-14-273-8]](/bjoc/content/figures/1860-5397-14-273-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Molecular structure of dication 6с.
Figure 8: Molecular structure of dication 6с.
Conclusion
In summary, we succeeded in preparing a new series of 1,8-bis(dimethylamino)naphthalene-ortho-ketimines with aromatic substituents containing strong electron donating OMe groups. Neutral compounds as well as their mono- and diprotonated species were studied. The obtained compounds demonstrated increased basicity of the imino nitrogen, which was able to effectively compete for the proton with the proton sponge core. Increasing the number of OMe groups facilitated the proton transfer to the imino function by up to 90%. As a result, studied compounds can be considered as superbasic imines. In one case, we were able to experimentally observe the simultaneous out-inversion and protonation of the 1-NMe2 group for the first time.
Supporting Information
| Supporting Information File 1: Experimental procedures and analytical data, copies of 1H NMR spectra of all studied compounds, 1H and 13C NMR spectra confirming the structure of new compounds, crystallographic data for 6a, 6b, 6c and 4a·HClO4. | ||
| Format: PDF | Size: 5.1 MB | Download |
| Supporting Information File 2: CIF file for compound 6a–c and 4a·HClO4 (CCDC 1867639-1867642). | ||
| Format: CIF | Size: 2.6 MB | Download |
Acknowledgements
This work was supported by the Russian Foundation for Basic Research (project 16-33-60030). Experimental measurements were performed in Centre for Magnetic Resonance, Chemical Analysis and Materials Research Centre, Centre for X-ray Diffraction Studies at St. Petersburg State University Research Park. The authors also thank Mr. Daniel Raith for proofreading the paper in terms of the English language.
References
-
Alder, R. W.; Bowman, P. S.; Steele, W. R. S.; Winterman, D. R. Chem. Commun. 1968, 723–724. doi:10.1039/c19680000723
Return to citation in text: [1] [2] -
Hibbert, F. J. Chem. Soc., Perkin Trans. 2 1974, 1862–1866. doi:10.1039/p29740001862
Return to citation in text: [1] [2] -
Kaljurand, I.; Kütt, A.; Sooväli, L.; Rodima, T.; Mäemets, V.; Leito, I.; Koppel, I. A. J. Org. Chem. 2005, 70, 1019–1028. doi:10.1021/jo048252w
Return to citation in text: [1] -
Benoit, R. L.; Lefebvre, D.; Fréchette, M. Can. J. Chem. 1987, 65, 996–1001. doi:10.1139/v87-170
Return to citation in text: [1] -
Pozharskii, A. F.; Ozeryanskii, V. A. Proton Sponges. In The Chemistry of Anilines; Rappoport, Z., Ed.; John Wiley & Sons, Ltd: Chichester, UK, 2007; pp 931–1026. doi:10.1002/9780470871737.ch17
Return to citation in text: [1] [2] [3] -
Pozharskii, A. F.; Ozeryanskii, V. A.; Vistorobskii, N. V. Russ. Chem. Bull. 2003, 52, 218–227. doi:10.1023/a:1022429322461
Return to citation in text: [1] -
Antonov, A. S.; Mikshiev, V. Y.; Pozharskii, A. F.; Ozeryanskii, V. A. Synthesis 2014, 46, 3273–3282. doi:10.1055/s-0034-1379008
Return to citation in text: [1] [2] [3] [4] -
Povalyakhina, M. A.; Antonov, A. S.; Dyablo, O. V.; Ozeryanskii, V. A.; Pozharskii, A. F. J. Org. Chem. 2011, 76, 7157–7166. doi:10.1021/jo201171z
Return to citation in text: [1] -
Afonin, A. V.; Vashchenko, A. V.; Sigalov, M. V. Org. Biomol. Chem. 2016, 14, 11199–11211. doi:10.1039/c6ob01604a
Return to citation in text: [1] -
Curtin, D. Y.; Grubbs, E. J.; McCarty, C. G. J. Am. Chem. Soc. 1966, 88, 2775–2786. doi:10.1021/ja00964a029
Return to citation in text: [1] -
Laurence, C.; Graton, J.; Berthelot, M.; Besseau, F.; Le Questel, J.-Y.; Luçon, M.; Ouvrard, C.; Planchat, A.; Renault, E. J. Org. Chem. 2010, 75, 4105–4123. doi:10.1021/jo100461z
Return to citation in text: [1] [2] -
Albert, A.; Serjeant, E. P. The Ionization Constants of Typical Acids and Bases. The Determination of Ionization Constants; Springer Netherlands: Dordrecht, 1984; pp 136–175. doi:10.1007/978-94-009-5548-6_9
Return to citation in text: [1] -
Alabugin, I. V.; Bresch, S.; dos Passos Gomes, G. J. Phys. Org. Chem. 2015, 28, 147–162. doi:10.1002/poc.3382
Return to citation in text: [1] -
Szemik-Hojniak, A.; Zwier, J. M.; Buma, W. J.; Bursi, R.; van der Waals, J. H. J. Am. Chem. Soc. 1998, 120, 4840–4844. doi:10.1021/ja974245w
Return to citation in text: [1] -
Ozeryanskii, V. A.; Pozharskii, A. F.; Antonov, A. S.; Filarowski, A. Org. Biomol. Chem. 2014, 12, 2360–2369. doi:10.1039/c3ob41986j
Return to citation in text: [1] [2] [3] -
Pozharskii, A. F.; Degtyarev, A. V.; Ryabtsova, O. V.; Ozeryanskii, V. A.; Kletskii, M. E.; Starikova, Z. A.; Sobczyk, L.; Filarowski, A. J. Org. Chem. 2007, 72, 3006–3019. doi:10.1021/jo062667v
Return to citation in text: [1] [2] -
Pozharskii, A. F.; Degtyarev, A. V.; Ozeryanskii, V. A.; Ryabtsova, O. V.; Starikova, Z. A.; Borodkin, G. S. J. Org. Chem. 2010, 75, 4706–4715. doi:10.1021/jo100384s
Return to citation in text: [1] [2] -
Pozharskii, A. F.; Ozeryanskii, V. A.; Mikshiev, V. Y.; Antonov, A. S.; Chernyshev, A. V.; Metelitsa, A. V.; Borodkin, G. S.; Fedik, N. S.; Dyablo, O. V. J. Org. Chem. 2016, 81, 5574–5587. doi:10.1021/acs.joc.6b00917
Return to citation in text: [1] -
Pozharskii, A. F.; Ryabtsova, O. V.; Ozeryanskii, V. A.; Degtyarev, A. V.; Starikova, Z. A.; Sobczyk, L.; Filarowski, A. Tetrahedron Lett. 2005, 46, 3973–3976. doi:10.1016/j.tetlet.2005.04.045
Return to citation in text: [1] -
Pozharskii, A. F.; Ryabtsova, O. V.; Ozeryanskii, V. A.; Degtyarev, A. V.; Kazheva, O. N.; Alexandrov, G. G.; Dyachenko, O. A. J. Org. Chem. 2003, 68, 10109–10122. doi:10.1021/jo035350t
Return to citation in text: [1] -
Antonov, A. S.; Pozharskii, A. F.; Ozeryanskii, V. A.; Filarowski, A.; Suponitsky, K. Y.; Tolstoy, P. M.; Vovk, M. A. Dalton Trans. 2015, 44, 17756–17766. doi:10.1039/c5dt02482j
Return to citation in text: [1]
| 1. | Alder, R. W.; Bowman, P. S.; Steele, W. R. S.; Winterman, D. R. Chem. Commun. 1968, 723–724. doi:10.1039/c19680000723 |
| 2. | Hibbert, F. J. Chem. Soc., Perkin Trans. 2 1974, 1862–1866. doi:10.1039/p29740001862 |
| 6. | Pozharskii, A. F.; Ozeryanskii, V. A.; Vistorobskii, N. V. Russ. Chem. Bull. 2003, 52, 218–227. doi:10.1023/a:1022429322461 |
| 13. | Alabugin, I. V.; Bresch, S.; dos Passos Gomes, G. J. Phys. Org. Chem. 2015, 28, 147–162. doi:10.1002/poc.3382 |
| 5. | Pozharskii, A. F.; Ozeryanskii, V. A. Proton Sponges. In The Chemistry of Anilines; Rappoport, Z., Ed.; John Wiley & Sons, Ltd: Chichester, UK, 2007; pp 931–1026. doi:10.1002/9780470871737.ch17 |
| 11. | Laurence, C.; Graton, J.; Berthelot, M.; Besseau, F.; Le Questel, J.-Y.; Luçon, M.; Ouvrard, C.; Planchat, A.; Renault, E. J. Org. Chem. 2010, 75, 4105–4123. doi:10.1021/jo100461z |
| 4. | Benoit, R. L.; Lefebvre, D.; Fréchette, M. Can. J. Chem. 1987, 65, 996–1001. doi:10.1139/v87-170 |
| 12. | Albert, A.; Serjeant, E. P. The Ionization Constants of Typical Acids and Bases. The Determination of Ionization Constants; Springer Netherlands: Dordrecht, 1984; pp 136–175. doi:10.1007/978-94-009-5548-6_9 |
| 3. | Kaljurand, I.; Kütt, A.; Sooväli, L.; Rodima, T.; Mäemets, V.; Leito, I.; Koppel, I. A. J. Org. Chem. 2005, 70, 1019–1028. doi:10.1021/jo048252w |
| 1. | Alder, R. W.; Bowman, P. S.; Steele, W. R. S.; Winterman, D. R. Chem. Commun. 1968, 723–724. doi:10.1039/c19680000723 |
| 2. | Hibbert, F. J. Chem. Soc., Perkin Trans. 2 1974, 1862–1866. doi:10.1039/p29740001862 |
| 9. | Afonin, A. V.; Vashchenko, A. V.; Sigalov, M. V. Org. Biomol. Chem. 2016, 14, 11199–11211. doi:10.1039/c6ob01604a |
| 11. | Laurence, C.; Graton, J.; Berthelot, M.; Besseau, F.; Le Questel, J.-Y.; Luçon, M.; Ouvrard, C.; Planchat, A.; Renault, E. J. Org. Chem. 2010, 75, 4105–4123. doi:10.1021/jo100461z |
| 7. | Antonov, A. S.; Mikshiev, V. Y.; Pozharskii, A. F.; Ozeryanskii, V. A. Synthesis 2014, 46, 3273–3282. doi:10.1055/s-0034-1379008 |
| 5. | Pozharskii, A. F.; Ozeryanskii, V. A. Proton Sponges. In The Chemistry of Anilines; Rappoport, Z., Ed.; John Wiley & Sons, Ltd: Chichester, UK, 2007; pp 931–1026. doi:10.1002/9780470871737.ch17 |
| 7. | Antonov, A. S.; Mikshiev, V. Y.; Pozharskii, A. F.; Ozeryanskii, V. A. Synthesis 2014, 46, 3273–3282. doi:10.1055/s-0034-1379008 |
| 8. | Povalyakhina, M. A.; Antonov, A. S.; Dyablo, O. V.; Ozeryanskii, V. A.; Pozharskii, A. F. J. Org. Chem. 2011, 76, 7157–7166. doi:10.1021/jo201171z |
| 7. | Antonov, A. S.; Mikshiev, V. Y.; Pozharskii, A. F.; Ozeryanskii, V. A. Synthesis 2014, 46, 3273–3282. doi:10.1055/s-0034-1379008 |
| 10. | Curtin, D. Y.; Grubbs, E. J.; McCarty, C. G. J. Am. Chem. Soc. 1966, 88, 2775–2786. doi:10.1021/ja00964a029 |
| 15. | Ozeryanskii, V. A.; Pozharskii, A. F.; Antonov, A. S.; Filarowski, A. Org. Biomol. Chem. 2014, 12, 2360–2369. doi:10.1039/c3ob41986j |
| 14. | Szemik-Hojniak, A.; Zwier, J. M.; Buma, W. J.; Bursi, R.; van der Waals, J. H. J. Am. Chem. Soc. 1998, 120, 4840–4844. doi:10.1021/ja974245w |
| 5. | Pozharskii, A. F.; Ozeryanskii, V. A. Proton Sponges. In The Chemistry of Anilines; Rappoport, Z., Ed.; John Wiley & Sons, Ltd: Chichester, UK, 2007; pp 931–1026. doi:10.1002/9780470871737.ch17 |
| 21. | Antonov, A. S.; Pozharskii, A. F.; Ozeryanskii, V. A.; Filarowski, A.; Suponitsky, K. Y.; Tolstoy, P. M.; Vovk, M. A. Dalton Trans. 2015, 44, 17756–17766. doi:10.1039/c5dt02482j |
| 15. | Ozeryanskii, V. A.; Pozharskii, A. F.; Antonov, A. S.; Filarowski, A. Org. Biomol. Chem. 2014, 12, 2360–2369. doi:10.1039/c3ob41986j |
| 16. | Pozharskii, A. F.; Degtyarev, A. V.; Ryabtsova, O. V.; Ozeryanskii, V. A.; Kletskii, M. E.; Starikova, Z. A.; Sobczyk, L.; Filarowski, A. J. Org. Chem. 2007, 72, 3006–3019. doi:10.1021/jo062667v |
| 17. | Pozharskii, A. F.; Degtyarev, A. V.; Ozeryanskii, V. A.; Ryabtsova, O. V.; Starikova, Z. A.; Borodkin, G. S. J. Org. Chem. 2010, 75, 4706–4715. doi:10.1021/jo100384s |
| 19. | Pozharskii, A. F.; Ryabtsova, O. V.; Ozeryanskii, V. A.; Degtyarev, A. V.; Starikova, Z. A.; Sobczyk, L.; Filarowski, A. Tetrahedron Lett. 2005, 46, 3973–3976. doi:10.1016/j.tetlet.2005.04.045 |
| 7. | Antonov, A. S.; Mikshiev, V. Y.; Pozharskii, A. F.; Ozeryanskii, V. A. Synthesis 2014, 46, 3273–3282. doi:10.1055/s-0034-1379008 |
| 20. | Pozharskii, A. F.; Ryabtsova, O. V.; Ozeryanskii, V. A.; Degtyarev, A. V.; Kazheva, O. N.; Alexandrov, G. G.; Dyachenko, O. A. J. Org. Chem. 2003, 68, 10109–10122. doi:10.1021/jo035350t |
| 15. | Ozeryanskii, V. A.; Pozharskii, A. F.; Antonov, A. S.; Filarowski, A. Org. Biomol. Chem. 2014, 12, 2360–2369. doi:10.1039/c3ob41986j |
| 16. | Pozharskii, A. F.; Degtyarev, A. V.; Ryabtsova, O. V.; Ozeryanskii, V. A.; Kletskii, M. E.; Starikova, Z. A.; Sobczyk, L.; Filarowski, A. J. Org. Chem. 2007, 72, 3006–3019. doi:10.1021/jo062667v |
| 17. | Pozharskii, A. F.; Degtyarev, A. V.; Ozeryanskii, V. A.; Ryabtsova, O. V.; Starikova, Z. A.; Borodkin, G. S. J. Org. Chem. 2010, 75, 4706–4715. doi:10.1021/jo100384s |
| 18. | Pozharskii, A. F.; Ozeryanskii, V. A.; Mikshiev, V. Y.; Antonov, A. S.; Chernyshev, A. V.; Metelitsa, A. V.; Borodkin, G. S.; Fedik, N. S.; Dyablo, O. V. J. Org. Chem. 2016, 81, 5574–5587. doi:10.1021/acs.joc.6b00917 |
© 2018 Antonov et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)