Abstract
In recent years, the highly polar C–F bond has been utilised in activation chemistry despite its low reactivity to traditional nucleophiles, when compared to other C–X halogen bonds. Paquin’s group has reported extensive studies on the C–F activation of benzylic fluorides for nucleophilic substitutions and Friedel–Crafts reactions, using a range of hydrogen bond donors such as water, triols or hexafluoroisopropanol (HFIP) as the activators. This study examines the stereointegrity of the C–F activation reaction through the use of an enantiopure isotopomer of benzyl fluoride to identify whether the reaction conditions favour a dissociative (SN1) or associative (SN2) pathway. [2H]-Isotopomer ratios in the reactions were assayed using the Courtieu 2H NMR method in a chiral liquid crystal (poly-γ-benzyl-L-glutamate) matrix and demonstrated that both associative and dissociative pathways operate to varying degrees, according to the nature of the nucleophile and the hydrogen bond donor.

Graphical Abstract
Introduction
The C–F bond is the strongest carbon–halogen bond known [1]. Its low reactivity, in comparison to other C–X bonds, means that it is inert to all but the most harsh reaction conditions, and fluorine can generally be carried through multistep syntheses without concern over side reactions (the exception being SNAr reactions). In recent years, there has been an increasing interest in C–F bond activation [2], with a view to using organic bound fluoride as a leaving group in substitution reactions that typically require more activated leaving groups. Such an approach could circumvent the requirement for protecting groups in multistep synthesis by capitalizing on the low reactivity of the C–F bond. Paquin et al. have published extensively on non-metal based methods for benzylic C–F bond activation [3-7]. The reactivity relies on protic activation driven by the capacity of organic fluoride to form hydrogen bonds [8,9]. Protocols using water/isopropanol [3], optimally coordinated triols [4,5], and hexafluoroisopropanol (HFIP) [6,7] as the corresponding hydrogen bond donors have shown considerable success. This mode of activation has been demonstrated for amination [3-5] and Friedel–Crafts reactions [6,7] on benzylic fluoride substrates (Figure 1), producing the corresponding substituted products in moderate to good yields. The water/isopropanol system was also shown to be amenable to phenolate and thiolate nucleophiles [3].
![[1860-5397-14-6-1]](/bjoc/content/figures/1860-5397-14-6-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: C–F activation of benzylic fluorides to generate benzylamine or diarylmethane products.
Figure 1: C–F activation of benzylic fluorides to generate benzylamine or diarylmethane products.
Previously, Paquin et al. undertook density functional theory (DFT) studies on the mechanism of C–F amination reactions employing water/isopropanol [3] and triols [4,5] as hydrogen-bond donor activators. Through these studies, the authors suggested that multiple donors (even when using a triol) surround the fluorine atom of the benzyl fluoride, thus stabilising the transition state through substantial F···HOR hydrogen bond interactions, rather than through electrostatic stabilisation only [3]. This stabilisation was suggested to lead to a purely associative bimolecular (SN2) mechanism. The authors also studied the C–F activated Friedel–Crafts reactions [6,7] using very strong hydrogen bond donors, namely HFIP, in the presence or absence of trifluoroacetic acid (TFA). For both of these activators, Paquin et al. proposed a dissociative unimolecular (SN1) mechanism, whereby the strong hydrogen bond donor associates with the benzyl fluoride, leading to ionisation of the molecule, generating a benzylic carbocation and a formal equivalent of HF (which behaves in an autocatalytic manner as a stronger hydrogen bond donor than HFIP or TFA).
Overall, there are three possible mechanistic pathways that these C–F activation reactions could follow: SN1, SN2, and a mixed SN1/SN2 pathway. Typically, benzylic substitutions would be expected to display a significant level of SN1 character. However, given the particularly poor properties of fluoride as a leaving group, developing a better understanding of the dissociative nature of these transformations remains of considerable interest. A direct bimolecular SN2 substitution would result in a complete inversion of configuration of the stereocenter and perfect enantiospecificity, while an SN1 mechanism would yield a fully racemized product. Any mixed pathway would generate products with partially racemized stereocenters. In this context, we decided to explore the stereointegrity of the aforementioned reactions using enantiopure 7-[2H1]-(R)-benzyl fluoride ((R)-1, Figure 2) as a primary, yet chiral electrophile [10].
![[1860-5397-14-6-2]](/bjoc/content/figures/1860-5397-14-6-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 7-[2H1]-(R)-Benzyl fluoride ((R)-1).
Figure 2: 7-[2H1]-(R)-Benzyl fluoride ((R)-1).
Substitution reactions of benzyl fluoride (1) will generate substituted products that retain the deuterium atom, and the degree of stereointegrity can be determined by examining the enantiopurity of the isotopically labelled product. Quadrupolar 2H-nuclei can serve as a particularly useful NMR probe for assaying enantiopurity. If the 2H NMR is recorded in a lyotropic liquid crystalline solvent, where tumbling of the solute is restricted, then the 2H NMR signal splits into a doublet due to differential interactions of the quadrupolar nuclei with the electric field gradient associated with the oriented media [10]. When placed in an enantiomerically enriched liquid crystalline environment, the enantiomeric isotopomers interact unequally with the electric field gradients associated with the orientated media, creating anisotropy and resolving into two sets of doublets. If there is sufficient resolution between these quadrupolar couplings, then the enantiomeric ratio can be recorded. We have used poly-γ-benzyl-L-glutamate (PBLG) previously as the liquid-crystalline matrix for the determination of ee of samples of deuterated benzyl alcohols, benzyl fluorides, and esters of fluoroacetic acid [11] by 2H NMR and found it to be effective for the resolution of enantiomers. In this study, we explore various nucleophilic substitutions and a Friedel–Crafts reaction on enantiomerically labelled [2H1]benzyl fluoride.
Results and Discussion
Highly enantiomerically enriched 7-[2H1]-(R)-benzyl fluoride ((R)-1) was synthesised in two steps from benzaldehyde (2), as described previously [11]; the procedure is summarised in Scheme 1.
![[1860-5397-14-6-i1]](/bjoc/content/inline/1860-5397-14-6-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of enantioenriched 7-[2H1]-(R)-benzyl fluoride ((R)-1) from benzaldehyde (2).
Scheme 1: Synthesis of enantioenriched 7-[2H1]-(R)-benzyl fluoride ((R)-1) from benzaldehyde (2).
Aldehyde 2 was reduced under Noyori’s conditions [12] using (S,S)-Ru(DPEN)2 as catalyst and [2H2]-formic acid as the deuterium source. This afforded the corresponding 7-[2H1]-(S)-benzyl alcohol ((S)-3) in moderate yield (81%) and high ee (95%), as evidenced by 2H-PBLG-NMR. Benzyl alcohol 3 was converted to the corresponding benzyl fluoride (1) using a modification of Bio’s method [13,14] to promote the SN2 reaction exclusively, using TMS-morpholine and DAST, in moderate yield (51%) and high ee (94%).
The isotopically enriched [2H1]-benzyl fluoride ((R)-1, 95% ee) was then subjected to a range of C–F activation reactions using a mixture of nucleophiles (for direct substitutions) and aryls (for Friedel–Crafts reactions) to give products 5–9. The nucleophilic substitution reactions of 1 are shown in Table 1 and Table 2, and were all conducted using either a mixture of water/isopropanol, or tris(hydroxymethyl)propane as the activating hydrogen bond donor. In addition, three reactions of racemic substrates (Table 1, entries 1–3), were performed in order to ensure that sufficient resolution could be obtained in the 2H{1H}-PBLG-NMR, therefore allowing the ee of the products to be determined. A representative example of the 2H NMR spectra (107.5 MHz) is displayed in Figure 3, using N-methylaniline as a nucleophile, showing the spectra of both a racemic sample (Figure 3A) and an enantioenriched sample (Figure 3B) of 6. However, as evidenced by entry 3 (Table 1), and entries 3 and 7 (Table 2), the analysis revealed that some nucleophiles (such as N-methylbenzylamine and morpholine [not shown]) were unsuitable for this study, as the resulting products 7 could not be resolved in the 2H NMR with PBLG assay.
Table 1: Nucleophilic substitution reactions of racemic 7-[2H1]-benzyl bromide (4).
| Entry | Reaction | ee (%) |
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-6-i3.svg?max-width=637&scale=1.0)
|
racemic |
| 2 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-6-i4.svg?max-width=637&scale=1.0)
|
racemic |
| 3 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-6-i5.svg?max-width=637&scale=1.0)
|
nda |
aee could not be determined as a result of poor 2H{1H} NMR resolution.
Table 2: Nucleophilic substitution reactions of 7-[2H1]-(R)-benzyl fluoride ((R)-1).
| Entry | Reaction | ee (%) |
| 1 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-6-i6.svg?max-width=637&scale=1.0)
|
94 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-14-6-i7.svg?max-width=637&scale=1.0)
|
90 |
| 3 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-14-6-i8.svg?max-width=637&scale=1.0)
|
nda |
| 4 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-14-6-i9.svg?max-width=637&scale=1.0)
|
93 |
| 5 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-14-6-i10.svg?max-width=637&scale=1.0)
|
91 |
| 6 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-14-6-i11.svg?max-width=637&scale=1.0)
|
87 |
| 7 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-14-6-i12.svg?max-width=637&scale=1.0)
|
nda |
| 8 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-14-6-i13.svg?max-width=637&scale=1.0)
|
89 |
aee could not be determined as a result of poor 2H{1H} NMR resolution.
![[1860-5397-14-6-3]](/bjoc/content/figures/1860-5397-14-6-3.png?scale=1.84&max-width=1024&background=FFFFFF)
Figure 3: Partial 2H{1H} NMR (107.5 MHz) with PBLG in CHCl3 (13% w/w). (A) racemic sample of 6 (from Table 1, entry 2) and (B) enantioenriched sample of 6 (from Table 2, entry 2). The magnitude of the quadrupolar splittings for the (R)- and (S)-enantiomers are labelled ΔνQ(ent). The ee of each sample was determined by deconvolution of the line shapes and subsequent integration.
Figure 3: Partial 2H{1H} NMR (107.5 MHz) with PBLG in CHCl3 (13% w/w). (A) racemic sample of 6 (from Table 1, entry ...
Two different activator systems were investigated for the nucleophilic substitution of 1: a mixture of water and isopropanol (Table 2, entries 1–5) and tris(hydroxymethyl)propane (Table 2, entries 6–8). Using water/isopropanol as the activator afforded the benzylated products 5–9 in moderate yields after 18 h. The ee values of all of the resulting products was very close to that of the original benzyl fluoride ((R)-1, 95%), indicating that a highly associative SN2-like pathway was operating, where the incoming nucleophile must have approached on a coordinate anti to the C–F bond resulting in an inversion of the configuration. These results are in good agreement with the transition state proposed by Paquin [3-5]. Unfortunately, N-methylbenzylamine (Table 2, entry 3) afforded a product 7 that did not resolve by 2H NMR, and thus the ee could not be determined.
Changing the activator from water/isopropanol to tris(hydroxymethyl)propane was anticipated to increase the stability of the triol–benzyl fluoride complex, and hence a tendancy towards an associative mechanism was expected. However, on performing the reactions with nitrogen nucleophiles (Table 2, entries 6 and 8) and the triol as the hydrogen bond donor, slightly lower ee’s were obtained relative to those obtained using the same nucleophiles with the water/isopropanol system (Table 2, entries 2 and 5). These minor differences in ee may be due to the higher temperature leading to a minor, but noticeable dissociative pathway. Once again, using N-methylbenzylamine as the nucleophile (Table 2, entry 7) afforded 7, which could not be resolved by 2H NMR. Overall, the nucleophilic substitution of 1, using either of the described hydrogen bond activating systems, afforded enantioenriched benzylated products with little erosion in stereointegrity.
In contrast to the above nucleophilic substitutions, which all proceeded with good stereointegrity, the Friedel–Crafts reactions of 1 with p-xylene gave very different results, as shown in Table 3.
![[Graphic 12]](/bjoc/content/inline/1860-5397-14-6-i14.svg?max-width=637&scale=1.0)
![[Graphic 13]](/bjoc/content/inline/1860-5397-14-6-i15.svg?max-width=637&scale=1.0)
![[Graphic 14]](/bjoc/content/inline/1860-5397-14-6-i16.svg?max-width=637&scale=1.0)
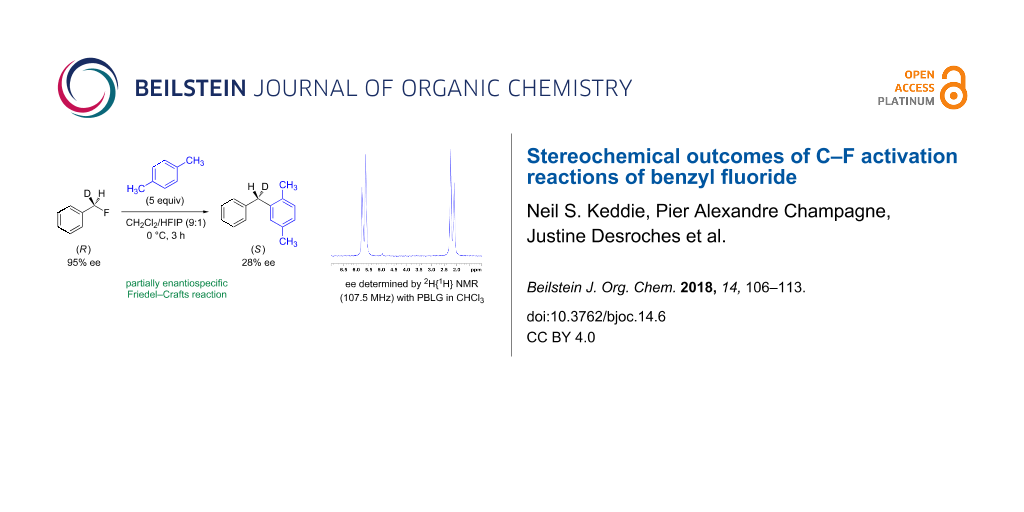
At room temperature (Table 3, entry 1), benzyl fluoride (R)-1 was activated by HFIP, affording biarylmethane 10 in a good yield (88%) after 18 h. The ee of the product was low (24%), but not racemic. The proposed stereochemistry of the product was verified by independent synthesis of the (S)-isomer from the unsymmetric diphenyl ketone 11 (Scheme 2).
![[1860-5397-14-6-i2]](/bjoc/content/inline/1860-5397-14-6-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of enantioenriched (S)-diarylmethane 10 from diaryl ketone 11 and confirmation of configuration of (R)-13 by single crystal X-ray structure.
Scheme 2: Synthesis of enantioenriched (S)-diarylmethane 10 from diaryl ketone 11 and confirmation of configu...
Corey–Bakshi–Shibata reduction of diaryl ketone 11, afforded the (R)-alcohol 12 in moderate to good yield and moderate ee [15,16]. The absolute stereochemistry of 12 was confirmed by X-ray crystallography of the 4-bromophenyl ester derivative 13. Alcohol 12 was activated as the tosyl ester at −20 °C, and then immediately displaced by LiAlD4 [17], inverting the stereocenter to afford the (S)-diarylmethane 10 isotopomer in 18% ee. 2H{1H} NMR in a PBLG matrix indicated that the dominant isomer was the same as was produced in entry 1, Table 3. Therefore, this analysis showed that the dominant enantiomer of 10 arose from an inversion, rather than retention, of configuration of the original stereocenter of 1.
There may be four different reaction mechanisms operating in these Friedel–Crafts reactions as shown in Figure 4. (A) Coordination of the fluorine atom with the hydrogen bond donor, followed by backside attack of the nucleophile leads to SN2 reaction and inversion of configuration. (B) Hydrogen bond donor coordination to fluorine leads to ionisation of 1, producing an intimate ion pair, which only permits backside attack of the nucleophile on the benzylic cation. (C) If the nucleophile is poor and k4 > k3, a solvent-separated ion pair will be formed, where the HBD-coordinated fluorine atom is loosely associated with the solvated cation, allowing a nucelophilic attack to occur from more trajectories, leading to a mixture of inversion (predominant) and retention products. (D) Fully solvated cation, where attack of the nucleophile can freely occur from either face, leading to racemization of the product in an SN1 reaction.
![[1860-5397-14-6-4]](/bjoc/content/figures/1860-5397-14-6-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Possible reactive intermediates for C–F activation of benzyl fluoride 1 with strong hydrogen bond donors.
Figure 4: Possible reactive intermediates for C–F activation of benzyl fluoride 1 with strong hydrogen bond d...
We propose that the actual attack of the nucleophile does not occur on the coordinated benzyl fluoride (A), or the fully solvated carbocation (D), as these scenarios would incur 100% or 0% enantiospecificity, respectively. Rather, the data suggest that attack occurs on a mixture of intimate (B) and solvent-separated (C) ion pairs. The partial racemization observed in Table 3 suggests that the solvent-separated ion-pair intermediate (C) is most likely the reactive species, as it would naturally lead to a partial racemization of the substrate stereocenter.
When the activator was changed from HFIP to a mixed system of HFIP and TFA (3 mol %, Table 3, entries 2 and 3), the reactions were complete in a significantly shorter time, i.e., the initial induction period observed when only HFIP was used [7] disappeared in each case. The ee of entry 2 was lower (19%), showing that the increased hydrogen bonding strength of the TFA, and thus the more rapid generation of HF (vide infra), promotes a dissociative pathway via the solvent-separated ion pair. The greater ionic strength of the solution may also play a part in stabilising the partially dissociated carbocation. Pleasingly, decreasing the temperature (Table 3, entry 3) did not slow the reaction down, and it completed after 3 h. However, the decreased temperature lead to a slightly higher ee (28%) for 10, suggesting that at lower temperatures the separation of the ions is less favoured in solution, presumably for entropic reasons.
The nature of the poorer nucleophile, coupled with the stronger hydrogen bond donor in the Friedel–Crafts reaction allows the solvent-separated ion-pair mechanism to predominate, significantly eroding the stereointegrity of the biarylmethane products 10. However, the products were not racemic, showing that the nucleophilic attack also occurs via an associated ion pair, rather than the fully solvated carbocation.
Conclusion
In summary, we have analyzed the stereochemical outcomes of substitution and Friedel–Crafts reactions of 7-[2H1]-(R)-benzyl fluoride ((R)-1), mediated by C–F activation using hydrogen-bond donors. When strong nucleophiles are used in conjunction with hydroxyl-based donors, an associative SN2-like reaction mechanism predominates, with almost complete inversion of the configuration at the stereogenic center. Poorer aryl nucleophiles can be used for Friedel–Crafts reactions if strong hydrogen bond donors (such as HFIP or TFA) are used to activate the C–F bond. In these cases, a dissociative mechanism operates, probably via a solvent-separated ion pair, rather than a fully solvated benzylic carbocation. The products arising from this mechanism are only partially enantioenriched, suggesting that there is still a steric influence for backside attack of the nucleophile in the solvent-separated ion pair, arising from the large, congested hydrogen bond networks around the fluorine atom.
Supporting Information
The Supporting Information features experimental protocols and 1H, 19F (where appropriate) and 2H{1H} NMR spectra of benzyl fluoride 1 and adducts 5–10. The methods for measurement of the ee by 2H{1H} NMR are also described.
| Supporting Information File 1: Experimental protocols. | ||
| Format: PDF | Size: 1.5 MB | Download |
| Supporting Information File 2: 2H NMR analysis of enantiopurity. | ||
| Format: PDF | Size: 1.3 MB | Download |
Acknowledgements
NK and DOH acknowledge support from the University of St Andrews, Engineering and Physical Sciences Research Council (EPSRC, Grant No.: EP/L017911/1), and the EPSRC UK National Mass Spectrometry Facility at Swansea University. This work was also supported by the Natural Sciences and Engineering Research Council of Canada (NSERC), the FRQNT Centre in Green Chemistry and Catalysis (CGCC), and the Université Laval.
References
-
Blanksby, S. J.; Ellison, G. B. Acc. Chem. Res. 2003, 36, 255–263. doi:10.1021/ar020230d
Return to citation in text: [1] -
Amii, H.; Uneyama, K. Chem. Rev. 2009, 109, 2119–2183. doi:10.1021/cr800388c
Return to citation in text: [1] -
Champagne, P. A.; Pomarole, J.; Thérien, M.-E.; Benhassine, Y.; Beaulieu, S.; Legault, C. Y.; Paquin, J.-F. Org. Lett. 2013, 15, 2210–2213. doi:10.1021/ol400765a
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Champagne, P. A.; Saint-Martin, A.; Drouin, M.; Paquin, J.-F. Beilstein J. Org. Chem. 2013, 9, 2451–2456. doi:10.3762/bjoc.9.283
Return to citation in text: [1] [2] [3] [4] [5] -
Champagne, P. A.; Drouin, M.; Legault, C. Y.; Audubert, C.; Paquin, J.-F. J. Fluorine Chem. 2015, 171, 113–119. doi:10.1016/j.jfluchem.2014.08.018
Return to citation in text: [1] [2] [3] [4] [5] -
Champagne, P. A.; Benhassine, Y.; Desroches, J.; Paquin, J.-F. Angew. Chem., Int. Ed. 2014, 53, 13835–13839. doi:10.1002/anie.201406088
Return to citation in text: [1] [2] [3] [4] -
Hemelaere, R.; Champagne, P. A.; Desroches, J.; Paquin, J.-F. J. Fluorine Chem. 2016, 190, 1–6. doi:10.1016/j.jfluchem.2016.08.003
Return to citation in text: [1] [2] [3] [4] [5] -
Schneider, H.-J. Chem. Sci. 2012, 3, 1381–1394. doi:10.1039/c2sc00764a
Return to citation in text: [1] -
Champagne, P. A.; Desroches, J.; Paquin, J.-F. Synthesis 2015, 47, 306–322. doi:10.1055/s-0034-1379537
Return to citation in text: [1] -
Canet, I.; Courtieu, J.; Loewenstein, A.; Meddour, A.; Pechine, J. M. J. Am. Chem. Soc. 1995, 117, 6520–6526. doi:10.1021/ja00129a015
Return to citation in text: [1] [2] -
Wadoux, R. D. P.; Lin, X.; Keddie, N. S.; O’Hagan, D. Tetrahedron: Asymmetry 2013, 24, 719–723. doi:10.1016/j.tetasy.2013.05.001
Return to citation in text: [1] [2] -
Yamada, I.; Noyori, R. Org. Lett. 2000, 2, 3425–3427. doi:10.1021/ol0002119
Return to citation in text: [1] -
Bio, M. M.; Waters, M.; Javadi, G.; Song, Z. J.; Zhang, F.; Thomas, D. Synthesis 2008, 891–896. doi:10.1055/s-2008-1032181
Return to citation in text: [1] -
Bresciani, S.; O’Hagan, D. Tetrahedron Lett. 2010, 51, 5795–5797. doi:10.1016/j.tetlet.2010.08.104
Return to citation in text: [1] -
Corey, E. J.; Helal, C. J. Tetrahedron Lett. 1996, 37, 5675–5678. doi:10.1016/0040-4039(96)01198-7
Return to citation in text: [1] -
The enantiomeric excess varied slightly when the reaction was run on different scales, due to variations in temperature.
Return to citation in text: [1] -
Bolshan, Y.; Chen, C.-y.; Chilenski, J. R.; Gosselin, F.; Mathre, D. J.; O'Shea, P. D.; Roy, A.; Tillyer, R. D. Org. Lett. 2003, 6, 111–114. doi:10.1021/ol0361655
Return to citation in text: [1]
| 11. | Wadoux, R. D. P.; Lin, X.; Keddie, N. S.; O’Hagan, D. Tetrahedron: Asymmetry 2013, 24, 719–723. doi:10.1016/j.tetasy.2013.05.001 |
| 11. | Wadoux, R. D. P.; Lin, X.; Keddie, N. S.; O’Hagan, D. Tetrahedron: Asymmetry 2013, 24, 719–723. doi:10.1016/j.tetasy.2013.05.001 |
| 1. | Blanksby, S. J.; Ellison, G. B. Acc. Chem. Res. 2003, 36, 255–263. doi:10.1021/ar020230d |
| 3. | Champagne, P. A.; Pomarole, J.; Thérien, M.-E.; Benhassine, Y.; Beaulieu, S.; Legault, C. Y.; Paquin, J.-F. Org. Lett. 2013, 15, 2210–2213. doi:10.1021/ol400765a |
| 10. | Canet, I.; Courtieu, J.; Loewenstein, A.; Meddour, A.; Pechine, J. M. J. Am. Chem. Soc. 1995, 117, 6520–6526. doi:10.1021/ja00129a015 |
| 8. | Schneider, H.-J. Chem. Sci. 2012, 3, 1381–1394. doi:10.1039/c2sc00764a |
| 9. | Champagne, P. A.; Desroches, J.; Paquin, J.-F. Synthesis 2015, 47, 306–322. doi:10.1055/s-0034-1379537 |
| 10. | Canet, I.; Courtieu, J.; Loewenstein, A.; Meddour, A.; Pechine, J. M. J. Am. Chem. Soc. 1995, 117, 6520–6526. doi:10.1021/ja00129a015 |
| 3. | Champagne, P. A.; Pomarole, J.; Thérien, M.-E.; Benhassine, Y.; Beaulieu, S.; Legault, C. Y.; Paquin, J.-F. Org. Lett. 2013, 15, 2210–2213. doi:10.1021/ol400765a |
| 4. | Champagne, P. A.; Saint-Martin, A.; Drouin, M.; Paquin, J.-F. Beilstein J. Org. Chem. 2013, 9, 2451–2456. doi:10.3762/bjoc.9.283 |
| 5. | Champagne, P. A.; Drouin, M.; Legault, C. Y.; Audubert, C.; Paquin, J.-F. J. Fluorine Chem. 2015, 171, 113–119. doi:10.1016/j.jfluchem.2014.08.018 |
| 6. | Champagne, P. A.; Benhassine, Y.; Desroches, J.; Paquin, J.-F. Angew. Chem., Int. Ed. 2014, 53, 13835–13839. doi:10.1002/anie.201406088 |
| 7. | Hemelaere, R.; Champagne, P. A.; Desroches, J.; Paquin, J.-F. J. Fluorine Chem. 2016, 190, 1–6. doi:10.1016/j.jfluchem.2016.08.003 |
| 3. | Champagne, P. A.; Pomarole, J.; Thérien, M.-E.; Benhassine, Y.; Beaulieu, S.; Legault, C. Y.; Paquin, J.-F. Org. Lett. 2013, 15, 2210–2213. doi:10.1021/ol400765a |
| 7. | Hemelaere, R.; Champagne, P. A.; Desroches, J.; Paquin, J.-F. J. Fluorine Chem. 2016, 190, 1–6. doi:10.1016/j.jfluchem.2016.08.003 |
| 6. | Champagne, P. A.; Benhassine, Y.; Desroches, J.; Paquin, J.-F. Angew. Chem., Int. Ed. 2014, 53, 13835–13839. doi:10.1002/anie.201406088 |
| 7. | Hemelaere, R.; Champagne, P. A.; Desroches, J.; Paquin, J.-F. J. Fluorine Chem. 2016, 190, 1–6. doi:10.1016/j.jfluchem.2016.08.003 |
| 6. | Champagne, P. A.; Benhassine, Y.; Desroches, J.; Paquin, J.-F. Angew. Chem., Int. Ed. 2014, 53, 13835–13839. doi:10.1002/anie.201406088 |
| 7. | Hemelaere, R.; Champagne, P. A.; Desroches, J.; Paquin, J.-F. J. Fluorine Chem. 2016, 190, 1–6. doi:10.1016/j.jfluchem.2016.08.003 |
| 3. | Champagne, P. A.; Pomarole, J.; Thérien, M.-E.; Benhassine, Y.; Beaulieu, S.; Legault, C. Y.; Paquin, J.-F. Org. Lett. 2013, 15, 2210–2213. doi:10.1021/ol400765a |
| 15. | Corey, E. J.; Helal, C. J. Tetrahedron Lett. 1996, 37, 5675–5678. doi:10.1016/0040-4039(96)01198-7 |
| 16. | The enantiomeric excess varied slightly when the reaction was run on different scales, due to variations in temperature. |
| 3. | Champagne, P. A.; Pomarole, J.; Thérien, M.-E.; Benhassine, Y.; Beaulieu, S.; Legault, C. Y.; Paquin, J.-F. Org. Lett. 2013, 15, 2210–2213. doi:10.1021/ol400765a |
| 4. | Champagne, P. A.; Saint-Martin, A.; Drouin, M.; Paquin, J.-F. Beilstein J. Org. Chem. 2013, 9, 2451–2456. doi:10.3762/bjoc.9.283 |
| 5. | Champagne, P. A.; Drouin, M.; Legault, C. Y.; Audubert, C.; Paquin, J.-F. J. Fluorine Chem. 2015, 171, 113–119. doi:10.1016/j.jfluchem.2014.08.018 |
| 4. | Champagne, P. A.; Saint-Martin, A.; Drouin, M.; Paquin, J.-F. Beilstein J. Org. Chem. 2013, 9, 2451–2456. doi:10.3762/bjoc.9.283 |
| 5. | Champagne, P. A.; Drouin, M.; Legault, C. Y.; Audubert, C.; Paquin, J.-F. J. Fluorine Chem. 2015, 171, 113–119. doi:10.1016/j.jfluchem.2014.08.018 |
| 17. | Bolshan, Y.; Chen, C.-y.; Chilenski, J. R.; Gosselin, F.; Mathre, D. J.; O'Shea, P. D.; Roy, A.; Tillyer, R. D. Org. Lett. 2003, 6, 111–114. doi:10.1021/ol0361655 |
| 6. | Champagne, P. A.; Benhassine, Y.; Desroches, J.; Paquin, J.-F. Angew. Chem., Int. Ed. 2014, 53, 13835–13839. doi:10.1002/anie.201406088 |
| 7. | Hemelaere, R.; Champagne, P. A.; Desroches, J.; Paquin, J.-F. J. Fluorine Chem. 2016, 190, 1–6. doi:10.1016/j.jfluchem.2016.08.003 |
| 13. | Bio, M. M.; Waters, M.; Javadi, G.; Song, Z. J.; Zhang, F.; Thomas, D. Synthesis 2008, 891–896. doi:10.1055/s-2008-1032181 |
| 14. | Bresciani, S.; O’Hagan, D. Tetrahedron Lett. 2010, 51, 5795–5797. doi:10.1016/j.tetlet.2010.08.104 |
| 4. | Champagne, P. A.; Saint-Martin, A.; Drouin, M.; Paquin, J.-F. Beilstein J. Org. Chem. 2013, 9, 2451–2456. doi:10.3762/bjoc.9.283 |
| 5. | Champagne, P. A.; Drouin, M.; Legault, C. Y.; Audubert, C.; Paquin, J.-F. J. Fluorine Chem. 2015, 171, 113–119. doi:10.1016/j.jfluchem.2014.08.018 |
| 3. | Champagne, P. A.; Pomarole, J.; Thérien, M.-E.; Benhassine, Y.; Beaulieu, S.; Legault, C. Y.; Paquin, J.-F. Org. Lett. 2013, 15, 2210–2213. doi:10.1021/ol400765a |
| 3. | Champagne, P. A.; Pomarole, J.; Thérien, M.-E.; Benhassine, Y.; Beaulieu, S.; Legault, C. Y.; Paquin, J.-F. Org. Lett. 2013, 15, 2210–2213. doi:10.1021/ol400765a |
| 4. | Champagne, P. A.; Saint-Martin, A.; Drouin, M.; Paquin, J.-F. Beilstein J. Org. Chem. 2013, 9, 2451–2456. doi:10.3762/bjoc.9.283 |
| 5. | Champagne, P. A.; Drouin, M.; Legault, C. Y.; Audubert, C.; Paquin, J.-F. J. Fluorine Chem. 2015, 171, 113–119. doi:10.1016/j.jfluchem.2014.08.018 |
© 2018 Keddie et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)