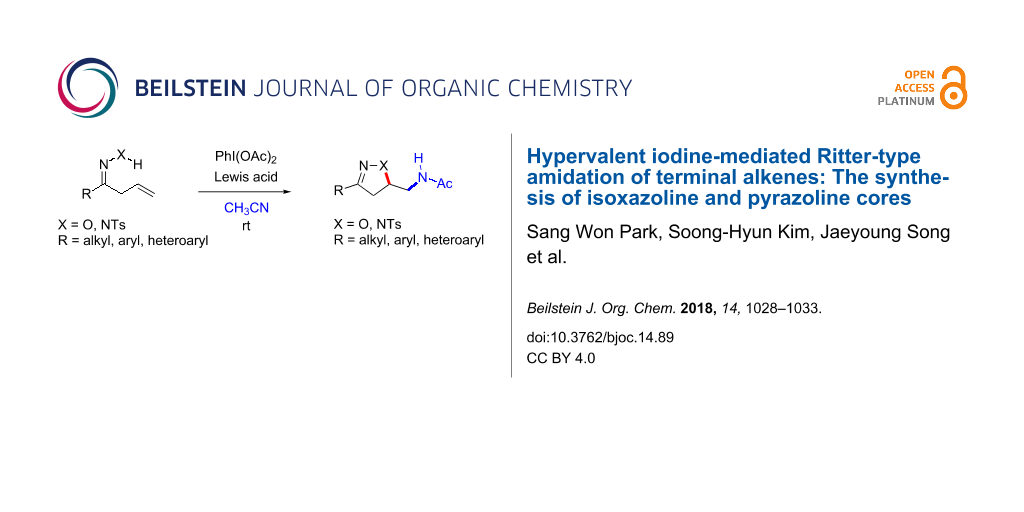
Abstract
Hypervalent iodine-mediated olefin functionalization provides a rapid gateway towards accessing both various heterocyclic cores and functional groups. In this regard, we have developed a Ritter-type alkene functionalization utilizing a PhI(OAc)2 ((diacetoxyiodo)benzene, PIDA)/Lewis acid combination in order to access isoxazoline and pyrazoline cores. Based on allyl ketone oximes and allyl ketone tosylhydrazones, we have developed an alkene oxyamidation and amido-amidation protocol en route to accessing both isoxazoline and pyrazoline cores. Additionally, acetonitrile serves as both the solvent and an amine source in the presence of this PIDA/Lewis acid combination. This operationally straightforward and metal-free protocol provides an easy access to isoxazoline and pyrazoline derivatives.

Graphical Abstract
Introduction
Isoxazoline and pyrazoline-containing heterocycles are abundant in natural products and biologically active molecules [1-5]. Thus, these scaffolds are also important from the standpoint of pharmaceutical and medicinal chemistry [6-11]. Not surprisingly, the construction of diverse heterocyclic cores including isoxazolines and pyrazolines has garnered much attention from synthetic chemists [12-15]. Among precedent synthetic methods, the functionalization of unactivated olefins provides a rapid construction of different heterocycles [16,17]. More specifically, the formation of isoxazoline and pyrazoline cores via alkene heterofunctionalization of allyl ketone oximes and/or allyl ketone tosylhydrazones has been well documented [18-22]. For example, diverse halonium sources have been utilized for the synthesis of isoxazolines via halocyclization. Furthermore, transition metal-, visible light, and hypervalent iodine-mediated oxidative cyclization protocols provide isoxazoline backbones bearing diverse substituents such as –SR, -CF3, -OH and halogens [23-27].
Results and Discussion
As depicted in Scheme 1, we have previously reported an inter-/intramolecular alkene diamination using either N-iodosuccinimide (NIS) or a phenyliodine diacetate (PIDA)/halide additive combination [28-30]. Vinylanilines and vinylaminopyridines in combination with electron-rich, Brønsted basic amines were converted to their corresponding indoline and azaindoline derivatives.
![[1860-5397-14-89-i1]](/bjoc/content/inline/1860-5397-14-89-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Hypervalent iodine-mediated heterofunctionalization of terminal alkenes.
Scheme 1: Hypervalent iodine-mediated heterofunctionalization of terminal alkenes.
However, an attempted expansion of this methodology to allyl ketone oximes and allyl ketone tosylhydrazones proved unsuccessful under the previously reported reaction conditions (see Supporting Information File 1, Table S1). Upon optimization with various oxidants and additives screened, it was found that a Lewis acid additive can promote the olefin heterofunctionalization via a Ritter-type amidation using acetonitrile as both the solvent and the amine source. Interestingly, this hypervalent iodine-mediated Ritter-type oxyamidation of 1a proved less efficient in the presence of solvent combinations with acetonitrile, despite acetonitrile being used in vast excess (see Supporting Information File 1, Table S1). Herein, we entail the first example of a hypervalent iodine(III)-mediated Ritter-type oxyamidation and amido-amidation of terminal alkenes in the presence of a Lewis acid.
Optimization studies of this Ritter-type oxyamidation commenced with oxidant screening in the presence of a Lewis acid (Table 1). Without oxidant, the Ritter-type oxyamidation still proceeded to give 3a albeit in low yield (Table 1, entry 1). The background reaction mediated by a Lewis acid seemed plausible via an electrophilic activation of the double bond. When the reaction is performed in the presence of hypervalent iodine reagents such as PIFA ([bis(trifluoroacetoxy)iodo]benzene), PhI(NPhth)2 and PIDP (bis(tert-butylcarbonyloxy)iodobenzene) much better yields were obtained (Table 1, entries 2–4), with PhI(OAc)2 proving to be the best (Table 1, entry 5). Refluxing conditions further improved the yield (Table 1, entry 6). Additionally, other cyclic hypervalent iodine oxidants such as IBX (2-iodoxybenzoic acid) and DMP (Dess–Martin periodinane) (Table 1, entries 7 and 8) gave similar yields to the background reaction. Lastly, a Lewis acid screen (Table 1, entries 9–12) was performed. Among the tested Lewis acids, AlCl3, SnCl4, TiCl4, TMSOTf and BF3·Et2O, the latter was found to be the best choice of additive. Remarkably, the activation of PIDA by a Lewis acid (BF3·OEt2) seemed to be crucial for this Ritter-type oxyamidation to proceed (Table 1, entry 13). Based on these experiments, we chose the conditions outlined in Table 1, entry 5 for our further investigations due to the mild (room temperature) reaction conditions.
Table 1: Hypervalent iodine-mediated Ritter-type alkene oxyamidation.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-89-i5.svg?max-width=637&scale=1.0)
|
||||
| entrya | oxidant (equiv) | additive (equiv) | T (°C) | yield of 3a (%)b |
| 1 | – | BF3·OEt2 (1.0) | 25 | 10 |
| 2 | PhI(OCOCF3)2 (1.0) | BF3·OEt2 (1.0) | 25 | 35 |
| 3 | PhI(NPhth)2 (1.0) | BF3·OEt2 (1.0) | 25 | 42 |
| 4 | PIDP (1.0) | BF3·OEt2 (1.0) | 25 | 49 |
| 5 | PhI(OAc)2 (1.0) | BF3·OEt2 (1.0) | 25 | 55 |
| 6 | PhI(OAc)2 (1.0) | BF3·OEt2 (1.0) | reflux | 60 |
| 7 | IBX (1.0) | BF3·OEt2 (1.0) | 25 | 14 |
| 8 | DMP (1.0) | BF3·OEt2 (1.0) | 25 | 14 |
| 9 | PhI(OAc)2 (1.0) | AlCl3 (1.0) | 25 | 0 |
| 10 | PhI(OAc)2 (1.0) | SnCl4 (1.0) | 25 | 0 |
| 11 | PhI(OAc)2 (1.0) | TiCl4 (1.0) | 25 | 12 |
| 12 | PhI(OAc)2 (1.0) | TMSOTf (1.0) | 25 | 45 |
| 13 | PhI(OAc)2 (1.0) | – | 25 | 0 |
aAll reactions were performed on a 0.21 mmol scale (0.1 M) and with a standard 18 h reaction time. bIsolated yield.
Next a series of allyl ketone oximes 1 were subjected to the optimized reaction conditions and the results are summarized in Scheme 2. Phenyl and electron-deficient aryl allyl ketone oximes showed robust reactivity as the corresponding products were obtained in good yields (3a–c, 3f) [31]. However, electron-rich aryl allyl ketone oximes such as 1d, 1e and 1g proved inferior. Also the furan-substituted allyl ketone oxime delivered the desired product 3h albeit in a moderate yield. In addition, various alkyl allyl ketone oximes were investigated. While cyclopropyl allyl ketone oxime 1m was converted to the corresponding isoxazoline 3m in 20% yield, other alkyl allyl ketone oximes afforded higher yields of the desired products. This observation is ascribed differences in reactivity due to an increased steric bulk at the α-position (t-Bu > c-Hex > n-Oct > n-Bu).
![[1860-5397-14-89-i2]](/bjoc/content/inline/1860-5397-14-89-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope of the Ritter-type oxyamidation: isoxazoline synthesis. All reactions were performed on a 0.21 mmol scale (0.1 M) and with a standard 18 h reaction time.
Scheme 2: Substrate scope of the Ritter-type oxyamidation: isoxazoline synthesis. All reactions were performe...
We next explored the hypervalent iodine-mediated Ritter-type amido-amidation reaction in the presence of a Lewis acid in order to access pyrazoline cores (Scheme 3). A series of allyl ketone tosylhydrazones 4 were subjected to the same reaction conditions. In general, the yields of the pyrazoline cores decreased marginally relative to the Ritter-type oxyamidation reaction. Monosubstituted aryl allyl ketone tosylhydrazones showed good reactivity and provided the pyrazoline heterocycles 5a–c in moderate yields (24–47%). On the other hand, disubstituted aryl allyl ketone tosylhydrazones 4d and 4e yielded the corresponding products in 28% and 26%, respectively. The reaction of heteroaryl allyl ketone tosylhydrazones such as 3-furyl allyl ketone tosylhydrazone 4f and 3-thiophenyl allyl ketone tosylhydrazone 4g provided their desired products in low yields. Lastly, alkyl allyl ketone tosylhydrazones 4h and 4i seemed to maintain of the trend in which increased reactivity is the result of increasing size of the alkyl side chain at the α-position (t-Bu > c-Hex).
![[1860-5397-14-89-i3]](/bjoc/content/inline/1860-5397-14-89-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Substrate scope of Ritter-type amido-amidation: pyrazoline synthesis. All reactions were performed on a 0.21 mmol scale (0.1 M) and with a standard 18 h reaction time.
Scheme 3: Substrate scope of Ritter-type amido-amidation: pyrazoline synthesis. All reactions were performed ...
Based on these experimental data and previous reports [32,33], a plausible mechanism of the Ritter-type oxyamidation and amido-amidation is proposed in Scheme 4. First, an activation of hypervalent iodine(III) by the Lewis acid generates the active iodine(III) species A in situ. The resulting iodine(III) then, in turn, forms the electrophilic iodonium intermediate B with the terminal alkene of the allyl ketone oxime or allyl ketone tosylhydrazone. The subsequent 5-exo-type cyclization by nucleophilic attack on the iodonium then leads to the isoxazoline or pyrazoline cores (C) bearing the hypervalent iodine(III) group. Following iodine activation by the Lewis acid, the iodonium ion D undergoes nucleophilic substitution with excess acetonitrile to form intermediate E. Then water can add to the corresponding nitrilium and subsequent tautomerization delivers isoxazoline 3 (X = O) and pyrazoline 5 (X = NTs) via a Ritter-type oxyamidation and amido-amidation.
![[1860-5397-14-89-i4]](/bjoc/content/inline/1860-5397-14-89-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Plausible mechanism of the hypervalent iodine(III)-mediated Ritter-type oxyamidation/amido-amidation in the presence of a Lewis acid.
Scheme 4: Plausible mechanism of the hypervalent iodine(III)-mediated Ritter-type oxyamidation/amido-amidatio...
Conclusion
In summary, we have developed a hypervalent iodine(III)-mediated inter-/intramolecular Ritter-type oxyamidation and amido-amidation protocol in the presence of a Lewis acid. This transformation provides direct access to diverse 5-acetaminomethyl substituted 2-isoxazoline/2-pyrazoline derivatives using acetonitrile as both the solvent and amine source.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, and copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 3.7 MB | Download |
References
-
Kiss, L.; Nonn, M.; Fülöp, F. Synthesis 2012, 44, 1951–1963. doi:10.1055/s-0031-1290373
Return to citation in text: [1] -
Kaur, K.; Kumar, V.; Sharma, A. K.; Gupta, G. K. Eur. J. Med. Chem. 2014, 77, 121–133. doi:10.1016/j.ejmech.2014.02.063
Return to citation in text: [1] -
Xue, C.-B.; Wityak, J.; Sielecki, T. M.; Pinto, D. J.; Batt, D. G.; Cain, G. A.; Sworin, M.; Rockwell, A. L.; Roderick, J. J.; Wang, S.; Orwat, M. J.; Frietze, W. E.; Bostrom, L. L.; Liu, J.; Higley, C. A.; Rankin, F. W.; Tobin, A. E.; Emmett, G.; Lalka, G. K.; Sze, J. Y.; Di Meo, S. V.; Mousa, S. A.; Thoolen, M. J.; Racanelli, A. L.; Hausner, E. A.; Reilly, T. M.; DeGrado, W. F.; Wexler, R. R.; Olson, R. E. J. Med. Chem. 1997, 40, 2064–2084. doi:10.1021/jm960799i
Return to citation in text: [1] -
Poutiainen, P. K.; Venäläinen, T. A.; Peräkylä, M.; Matilainen, J. M.; Väisänen, S.; Honkakoski, P.; Laatikainen, R.; Pulkkinen, J. T. Bioorg. Med. Chem. 2010, 18, 3437–3447. doi:10.1016/j.bmc.2010.04.007
Return to citation in text: [1] -
Castellano, S.; Kuck, D.; Viviano, M.; Yoo, J.; López-Vallejo, F.; Conti, P.; Tamborini, L.; Pinto, A.; Medina-Franco, J. L.; Sbardella, G. J. Med. Chem. 2011, 54, 7663–7677. doi:10.1021/jm2010404
Return to citation in text: [1] -
Lee, M.; Brockway, O.; Dandavati, A.; Tzou, S.; Sjoholm, R.; Satam, V.; Westbrook, C.; Mooberry, S. L.; Zeller, M.; Babu, B.; Lee, M. Eur. J. Med. Chem. 2011, 46, 3099–3104. doi:10.1016/j.ejmech.2011.03.064
Return to citation in text: [1] -
Padmavathi, V.; Thriveni, P.; Reddy, G. S.; Deepti, D. Eur. J. Med. Chem. 2008, 43, 917–924. doi:10.1016/j.ejmech.2007.06.011
Return to citation in text: [1] -
Acharya, B. N.; Saraswat, D.; Tiwari, M.; Shrivastava, A. K.; Ghorpade, R.; Bapna, S.; Kaushik, M. P. Eur. J. Med. Chem. 2010, 45, 430–438. doi:10.1016/j.ejmech.2009.10.023
Return to citation in text: [1] -
Dardić, D.; Lauro, G.; Bifulco, G.; Laboudie, P.; Sakhaii, P.; Bauer, A.; Vilcinskas, A.; Hammann, P. E.; Plaza, A. J. Org. Chem. 2017, 82, 6032–6043. doi:10.1021/acs.joc.7b00228
Return to citation in text: [1] -
Buhrlage, S. J.; Bates, C. A.; Rowe, S. P.; Minter, A. R.; Brennan, B. B.; Majmudar, C. Y.; Wemmer, D. E.; Al-Hashimi, H.; Mapp, A. K. ACS Chem. Biol. 2009, 4, 335–344. doi:10.1021/cb900028j
Return to citation in text: [1] -
Liu, X.-H.; Ruan, B.-F.; Li, J.; Chen, F.-H.; Song, B.-A.; Zhu, H.-L.; Bhadury, P. S.; Zhao, J. Mini-Rev. Med. Chem. 2011, 11, 771–821. doi:10.2174/138955711796355285
Return to citation in text: [1] -
Kozikowski, A. P. Acc. Chem. Res. 1984, 17, 410–416. doi:10.1021/ar00108a001
Return to citation in text: [1] -
Jiang, D.; Peng, J.; Chen, Y. Org. Lett. 2008, 10, 1695–1698. doi:10.1021/ol8002173
Return to citation in text: [1] -
Zhu, M.-K.; Zhao, J.-F.; Loh, T.-P. J. Am. Chem. Soc. 2010, 132, 6284–6285. doi:10.1021/ja100716x
Return to citation in text: [1] -
He, Y.-T.; Li, L.-H.; Yang, Y.-F.; Wang, Y.-Q.; Luo, J.-Y.; Liu, X.-Y.; Liang, Y.-M. Chem. Commun. 2013, 49, 5687–5689. doi:10.1039/C3CC42588F
Return to citation in text: [1] -
Han, B.; Yang, X.-L.; Fang, R.; Yu, W.; Wang, C.; Duan, X.-Y.; Liu, S. Angew. Chem., Int. Ed. 2012, 51, 8816–8820. doi:10.1002/anie.201203799
Return to citation in text: [1] -
Peng, X.-X.; Deng, Y.-J.; Yang, X.-L.; Zhang, L.; Yu, W.; Han, B. Org. Lett. 2014, 16, 4650–4653. doi:10.1021/ol502258n
Return to citation in text: [1] -
Tripathi, C. B.; Mukherjee, S. Angew. Chem., Int. Ed. 2013, 52, 8450–8453. doi:10.1002/anie.201304173
Return to citation in text: [1] -
Tiecco, M.; Testaferri, L.; Bagnoli, L.; Purgatorio, V.; Temperini, A.; Marini, F.; Santi, C. Tetrahedron: Asymmetry 2001, 12, 3297–3304. doi:10.1016/S0957-4166(02)00013-7
Return to citation in text: [1] -
Dondas, H. A.; Grigg, R.; Hadjisoteriou, M.; Markandu, J.; Kennewell, P.; Thornton-Pett, M. Tetrahedron 2001, 57, 1119–1128. doi:10.1016/S0040-4020(00)01084-X
Return to citation in text: [1] -
Karapetyan, V.; Mkrtchyan, S.; Dang, T. T.; Villinger, A.; Reinke, H.; Langer, P. Tetrahedron 2008, 64, 8010–8015. doi:10.1016/j.tet.2008.05.116
Return to citation in text: [1] -
Mosher, M. D.; Norman, A. L.; Shurrush, K. A. Tetrahedron Lett. 2009, 50, 5647–5648. doi:10.1016/j.tetlet.2009.07.106
Return to citation in text: [1] -
Zhu, L.; Yu, H.; Xu, Z.; Jiang, X.; Lin, L.; Wang, R. Org. Lett. 2014, 16, 1562–1565. doi:10.1021/ol403687k
Return to citation in text: [1] -
Zhu, L.; Wang, G.; Guo, Q.; Xu, Z.; Zhang, D.; Wang, R. Org. Lett. 2014, 16, 5390–5393. doi:10.1021/ol502624z
Return to citation in text: [1] -
Wei, Q.; Chen, J.-R.; Hu, X.-Q.; Yang, X.-C.; Lu, B.; Xiao, W.-J. Org. Lett. 2015, 17, 4464–4667. doi:10.1021/acs.orglett.5b02118
Return to citation in text: [1] -
Hu, X.-Q.; Chen, J.-R.; Wei, Q.; Liu, F.-L.; Deng, Q.-H.; Beauchemin, A. M.; Xiao, W.-J. Angew. Chem., Int. Ed. 2014, 53, 12163–12167. doi:10.1002/anie.201406491
Return to citation in text: [1] -
Li, W.; Jia, P.; Han, B.; Li, D.; Yu, W. Tetrahedron 2013, 69, 3274–3280. doi:10.1016/j.tet.2013.02.032
Return to citation in text: [1] -
Hong, K. B.; Johnston, J. N. Org. Lett. 2014, 16, 3804–3807. doi:10.1021/ol501693j
Return to citation in text: [1] -
Danneman, M. W.; Hong, K. B.; Johnston, J. N. Org. Lett. 2015, 17, 2558–2561. doi:10.1021/acs.orglett.5b01177
Return to citation in text: [1] -
Danneman, M. W.; Hong, K. B.; Johnston, J. N. Org. Lett. 2015, 17, 3806–3809. doi:10.1021/acs.orglett.5b01783
Return to citation in text: [1] -
CCDC 1815928 and contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from TheCambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Reddy, B. V. S.; Reddy, N. S.; Madan, C.; Yadav, J. S. Tetrahedron Lett. 2010, 51, 4827–4829. doi:10.1016/j.tetlet.2010.07.032
Return to citation in text: [1] -
Cui, H.; Liu, X.; Wei, W.; Yang, D.; He, C.; Zhang, T.; Wang, H. J. Org. Chem. 2016, 81, 2252–2260. doi:10.1021/acs.joc.5b02579
Return to citation in text: [1]
| 1. | Kiss, L.; Nonn, M.; Fülöp, F. Synthesis 2012, 44, 1951–1963. doi:10.1055/s-0031-1290373 |
| 2. | Kaur, K.; Kumar, V.; Sharma, A. K.; Gupta, G. K. Eur. J. Med. Chem. 2014, 77, 121–133. doi:10.1016/j.ejmech.2014.02.063 |
| 3. | Xue, C.-B.; Wityak, J.; Sielecki, T. M.; Pinto, D. J.; Batt, D. G.; Cain, G. A.; Sworin, M.; Rockwell, A. L.; Roderick, J. J.; Wang, S.; Orwat, M. J.; Frietze, W. E.; Bostrom, L. L.; Liu, J.; Higley, C. A.; Rankin, F. W.; Tobin, A. E.; Emmett, G.; Lalka, G. K.; Sze, J. Y.; Di Meo, S. V.; Mousa, S. A.; Thoolen, M. J.; Racanelli, A. L.; Hausner, E. A.; Reilly, T. M.; DeGrado, W. F.; Wexler, R. R.; Olson, R. E. J. Med. Chem. 1997, 40, 2064–2084. doi:10.1021/jm960799i |
| 4. | Poutiainen, P. K.; Venäläinen, T. A.; Peräkylä, M.; Matilainen, J. M.; Väisänen, S.; Honkakoski, P.; Laatikainen, R.; Pulkkinen, J. T. Bioorg. Med. Chem. 2010, 18, 3437–3447. doi:10.1016/j.bmc.2010.04.007 |
| 5. | Castellano, S.; Kuck, D.; Viviano, M.; Yoo, J.; López-Vallejo, F.; Conti, P.; Tamborini, L.; Pinto, A.; Medina-Franco, J. L.; Sbardella, G. J. Med. Chem. 2011, 54, 7663–7677. doi:10.1021/jm2010404 |
| 18. | Tripathi, C. B.; Mukherjee, S. Angew. Chem., Int. Ed. 2013, 52, 8450–8453. doi:10.1002/anie.201304173 |
| 19. | Tiecco, M.; Testaferri, L.; Bagnoli, L.; Purgatorio, V.; Temperini, A.; Marini, F.; Santi, C. Tetrahedron: Asymmetry 2001, 12, 3297–3304. doi:10.1016/S0957-4166(02)00013-7 |
| 20. | Dondas, H. A.; Grigg, R.; Hadjisoteriou, M.; Markandu, J.; Kennewell, P.; Thornton-Pett, M. Tetrahedron 2001, 57, 1119–1128. doi:10.1016/S0040-4020(00)01084-X |
| 21. | Karapetyan, V.; Mkrtchyan, S.; Dang, T. T.; Villinger, A.; Reinke, H.; Langer, P. Tetrahedron 2008, 64, 8010–8015. doi:10.1016/j.tet.2008.05.116 |
| 22. | Mosher, M. D.; Norman, A. L.; Shurrush, K. A. Tetrahedron Lett. 2009, 50, 5647–5648. doi:10.1016/j.tetlet.2009.07.106 |
| 16. | Han, B.; Yang, X.-L.; Fang, R.; Yu, W.; Wang, C.; Duan, X.-Y.; Liu, S. Angew. Chem., Int. Ed. 2012, 51, 8816–8820. doi:10.1002/anie.201203799 |
| 17. | Peng, X.-X.; Deng, Y.-J.; Yang, X.-L.; Zhang, L.; Yu, W.; Han, B. Org. Lett. 2014, 16, 4650–4653. doi:10.1021/ol502258n |
| 12. | Kozikowski, A. P. Acc. Chem. Res. 1984, 17, 410–416. doi:10.1021/ar00108a001 |
| 13. | Jiang, D.; Peng, J.; Chen, Y. Org. Lett. 2008, 10, 1695–1698. doi:10.1021/ol8002173 |
| 14. | Zhu, M.-K.; Zhao, J.-F.; Loh, T.-P. J. Am. Chem. Soc. 2010, 132, 6284–6285. doi:10.1021/ja100716x |
| 15. | He, Y.-T.; Li, L.-H.; Yang, Y.-F.; Wang, Y.-Q.; Luo, J.-Y.; Liu, X.-Y.; Liang, Y.-M. Chem. Commun. 2013, 49, 5687–5689. doi:10.1039/C3CC42588F |
| 6. | Lee, M.; Brockway, O.; Dandavati, A.; Tzou, S.; Sjoholm, R.; Satam, V.; Westbrook, C.; Mooberry, S. L.; Zeller, M.; Babu, B.; Lee, M. Eur. J. Med. Chem. 2011, 46, 3099–3104. doi:10.1016/j.ejmech.2011.03.064 |
| 7. | Padmavathi, V.; Thriveni, P.; Reddy, G. S.; Deepti, D. Eur. J. Med. Chem. 2008, 43, 917–924. doi:10.1016/j.ejmech.2007.06.011 |
| 8. | Acharya, B. N.; Saraswat, D.; Tiwari, M.; Shrivastava, A. K.; Ghorpade, R.; Bapna, S.; Kaushik, M. P. Eur. J. Med. Chem. 2010, 45, 430–438. doi:10.1016/j.ejmech.2009.10.023 |
| 9. | Dardić, D.; Lauro, G.; Bifulco, G.; Laboudie, P.; Sakhaii, P.; Bauer, A.; Vilcinskas, A.; Hammann, P. E.; Plaza, A. J. Org. Chem. 2017, 82, 6032–6043. doi:10.1021/acs.joc.7b00228 |
| 10. | Buhrlage, S. J.; Bates, C. A.; Rowe, S. P.; Minter, A. R.; Brennan, B. B.; Majmudar, C. Y.; Wemmer, D. E.; Al-Hashimi, H.; Mapp, A. K. ACS Chem. Biol. 2009, 4, 335–344. doi:10.1021/cb900028j |
| 11. | Liu, X.-H.; Ruan, B.-F.; Li, J.; Chen, F.-H.; Song, B.-A.; Zhu, H.-L.; Bhadury, P. S.; Zhao, J. Mini-Rev. Med. Chem. 2011, 11, 771–821. doi:10.2174/138955711796355285 |
| 32. | Reddy, B. V. S.; Reddy, N. S.; Madan, C.; Yadav, J. S. Tetrahedron Lett. 2010, 51, 4827–4829. doi:10.1016/j.tetlet.2010.07.032 |
| 33. | Cui, H.; Liu, X.; Wei, W.; Yang, D.; He, C.; Zhang, T.; Wang, H. J. Org. Chem. 2016, 81, 2252–2260. doi:10.1021/acs.joc.5b02579 |
| 31. | CCDC 1815928 and contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from TheCambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 28. | Hong, K. B.; Johnston, J. N. Org. Lett. 2014, 16, 3804–3807. doi:10.1021/ol501693j |
| 29. | Danneman, M. W.; Hong, K. B.; Johnston, J. N. Org. Lett. 2015, 17, 2558–2561. doi:10.1021/acs.orglett.5b01177 |
| 30. | Danneman, M. W.; Hong, K. B.; Johnston, J. N. Org. Lett. 2015, 17, 3806–3809. doi:10.1021/acs.orglett.5b01783 |
| 23. | Zhu, L.; Yu, H.; Xu, Z.; Jiang, X.; Lin, L.; Wang, R. Org. Lett. 2014, 16, 1562–1565. doi:10.1021/ol403687k |
| 24. | Zhu, L.; Wang, G.; Guo, Q.; Xu, Z.; Zhang, D.; Wang, R. Org. Lett. 2014, 16, 5390–5393. doi:10.1021/ol502624z |
| 25. | Wei, Q.; Chen, J.-R.; Hu, X.-Q.; Yang, X.-C.; Lu, B.; Xiao, W.-J. Org. Lett. 2015, 17, 4464–4667. doi:10.1021/acs.orglett.5b02118 |
| 26. | Hu, X.-Q.; Chen, J.-R.; Wei, Q.; Liu, F.-L.; Deng, Q.-H.; Beauchemin, A. M.; Xiao, W.-J. Angew. Chem., Int. Ed. 2014, 53, 12163–12167. doi:10.1002/anie.201406491 |
| 27. | Li, W.; Jia, P.; Han, B.; Li, D.; Yu, W. Tetrahedron 2013, 69, 3274–3280. doi:10.1016/j.tet.2013.02.032 |
© 2018 Park et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)