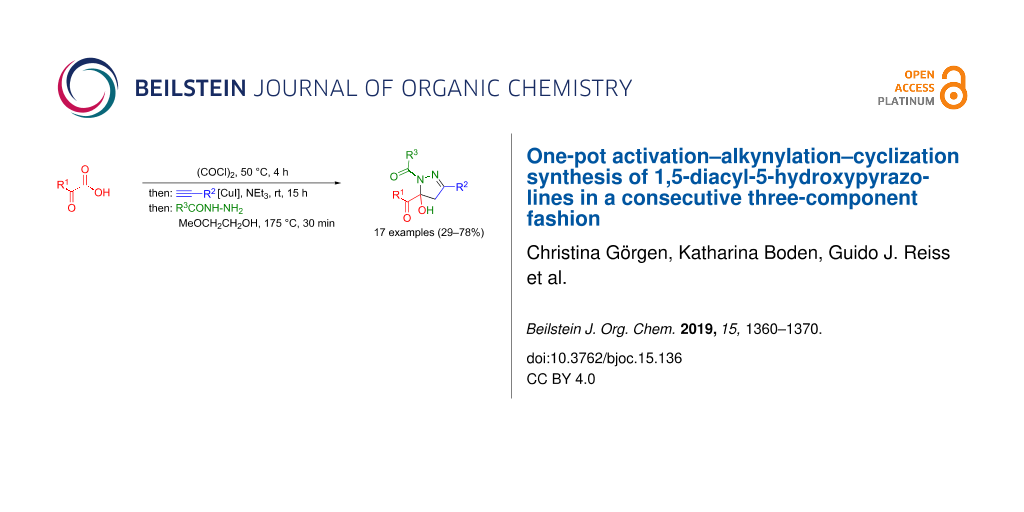
Abstract
A consecutive three-component activation–alkynylation–cyclization reaction of (hetero)aryl glyoxylic acids, oxalyl chloride, arylacetylenes, and hydrazides efficiently forms 1,5-diacyl-5-hydroxypyrazolines in moderate to good yields. The structures were unambiguously corroborated by comprehensive NMR spectroscopy and X-ray structure analyses of selected derivatives.

Graphical Abstract
Introduction
Pyrazoles [1,2] and pyrazolines [3-5] are privileged 1,2-diazole derivatives in a broad range of application, both in life and materials sciences. While the former are fully conjugated and can be considered as heteroaromatic 6π-systems with interesting properties as crop-protecting agents [6,7], as pharmaceutically active ingredients [8-11], as ligands [12,13], and as chromophores [14-16], the partially unsaturated 2H-pyrazolines have particularly attracted attention for instance as antibacterial [17], anti-inflammatory [18], antidiabetic [19], and antidepressive [20] agents. Especially, 1-acylpyrazolines have shown nanomolar in vitro activities against chloroquine-sensitive and resistant strains of Plasmodium falciparum and can therefore be considered for the treatment of malaria [21]. Furthermore, similar derivatives have shown micromolar and submicromolar activity against 60 selected cancer cell lines, presumably by inhibition of microtubuli formation in cancer cells [22]. More specifically, a series of 60 1,3-diaryl-1-acylpyrazolines was tested as xanthine oxygenase inhibitors that can be efficacious against articular gout, cancer, and inflammation, with IC50 values of four derivatives in the range of 5.3–15.2 μM (Figure 1) [23].
![[1860-5397-15-136-1]](/bjoc/content/figures/1860-5397-15-136-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Selected anticancer active 3,5-diaryl-1-acylpyrazoline (left) and xanthine oxygenase inhibitors (center and right).
Figure 1: Selected anticancer active 3,5-diaryl-1-acylpyrazoline (left) and xanthine oxygenase inhibitors (ce...
1-Acyl-5-hydroxypyrazolines have been shown to be analgesics with a slightly improved pain-relieving efficacy than Aspirin® [24,25], and 5-nitro-2-furyl-substituted derivatives are active antibacterials against the strains S. aureus, A. aerogenes, E. coli and B. subtilis (Figure 2) [26,27].
![[1860-5397-15-136-2]](/bjoc/content/figures/1860-5397-15-136-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Selected 1-acyl-5-hydroxypyrazolines with analgesic (left, center) and antibacterial activity (center and right).
Figure 2: Selected 1-acyl-5-hydroxypyrazolines with analgesic (left, center) and antibacterial activity (cent...
In addition, 1-acyl-5-hydroxypyrazolines are bidentate ligands for zinc complexes and by virtue of being ring tautomers of β-enolhydrazones they can also act as tridentate ligands for nickel [28] and tin [29,30] complexes. In contrast, dimethylzinc forms dimeric complexes where the 1-acyl-5-hydroxypyrazoline acts as a bidentate ligand [31]. Upon treatment with TMEDA mononuclear complexes with concomitant ring opening to give a seven-membered bidentate chelate are generated.
Although numerous syntheses of pyrazolines [3-5] in general and 1-acyl-5-hydroxypyrazolines [24-26] specifically have been published employing a cyclizing addition of an acylhydrazone to the carbonyl group as a ring-forming reaction [32-40], their diversity-oriented one-pot synthesis in a multicomponent approach has remained unexplored to date. In the course of our program directed to develop multicomponent syntheses of heterocycles by transition-metal catalysis [41,42] we conceptualized catalytic entries to alkynones and alkynediones as suitable intermediates in addition–cyclocondensation syntheses of numerous heterocycles, which can indeed be prepared by consecutive multicomponent reactions [43-47]. Particularly interesting are alkynediones, because, as densely functionalized trielectrophiles, the alkyne, ynone and dicarbonyl functionalities can be selectively addressed. We have established two complementary one-pot pathways to alkynediones, a glyoxylation–alkynylation (GA) [48] and an activation–alkynylation (AA) [49] sequence, which both take advantage of a copper-catalyzed alkynylation of the intermediary formed (hetero)arylglyoxyl chloride (Scheme 1). The alkynediones can be subsequently transformed, still in the same reaction vessel, to quinoxalines [48,50-52], pyrimidines [48,49], and 5-acylpyrazoles [48,49]. The latter 5-acylpyrazole arose after work-up from the three-component AA–cyclocondensation synthesis employing Boc-hydrazine as a binucleophilic hydrazide substrate. Based on our attempts to isolate potential 1,5-diacylpyrazole precursors we discovered that 1,5-diacyl-5-hydroxypyrazolines are the intermediary products. Here, we report on the novel three-component AA–condensation–cyclization synthesis of 1,5-diacyl-5-hydroxypyrazolines.
![[1860-5397-15-136-i1]](/bjoc/content/inline/1860-5397-15-136-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Glyoxylation–alkynylation (GA) and activation–alkynylation (AA) synthesis of alkynediones in a one-pot fashion.
Scheme 1: Glyoxylation–alkynylation (GA) and activation–alkynylation (AA) synthesis of alkynediones in a one-...
Results and Discussion
In our initial study [49], the three-component AA–cyclocondensation synthesis, starting from phenylglyoxylic acid (1a), phenylacetylene (2a), and Boc-hydrazide (4a) through the formation of 1,4-diphenylbut-3-yne-1,2-dione (3a), with subsequent N-deacylation as the consequence of basic work-up (Scheme 2), furnished 5-benzoyl-3-phenyl-1H-pyrazole (6a) in 41% isolated yield.
![[1860-5397-15-136-i2]](/bjoc/content/inline/1860-5397-15-136-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Consecutive three-component synthesis to give 5-benzoyl-3-phenyl-1H-pyrazole (6a) after alkaline deacylation of intermediate 5a.
Scheme 2: Consecutive three-component synthesis to give 5-benzoyl-3-phenyl-1H-pyrazole (6a) after alkaline de...
In addition to spectroscopic assignment the structure of 6a has now been corroborated by an X-ray structure analysis showing infinite chains of molecules 6a formed by intermolecular hydrogen bonding between the pyrazole N1 and the carbonyl O1 (Figure 3) [53].
![[1860-5397-15-136-3]](/bjoc/content/figures/1860-5397-15-136-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: ORTEP plot of 5-benzoyl-3-phenyl-1H-pyrazole (6a) (thermal ellipsoids at 30% probability); the direction of intermolecular N−H···O hydrogen bonding is indicated by dashed lines.
Figure 3: ORTEP plot of 5-benzoyl-3-phenyl-1H-pyrazole (6a) (thermal ellipsoids at 30% probability); the dire...
The first assumption was that the tentative intermediate 5a could be a 1,5-diacylpyrazole. However, upon performing the terminal cyclization step starting from 1,4-diphenylbut-3-yne-1,2-dione (3a) and Boc-hydrazine (4a) under identical conditions 1-Boc-5-benzoyl-5-hydroxypyrazoline was isolated in 83% yield (Scheme 3).
![[1860-5397-15-136-i3]](/bjoc/content/inline/1860-5397-15-136-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Cyclization of 1,4-diphenylbut-3-yne-1,2-dione (3a) and Boc-hydrazine (4a) to give intermediate 5a.
Scheme 3: Cyclization of 1,4-diphenylbut-3-yne-1,2-dione (3a) and Boc-hydrazine (4a) to give intermediate 5a.
The molecular structure was additionally corroborated by X-ray structure analysis showing that the assignment of intermediate 5a was not a fully unsaturated pyrazole (Figure 4) [53].
![[1860-5397-15-136-4]](/bjoc/content/figures/1860-5397-15-136-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Ellipsoid plot of 1-Boc-5-benzoyl-5-hydroxypyrazoline 5a.
Figure 4: Ellipsoid plot of 1-Boc-5-benzoyl-5-hydroxypyrazoline 5a.
Therefore, we set out to optimize the one-pot synthesis of 1,5-diacyl-5-hydroxypyrazolines by choosing the model reaction of phenylglyoxylic acid (1a), phenylacetylene (2a), and benzoyl hydrazide (4b) giving 1,5-diacyl-5-hydroxypyrazoline 5b, where the reaction times t1 and t2, as well as the conditions of the cyclization step needed to be optimized (Scheme 4).
![[1860-5397-15-136-i4]](/bjoc/content/inline/1860-5397-15-136-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Model reaction for optimizing the activation–alkynylation–cyclization synthesis of 1,5-diacyl-5-hydroxypyrazoline 5b.
Scheme 4: Model reaction for optimizing the activation–alkynylation–cyclization synthesis of 1,5-diacyl-5-hyd...
A quick optimization screening of the activation–alkynylation synthesis of 1,4-diphenylbut-3-yne-1,2-dione (3a) revealed that the use of KOH dried triethylamine instead of the initial preconditioning (Na/benzophenone dried) led to a reduction of the reaction time t1 from 24 to 15 h (see Supporting Information File 1, Table S1). In addition, the concentration could be doubled and the obtained yield of diphenylbut-3-yne-1,2-dione (3a) increased from 63 to 76%.
The terminal cyclization step, consisting of a Michael addition of benzoyl hydrazide (4b) to diphenylbut-3-yne-1,2-dione (3a) followed by a cyclizing addition of the central hydrazide nitrogen atom to the carbonyl group, was monitored by GC–MS and optimized with respect to temperature T, reaction time t2, and the alcohol additive (Table 1).
Table 1: Optimization of the cyclization step of 1,5-diacyl-5-hydroxypyrazoline 5b.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-136-i6.svg?max-width=637&scale=1.0)
|
||||
| entry | ROH [mL] | T [°C] | t2 [min] | 1,5-diacyl-5-hydroxypyrazoline 5b (%)b |
| 1c,d | 2-methoxyethanol (0.2) | 100 | 60 | incomplete conversione (n.i.) |
| 2c,d | 2-methoxyethanol (0.2) | 150 | 60 | incomplete conversione (n.i.) |
| 3d,f | 2-methoxyethanol (0.2) | 150 | 60 | complete conversione (n.i.) |
| 4d,g | 2-methoxyethanol (0.2) | 150 | 60 | complete conversione (n.i.) |
| 5d,h | 2-methoxyethanol (0.2) | 150 | 60 | complete conversione (n.i.) |
| 6d,i | 2-methoxyethanol (0.2) | 150 | 60 | incomplete conversione (n.i.) |
| 7d,h | 2-methoxyethanol (0.2) | 150 | 30 | complete conversione (n.i.) |
| 8d,h | 2-methoxyethanol (0.2) | 150 | 15 | complete conversione (n.i.) |
| 9d,h | 2-methoxyethanol (0.2) | 150 | 5 | incomplete conversione (n.i.) |
| 10d,h | 2-methoxyethanol (0.2) | 100 | 10 | incomplete conversione (n.i.) |
| 11d,h | 2-methoxyethanol (0.2) | 125 | 10 | incomplete conversione (n.i.) |
| 12d,h,j | 2-methoxyethanol (0.2) | 175 | 5 | full conversione (94) |
| 13d,h,j | ethylene glycol (0.2) | 175 | 5 | full conversione (96) |
| 14d,h,j | ethanol (0.2) | 175 | 5 | full conversione (87) |
| 15k | 2-methoxyethanol (0.2) | 175 | 5 | full conversione (90) |
| 16h,l,j | 2-methoxyethanol (0.2) | 175 | 5 | full conversione (93) |
ac0(3a) = 0.17 M; 1,4-dioxane (1.0 mL). bIsolated yield (n.i. = not isolated). cc0(4b) = 0.17 M. dDielectric heating in a microwave cavity (T is the set temperature and t2 is the hold time). eAs monitored by GC–MS. fc0(4b) = 0.25 M. gc0(4b) = 0.21 M. hc0(4b) = 0.20 M. ic0(4b) = 0.18 M. jOn a 1.00 mmol scale (3a). kOn a 1.00 mmol scale (3a), c0(3a) = 0.34 M; c0(4b) = 0.40 M. 1,4-Dioxane (1.0 mL). lConductive heating in an oil bath at preheated temperature T.
A ratio of 1.2 equiv of hydrazide 4b to 1.0 equiv of 3a turned to be optimal for achieving full conversion (Table 1, entries 7–16) and at a reaction temperature of 175 °C the reaction time of 5 min was identified to achieve full conversion with very good to excellent yields of isolated 1,5-diacyl-5-hydroxypyrazoline 5b (Table 1, entries 12–16). Although ethylene glycol as a cosolvent (Table 1, entry 13) gave slightly higher yields and ethanol furnished slightly lower yields (Table 1, entry 14), 2-methoxyethanol not only gave high yields of 5b, but also proved to be practical with respect to work-up. Upon comparison between dielectric and conductive heating the reaction in the microwave cavity gave no detectable difference in reaction time and yield. All these optimized conditions were therefore directly employed in the consecutive one-pot sequence. However, some adjustments in the final step were necessary because an increase of pressure was detected under dielectric heating. Therefore, the consecutive process was optimized with respect to the terminal step (Table 2).
Table 2: Optimization of the consecutive three-component synthesis of 1,5-diacyl-5-hydroxypyrazoline 5b.
![[Graphic 2]](/bjoc/content/inline/1860-5397-15-136-i7.svg?max-width=637&scale=1.0)
|
|||
| entry | c0(1a) | t2 [min] | 1,5-diacyl-5-hydroxypyrazoline 5b, yield [%]a |
| 1b | 0.4 M | 5 | 37 |
| 2b | 0.25 M | 5 | 32 |
| 3b | 0.25 M | 10 | 35 |
| 4c | 0.4 M | 10 | no product formationd |
| 5e,f | 0.4 M | 5 | no product formationd |
| 6e | 0.4 M | 10 | 64 |
| 7e | 0.4 M | 20 | 69 |
| 8e | 0.4 M | 30 | 78 |
| 9e | 0.4 M | 45 | 79 |
aIsolated yield. bDielectric heating in a microwave cavity (T is set to 175 °C and t2 is the hold time). cDielectric heating in a microwave cavity (T is set to 150 °C and t2 is the hold time). dAs monitored by GC–MS. eConductive heating in an oil bath at preheated temperature T = 175 °C. f2.00 equiv of NEt3 were added.
In the sequence dielectric heating gave considerably lower yields (Table 2, entries 1–3) than in the separated process (Table 1, entries 12–15). However, conductive heating, which already gave comparable results in the terminal cyclization step (Table 1, entry 16), is obviously better suited to achieve full conversion and, ultimately, slightly longer heating also gives rise to good yields (Table 2, entries 6–9).
Taking into account the combined yield of 71% for both individually performed steps (ynedione formation with 76% and cyclization with 94%) is slightly lower than that of the one-pot sequence with 78% (Table 2, entry 8), the consecutive three-component process clearly is superior. With four bond-forming steps (activation, alkynylation, Michael addition, and cyclization) the average yield per bond-forming step accounts to 94%.
With the optimized conditions of the consecutive three-component synthesis in hand (hetero)arylglyoxylic acids 1, oxalyl chloride, arylacetylenes 2, and hydrazides 4 were reacted in 1,4-dioxane and in the presence of catalytic amounts of copper(I) iodide in a one-pot activation–alkynylation–cyclization sequence to give 1,5-diacyl-5-hydroxypyrazoline 5 after flash chromatography on silica gel in moderate to good yields (Scheme 5, Table 3).
![[1860-5397-15-136-i5]](/bjoc/content/inline/1860-5397-15-136-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: One-pot activation–alkynylation–cyclization synthesis of 1,5-diacyl-5-hydroxypyrazolines 5.
Scheme 5: One-pot activation–alkynylation–cyclization synthesis of 1,5-diacyl-5-hydroxypyrazolines 5.
Table 3: Consecutive three-component synthesis of 1,5-diacyl-5-hydroxypyrazolines 5.
| entry |
glyoxylic acid
R1COCO2H 1 |
alkyne
R2C≡CH 2 |
hydrazide
R3CONHNH2 4 |
1,5-diacyl-5-hydroxypyrazoline 5
yield |
| 1 | R1 = Ph (1a) | R2 = Ph (2a) | R3 = Ph (4b) |
![[Graphic 3]](/bjoc/content/inline/1860-5397-15-136-i8.svg?max-width=637&scale=1.0)
5b (78%) |
| 2a | 1a | 2a | R3 = p-MeC6H4 (4c) |
![[Graphic 4]](/bjoc/content/inline/1860-5397-15-136-i9.svg?max-width=637&scale=1.0)
5c (55%) |
| 3 | 1a | 2a | R3 = p-BrC6H4 (4d) |
![[Graphic 5]](/bjoc/content/inline/1860-5397-15-136-i10.svg?max-width=637&scale=1.0)
5d (41%) |
| 4 | 1a | 2a | R3 = 2-thienyl (4e) |
![[Graphic 6]](/bjoc/content/inline/1860-5397-15-136-i11.svg?max-width=637&scale=1.0)
5e (67%) |
| 5 | 1a | 2a | R3 = 2-furyl (4f) |
![[Graphic 7]](/bjoc/content/inline/1860-5397-15-136-i12.svg?max-width=637&scale=1.0)
5f (67%) |
| 6 | 1a | 2a | R3 = PhCH2 (4g) |
![[Graphic 8]](/bjoc/content/inline/1860-5397-15-136-i13.svg?max-width=637&scale=1.0)
5g (59%) |
| 7 | 1a | 2a | R3 = iPr (4h) |
![[Graphic 9]](/bjoc/content/inline/1860-5397-15-136-i14.svg?max-width=637&scale=1.0)
5h (66%) |
| 8 | 1a | 2a | R3 = cyclopropyl (4i) |
![[Graphic 10]](/bjoc/content/inline/1860-5397-15-136-i15.svg?max-width=637&scale=1.0)
5i (69%) |
| 9 | 1a | 2a | R3 = t-Bu (4j) |
![[Graphic 11]](/bjoc/content/inline/1860-5397-15-136-i16.svg?max-width=637&scale=1.0)
5j (58%) |
| 10 | 1a | 2a | R3 = n-Pr (4k) |
![[Graphic 12]](/bjoc/content/inline/1860-5397-15-136-i17.svg?max-width=637&scale=1.0)
5k (33%) |
| 11 | 1a | R2 = p-MeOC6H4 (2b) | 4b |
![[Graphic 13]](/bjoc/content/inline/1860-5397-15-136-i18.svg?max-width=637&scale=1.0)
5l (55%) |
| 12 | 1a | R2 = p-t-BuC6H4 (2c) | 4b |
![[Graphic 14]](/bjoc/content/inline/1860-5397-15-136-i19.svg?max-width=637&scale=1.0)
5m (69%) |
| 13 | 1a | R2 = p-FC6H4 (2d) | 4b |
![[Graphic 15]](/bjoc/content/inline/1860-5397-15-136-i20.svg?max-width=637&scale=1.0)
5n (66%) |
| 14 | 1a | R2 = p-NCC6H4 (2e) | 4b |
![[Graphic 16]](/bjoc/content/inline/1860-5397-15-136-i21.svg?max-width=637&scale=1.0)
5o (29%) |
| 15 | R1 = 2,4,6-Me3C6H2 (1b) | 2a | 4b |
![[Graphic 17]](/bjoc/content/inline/1860-5397-15-136-i22.svg?max-width=637&scale=1.0)
5p (47%) |
| 16 | R1 = 2-thienyl (1c) | 2a | 4b |
![[Graphic 18]](/bjoc/content/inline/1860-5397-15-136-i23.svg?max-width=637&scale=1.0)
5q (73%) |
| 17 | 1b | 2b | 4e |
![[Graphic 19]](/bjoc/content/inline/1860-5397-15-136-i24.svg?max-width=637&scale=1.0)
5r (38%) |
aReaction time t2 = 20 min.
The structures of the 1,5-diacyl-5-hydroxypyrazolines 5 were unambiguously assigned by 1H and 13C NMR spectroscopy, in selected cases by NOESY, HSQC, and HMBC experiments, as well as by EI mass spectrometry and the elemental composition was confirmed by combustion analyses. Additionally, the structure was corroborated by an X-ray structure analysis of compound 5r showing dimers held together by inter- and intramolecular hydrogen bonding (Figure 5) [53].
![[1860-5397-15-136-5]](/bjoc/content/figures/1860-5397-15-136-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: ORTEP plot and dimer of compound 5r (thermal ellipsoids at 30% probability).
Figure 5: ORTEP plot and dimer of compound 5r (thermal ellipsoids at 30% probability).
The three-component synthesis allows addressing three points of diversity and especially for the hydrazide substrate 4 all different types of (hetero)aromatic, aliphatic, and alicyclic substituents R3 are well tolerated in the sequence (Table 3, entries 1–10). The alkynes 2 can bear electron-donating and electron-withdrawing substituents R2 (Table 3, entries 1, 11–14), however, for the electron-poor cyano substituent a somewhat lower yield of the title compound is obtained (Table 3, entry 14). Finally, the substituents R1 of the glyoxylic acids 1 can be aromatic, heteroaromatic and even sterically demanding (Table 3, entries 1, 15–17).
All attempts to dehydrate 1,5-diacyl-5-hydroxypyrazoline 5b under alkaline or Brønsted acidic conditions were accompanied by simultaneous deacylation of substituent R3 finally furnishing 5-(hetero)aroyl-3-(hetero)aryl-1H-pyrazole 6a (for attempted dehydrative aromatization, see Supporting Information File 1, Table S5), as already reported for alkaline deprotection–aromatization [49]. However, compound 5b is stable against water and weakly basic conditions. This indicates that 1,5-diacyl-5-hydroxypyrazolines might act as acyl transferring agents under certain conditions.
Conclusion
In summary we could elucidate that the consecutive three-component activation–alkynylation–cyclization sequence of (hetero)arylglyoxylic acids, oxalyl chloride, arylacetylenes, and hydrazides does not form aromatic pyrazoles, but rather 1,5-diacyl-5-hydroxypyrazolines, i.e., the aromatizing elimination of water does not occur under these neutral conditions. This novel one-pot synthesis of 1,5-diacyl-5-hydroxypyrazolines is concise, highly efficient and diversity-oriented. The deacylating aromatization of the title compounds under weakly alkaline or acidic conditions indicates acyl-transfer ability. Furthermore, the peculiar reactivity of the ynedione intermediate calls for more sophisticated cyclizing processes, eventually in a one-pot fashion. Further studies exploring the dense electrophilic reactivity of ynediones in consecutive multicomponent reactions are still underway.
Experimental
Typical procedure for the three-component synthesis of compound 5b: In an oven-dried Schlenk flask equipped with a magnetic stirring bar and screw cap were placed glyoxylic acid 1a (150 mg, 1.00 mmol) and dry 1,4-dioxane (2.5 mL) under argon. Then, oxalyl chloride (0.09 mL, 1.00 mmol) was added dropwise at room temperature (external water bath) and the reaction mixture was stirred at 50 °C (preheated oil bath) for 4 h. After the mixture had cooled to room temperature, CuI (10 mg, 0.05 mmol), phenylacetylene (2a, 0.11 mL, 1.00 mmol), and dry triethylamine (0.42 mL, 3.00 mmol) were successively added. Stirring at room temperature (external water bath) was continued for 15 h. Then, phenylhydrazide (3b, 163 mg, 1.20 mmol) and 2-methoxyethanol (1.0 mL) were added and the reaction mixture was stirred at 175 °C (preheated oil bath) for 30 min. After cooling to room temperature deionized water (5 mL) was added and the mixture was extracted with dichloromethane (4 × 5 mL). The combined organic phases were dried with anhydrous sodium sulfate and the solvents were removed in vacuo. The crude product was adsorbed on celite© and purified by flash chromatography on silica gel (petroleum ether 40–60 °C/ethyl acetate 5:1) to give analytically pure 1,5-dibenzoyl-5-hydroxy-3-phenylpyrazoline (5b, 291 mg, 78%) as colorless solid. Rf = 0.15 (petroleum ether/ethyl acetate 5:1, detected with a hand-held UV lamp at 254 and 365 nm). Mp 152 °C; 1H NMR (CDCl3, 300 MHz) δ 3.54 (d, J = 18.5 Hz, 1H), 3.76 (d, J = 18.5 Hz, 1H), 5.60–6.08 (br, 1H), 7.36–7.62 (m, 9H), 7.72–7.83 (m, 2H), 7.90–8.05 (m, 4H); 13C NMR (CDCl3, 75 MHz) δ 45.6 (CH2), 92.2 (Cquat), 126.9 (CH), 127.8 (CH), 128.9 (CH)*, 129.0 (CH), 130.2 (CH), 130.7 (Cquat), 130.9 (CH), 131.7 (CH), 131.8 (Cquat), 132.9 (Cquat), 133.9 (CH), 153.1 (Cquat), 166.7 (Cquat), 193.4 (Cquat); *broadened signal; EIMS (m/z): 352 ([M − H2O])+, 2), 266 (11), 265 ([M − PhCO]+, 59), 248 ([M − PhCO − H2O]+, 20), 105 (PhCO+, 100), 77 (C6H5+, 34); IR (ATR), ![[Graphic 20]](/bjoc/content/inline/1860-5397-15-136-i25.svg?max-width=637&scale=1.18182) [cm−1]: 3333 (w), 1697 (m), 1626 (m), 1612 (m), 1566 (w), 1450 (m), 1427 (m), 1339 (m), 1315 (w), 1254 (w), 1202 (m), 1180 (m), 1113 (m), 1057 (w), 1028 (w), 922 (w), 895 (w), 866 (m), 845 (w), 791 (w), 762 (m), 708 (s), 689 (s), 669 (m), 627 (w); anal. calcd for C23H18N2O3 (370.4): C, 74.58; H, 4.90; N, 7.56; found: C, 74.67; H, 5.07; N, 7.79.
[cm−1]: 3333 (w), 1697 (m), 1626 (m), 1612 (m), 1566 (w), 1450 (m), 1427 (m), 1339 (m), 1315 (w), 1254 (w), 1202 (m), 1180 (m), 1113 (m), 1057 (w), 1028 (w), 922 (w), 895 (w), 866 (m), 845 (w), 791 (w), 762 (m), 708 (s), 689 (s), 669 (m), 627 (w); anal. calcd for C23H18N2O3 (370.4): C, 74.58; H, 4.90; N, 7.56; found: C, 74.67; H, 5.07; N, 7.79.
Supporting Information
For experimental details of the optimization studies on intermediate 3a, on the cyclization step of 3a and 4b (compound 5b), on the consecutive three-component synthesis of compound 5b, experimental details of general procedure of the consecutive three-component synthesis and analytical data of 1,5-diacyl-5-hydroxypyrazolines 5, experimental details on the attempted dehydrative aromatization of compound 5b, and NMR spectra of the compounds 5, and for summaries on the crystal structure analyses of 5a, 5r, and 6a see Supporting Information File 1.
| Supporting Information File 1: Experimental details, copies of NMR spectra and crystallographic data. | ||
| Format: PDF | Size: 4.9 MB | Download |
Acknowledgements
The support of this work by the Fonds der Chemischen Industrie is gratefully acknowledged.
Statement
The reported results have been summarized in the inaugural dissertation "Diversitätsorientierte katalytische Ein-Topf-Synthesen von ausgewählten Azolderivaten" by Dr. Christina Boersch, Heinrich Heine University Düsseldorf, 2014. Dr. Christina Görgen (née Boersch) is the first author of this article.
References
-
Yet, L. Pyrazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Ramsden, C. A.; Scriven, E. F.; Taylor, R. J., Eds.; Elsevier: Oxford, 2008; Vol. 4, pp 3–141. doi:10.1016/b978-008044992-0.00401-6
Return to citation in text: [1] -
Götzinger, A. C.; Müller, T. J. J. 12.1.5 Pyrazoles (Update 2017). In Science of Synthesis; Carreira, E. M.; Christmann, M.; Reissig, H.-U.; Schaumann, E., Eds.; Georg Thieme Verlag: Stuttgart, New York, 2017; pp 1–228. doi:10.1055/sos-sd-112-00112
Return to citation in text: [1] -
Stanovnik, B.; Svete, J. Product Class 1: Pyrazoles. In Category 2, Hetarenes and Related Ring Systems; Neier, R.; Bellus, D., Eds.; Science of Synthesis; Georg Thieme Verlag: Stuttgart, New York, 2002; pp 15–225. doi:10.1055/sos-sd-012-00002
Return to citation in text: [1] [2] -
Elguero, J.; Silva, A. M. S.; Tomé, A. C. Five-Membered Heterocycles: 1,2-Azoles. Part 1. Pyrazoles. In Modern Heterocyclic Chemistry; Alvarez-Builla, J.; Vaquero, J. J.; Barluenga, J., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp 635–725. doi:10.1002/9783527637737.ch8
Return to citation in text: [1] [2] -
Eicher, T.; Hauptmann, S. The Chemistry of Heterocycles, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2003; pp 186–189. doi:10.1002/352760183x
Return to citation in text: [1] [2] -
Motoba, K.; Nishizawa, H.; Suzuki, T.; Hamaguchi, H.; Uchida, M.; Funayama, S. Pestic. Biochem. Physiol. 2000, 67, 73–84. doi:10.1006/pest.2000.2477
Return to citation in text: [1] -
Beckmann, M.; Haack, K.-J. Chem. Unserer Zeit 2003, 37, 88–97. doi:10.1002/ciuz.200300268
Return to citation in text: [1] -
Orth, R. E. J. Pharm. Sci. 1968, 57, 537–556. doi:10.1002/jps.2600570401
Return to citation in text: [1] -
Chauhan, A.; Sharma, P. K.; Kaushik, N. Int. J. ChemTech Res. 2011, 3, 11–17.
Return to citation in text: [1] -
Ismail, M. A. H.; Lehmann, J.; Abou El Ella, D. A.; Albohy, A.; Abouzid, K. A. M. Med. Chem. Res. 2009, 18, 725–744. doi:10.1007/s00044-009-9163-2
Return to citation in text: [1] -
Stauffer, S. R.; Coletta, C. J.; Tedesco, R.; Nishiguchi, G.; Carlson, K.; Sun, J.; Katzenellenbogen, B. S.; Katzenellenbogen, J. A. J. Med. Chem. 2000, 43, 4934–4947. doi:10.1021/jm000170m
Return to citation in text: [1] -
Singer, R. A.; Caron, S.; McDermott, R. E.; Arpin, P.; Do, N. M. Synthesis 2003, 1727–1731. doi:10.1055/s-2003-40881
Return to citation in text: [1] -
Mukherjee, A.; Sarkar, A. Tetrahedron Lett. 2004, 45, 9525–9528. doi:10.1016/j.tetlet.2004.11.016
Return to citation in text: [1] -
Dorlars, A.; Schellhammer, C.-W.; Schroeder, J. Angew. Chem., Int. Ed. Engl. 1975, 14, 665–679. doi:10.1002/anie.197506651
Return to citation in text: [1] -
Catalan, J.; Fabero, F.; Claramunt, R. M.; Santa Maria, M. D.; de la Concepcion Foces-Foces, M.; Hernandez Cano, F.; Martinez-Ripoll, M.; Elguero, J.; Sastre, R. J. Am. Chem. Soc. 1992, 114, 5039–5048. doi:10.1021/ja00039a014
Return to citation in text: [1] -
Yang, Z.; Zhang, K.; Gong, F.; Li, S.; Chen, J.; Ma, J. S.; Sobenina, L. N.; Mikhaleva, A. I.; Trofimov, B. A.; Yang, G. J. Photochem. Photobiol., A 2011, 217, 29–34. doi:10.1016/j.jphotochem.2010.09.012
Return to citation in text: [1] -
Holla, B. S.; Akberali, P. M.; Shivananda, M. K. Farmaco 2000, 55, 256–263. doi:10.1016/s0014-827x(00)00030-6
Return to citation in text: [1] -
Bansal, E.; Srivastava, V. K.; Kumar, A. Eur. J. Med. Chem. 2001, 36, 81–92. doi:10.1016/s0223-5234(00)01179-x
Return to citation in text: [1] -
Ahn, J. H.; Kim, H.-M.; Jung, S. H.; Kang, S. K.; Kim, K. R.; Rhee, S. D.; Yang, S.-D.; Cheon, H. G.; Kim, S. S. Bioorg. Med. Chem. Lett. 2004, 14, 4461–4465. doi:10.1016/j.bmcl.2004.06.046
Return to citation in text: [1] -
Rajendra Prasad, Y.; Lakshmana Rao, A.; Prasoona, L.; Murali, K.; Ravi Kumar, P. Bioorg. Med. Chem. Lett. 2005, 15, 5030–5034. doi:10.1016/j.bmcl.2005.08.040
Return to citation in text: [1] -
Acharya, B. N.; Saraswat, D.; Tiwari, M.; Shrivastava, A. K.; Ghorpade, R.; Bapna, S.; Kaushik, M. P. Eur. J. Med. Chem. 2010, 45, 430–438. doi:10.1016/j.ejmech.2009.10.023
Return to citation in text: [1] -
Ciupa, A.; De Bank, P. A.; Mahon, M. F.; Wood, P. J.; Caggiano, L. Med. Chem. Commun. 2013, 4, 956–961. doi:10.1039/c3md00077j
Return to citation in text: [1] -
Nepali, K.; Singh, G.; Turan, A.; Agarwal, A.; Sapra, S.; Kumar, R.; Banerjee, U. C.; Verma, P. K.; Satti, N. K.; Gupta, M. K.; Suri, O. P.; Dhar, K. L. Bioorg. Med. Chem. 2011, 19, 1950–1958. doi:10.1016/j.bmc.2011.01.058
Return to citation in text: [1] -
Machado, P.; Rosa, F. A.; Rossatto, M.; da S. Sant'Anna, G.; Sauzem, P. D.; Siqueira da Silva, R. M.; Rubin, M. A.; Ferreira, J.; Bonacorso, H. G.; Zanatta, N.; Martins, M. A. P. ARKIVOC 2007, No. xvi, 281–297. doi:10.3998/ark.5550190.0008.g28
Return to citation in text: [1] [2] -
Sauzem, P. D.; Machado, P.; Rubin, M. A.; da S. Sant'Anna, G.; Faber, H. B.; de Souza, A. H.; Mello, C. F.; Beck, P.; Burrow, R. A.; Bonacorso, H. G.; Zanatta, N.; Martins, M. A. P. Eur. J. Med. Chem. 2008, 43, 1237–1247. doi:10.1016/j.ejmech.2007.07.018
Return to citation in text: [1] [2] -
Holla, B. S.; Udupa, K. V.; Sridhar, K. R. Bull. Chem. Soc. Jpn. 1989, 62, 3409–3411. doi:10.1246/bcsj.62.3409
Return to citation in text: [1] [2] -
Waldo, J. P.; Mehta, S.; Larock, R. C. J. Org. Chem. 2008, 73, 6666–6670. doi:10.1021/jo800789p
Return to citation in text: [1] -
Joshi, K. C.; Bohra, R.; Joshi, B. S. Inorg. Chem. 1992, 31, 598–603. doi:10.1021/ic00030a014
Return to citation in text: [1] -
Kumar Dey, D.; Lycka, A.; Mitra, S.; Rosair, G. M. J. Organomet. Chem. 2004, 689, 88–95. doi:10.1016/j.jorganchem.2003.09.035
Return to citation in text: [1] -
de Sousa, G. F.; Garcia, E.; Gatto, C. C.; Resck, I. S.; Deflon, V. M.; Ardisson, J. D. J. Mol. Struct. 2010, 981, 46–53. doi:10.1016/j.molstruc.2010.07.023
Return to citation in text: [1] -
Someya, C. I.; Inoue, S.; Irran, E.; Krackl, S.; Enthaler, S. Eur. J. Inorg. Chem. 2011, 2691–2697. doi:10.1002/ejic.201100248
Return to citation in text: [1] -
Sevenard, D. V.; Khomutov, O. G.; Kodess, M. I.; Pashkevich, K. I.; Loop, I.; Lork, E.; Röschenthaler, G.-V. Can. J. Chem. 2001, 79, 183–194. doi:10.1139/v01-003
Return to citation in text: [1] -
Zelenin, K. N.; Tugusheva, A. R.; Yakimovitch, S. I.; Alekseev, V. V.; Zerova, E. V. Chem. Heterocycl. Compd. 2002, 38, 668–676. doi:10.1023/a:1019909117505
Return to citation in text: [1] -
Zelenin, K. N.; Alekseyev, V. V.; Tygysheva, A. R.; Yakimovitch, S. I. Tetrahedron 1995, 51, 11251–11256. doi:10.1016/0040-4020(95)00672-u
Return to citation in text: [1] -
Rateb, L.; Azmy, B.; Nashed, M. A.; Iskander, M. F. Z. Naturforsch., B: Anorg. Chem., Org. Chem. 1978, 33, 1527–1534. doi:10.1515/znb-1978-1233
Return to citation in text: [1] -
Bonacorso, H. G.; Wiethan, C. W.; Porte, L. M. F.; Moraes, M. C.; Navarini, J.; Belo, C. R.; Luz, F. M.; Zanatta, N.; Martins, M. A. P. ARKIVOC 2013, No. iv, 291–305. doi:10.3998/ark.5550190.p008.164
Return to citation in text: [1] -
Martins, M. A. P.; Moreira, D. N.; Frizzo, C. P.; Longhi, K.; Zanatta, N.; Bonacorso, H. G. J. Braz. Chem. Soc. 2008, 19, 1361–1368. doi:10.1590/s0103-50532008000700019
Return to citation in text: [1] -
Moreira, D. N.; Frizzo, C. P.; Longhi, K.; Zanatta, N.; Bonacorso, H. G.; Martins, M. A. P. Monatsh. Chem. 2008, 139, 1049–1054. doi:10.1007/s00706-008-0874-8
Return to citation in text: [1] -
Martins, M. A. P.; Beck, P.; Machado, P.; Brondani, S.; Moura, S.; Zanatta, N.; Bonacorso, H. G.; Flores, A. F. C. J. Braz. Chem. Soc. 2006, 17, 408–411. doi:10.1590/s0103-50532006000200027
Return to citation in text: [1] -
Buriol, L.; Frizzo, C. P.; Marzari, M. R. B.; Moreira, D. N.; Prola, L. D. T.; Zanatta, N.; Bonacorso, H. G.; Martins, M. A. P. J. Braz. Chem. Soc. 2010, 21, 1037–1044. doi:10.1590/s0103-50532010000600012
Return to citation in text: [1] -
Lessing, T.; Müller, T. J. J. Appl. Sci. 2015, 5, 1803–1836. doi:10.3390/app5041803
Return to citation in text: [1] -
D'Souza, D. M.; Müller, T. J. J. Chem. Soc. Rev. 2007, 36, 1095–1108. doi:10.1039/b608235c
Return to citation in text: [1] -
Müller, T. J. J. Drug Discovery Today: Technol. 2018, 29, 19–26. doi:10.1016/j.ddtec.2018.06.003
Return to citation in text: [1] -
Gers-Panther, C. F.; Müller, T. J. J. Multicomponent Synthesis of Heterocycles Initiated by Catalytic Generation of Ynones and Ynediones. In Advances in Heterocyclic Chemistry; Scriven, E. F. V.; Ramsden, C. A., Eds.; Academic Press, 2016; Vol. 120, pp 67–98. doi:10.1016/bs.aihch.2016.04.007
Return to citation in text: [1] -
Müller, T. J. J. Top. Heterocycl. Chem. 2010, 25, 25–94. doi:10.1007/7081_2010_43
Return to citation in text: [1] -
Willy, B.; Müller, T. J. J. Curr. Org. Chem. 2009, 13, 1777–1790. doi:10.2174/138527209789630479
Return to citation in text: [1] -
Willy, B.; Müller, T. J. J. ARKIVOC 2008, No. i, 195–208. doi:10.3998/ark.5550190.0009.107
Return to citation in text: [1] -
Merkul, E.; Dohe, J.; Gers, C.; Rominger, F.; Müller, T. J. J. Angew. Chem., Int. Ed. 2011, 50, 2966–2969. doi:10.1002/anie.201007194
Return to citation in text: [1] [2] [3] [4] -
Boersch, C.; Merkul, E.; Müller, T. J. J. Angew. Chem., Int. Ed. 2011, 50, 10448–10452. doi:10.1002/anie.201103296
Return to citation in text: [1] [2] [3] [4] [5] -
Merkt, F. K.; Höwedes, S. P.; Gers-Panther, C. F.; Gruber, I.; Janiak, C.; Müller, T. J. J. Chem. – Eur. J. 2018, 24, 8114–8125. doi:10.1002/chem.201800079
Return to citation in text: [1] -
Gers-Panther, C. F.; Fischer, H.; Nordmann, J.; Seiler, T.; Behnke, T.; Würth, C.; Frank, W.; Resch-Genger, U.; Müller, T. J. J. J. Org. Chem. 2017, 82, 567–578. doi:10.1021/acs.joc.6b02581
Return to citation in text: [1] -
Gers, C. F.; Nordmann, J.; Kumru, C.; Frank, W.; Müller, T. J. J. J. Org. Chem. 2014, 79, 3296–3310. doi:10.1021/jo4025978
Return to citation in text: [1] -
CCDC 1902138 (5a), CCDC 1906570 (5r), and CCDC 1906571 (6a) contain the supplementary crystallographic data (excluding structure factors) for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
Return to citation in text: [1] [2] [3]
| 48. | Merkul, E.; Dohe, J.; Gers, C.; Rominger, F.; Müller, T. J. J. Angew. Chem., Int. Ed. 2011, 50, 2966–2969. doi:10.1002/anie.201007194 |
| 49. | Boersch, C.; Merkul, E.; Müller, T. J. J. Angew. Chem., Int. Ed. 2011, 50, 10448–10452. doi:10.1002/anie.201103296 |
| 49. | Boersch, C.; Merkul, E.; Müller, T. J. J. Angew. Chem., Int. Ed. 2011, 50, 10448–10452. doi:10.1002/anie.201103296 |
| 53. | CCDC 1902138 (5a), CCDC 1906570 (5r), and CCDC 1906571 (6a) contain the supplementary crystallographic data (excluding structure factors) for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre. |
| 1. | Yet, L. Pyrazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Ramsden, C. A.; Scriven, E. F.; Taylor, R. J., Eds.; Elsevier: Oxford, 2008; Vol. 4, pp 3–141. doi:10.1016/b978-008044992-0.00401-6 |
| 2. | Götzinger, A. C.; Müller, T. J. J. 12.1.5 Pyrazoles (Update 2017). In Science of Synthesis; Carreira, E. M.; Christmann, M.; Reissig, H.-U.; Schaumann, E., Eds.; Georg Thieme Verlag: Stuttgart, New York, 2017; pp 1–228. doi:10.1055/sos-sd-112-00112 |
| 12. | Singer, R. A.; Caron, S.; McDermott, R. E.; Arpin, P.; Do, N. M. Synthesis 2003, 1727–1731. doi:10.1055/s-2003-40881 |
| 13. | Mukherjee, A.; Sarkar, A. Tetrahedron Lett. 2004, 45, 9525–9528. doi:10.1016/j.tetlet.2004.11.016 |
| 26. | Holla, B. S.; Udupa, K. V.; Sridhar, K. R. Bull. Chem. Soc. Jpn. 1989, 62, 3409–3411. doi:10.1246/bcsj.62.3409 |
| 27. | Waldo, J. P.; Mehta, S.; Larock, R. C. J. Org. Chem. 2008, 73, 6666–6670. doi:10.1021/jo800789p |
| 8. | Orth, R. E. J. Pharm. Sci. 1968, 57, 537–556. doi:10.1002/jps.2600570401 |
| 9. | Chauhan, A.; Sharma, P. K.; Kaushik, N. Int. J. ChemTech Res. 2011, 3, 11–17. |
| 10. | Ismail, M. A. H.; Lehmann, J.; Abou El Ella, D. A.; Albohy, A.; Abouzid, K. A. M. Med. Chem. Res. 2009, 18, 725–744. doi:10.1007/s00044-009-9163-2 |
| 11. | Stauffer, S. R.; Coletta, C. J.; Tedesco, R.; Nishiguchi, G.; Carlson, K.; Sun, J.; Katzenellenbogen, B. S.; Katzenellenbogen, J. A. J. Med. Chem. 2000, 43, 4934–4947. doi:10.1021/jm000170m |
| 28. | Joshi, K. C.; Bohra, R.; Joshi, B. S. Inorg. Chem. 1992, 31, 598–603. doi:10.1021/ic00030a014 |
| 6. | Motoba, K.; Nishizawa, H.; Suzuki, T.; Hamaguchi, H.; Uchida, M.; Funayama, S. Pestic. Biochem. Physiol. 2000, 67, 73–84. doi:10.1006/pest.2000.2477 |
| 7. | Beckmann, M.; Haack, K.-J. Chem. Unserer Zeit 2003, 37, 88–97. doi:10.1002/ciuz.200300268 |
| 23. | Nepali, K.; Singh, G.; Turan, A.; Agarwal, A.; Sapra, S.; Kumar, R.; Banerjee, U. C.; Verma, P. K.; Satti, N. K.; Gupta, M. K.; Suri, O. P.; Dhar, K. L. Bioorg. Med. Chem. 2011, 19, 1950–1958. doi:10.1016/j.bmc.2011.01.058 |
| 3. | Stanovnik, B.; Svete, J. Product Class 1: Pyrazoles. In Category 2, Hetarenes and Related Ring Systems; Neier, R.; Bellus, D., Eds.; Science of Synthesis; Georg Thieme Verlag: Stuttgart, New York, 2002; pp 15–225. doi:10.1055/sos-sd-012-00002 |
| 4. | Elguero, J.; Silva, A. M. S.; Tomé, A. C. Five-Membered Heterocycles: 1,2-Azoles. Part 1. Pyrazoles. In Modern Heterocyclic Chemistry; Alvarez-Builla, J.; Vaquero, J. J.; Barluenga, J., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp 635–725. doi:10.1002/9783527637737.ch8 |
| 5. | Eicher, T.; Hauptmann, S. The Chemistry of Heterocycles, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2003; pp 186–189. doi:10.1002/352760183x |
| 24. | Machado, P.; Rosa, F. A.; Rossatto, M.; da S. Sant'Anna, G.; Sauzem, P. D.; Siqueira da Silva, R. M.; Rubin, M. A.; Ferreira, J.; Bonacorso, H. G.; Zanatta, N.; Martins, M. A. P. ARKIVOC 2007, No. xvi, 281–297. doi:10.3998/ark.5550190.0008.g28 |
| 25. | Sauzem, P. D.; Machado, P.; Rubin, M. A.; da S. Sant'Anna, G.; Faber, H. B.; de Souza, A. H.; Mello, C. F.; Beck, P.; Burrow, R. A.; Bonacorso, H. G.; Zanatta, N.; Martins, M. A. P. Eur. J. Med. Chem. 2008, 43, 1237–1247. doi:10.1016/j.ejmech.2007.07.018 |
| 19. | Ahn, J. H.; Kim, H.-M.; Jung, S. H.; Kang, S. K.; Kim, K. R.; Rhee, S. D.; Yang, S.-D.; Cheon, H. G.; Kim, S. S. Bioorg. Med. Chem. Lett. 2004, 14, 4461–4465. doi:10.1016/j.bmcl.2004.06.046 |
| 21. | Acharya, B. N.; Saraswat, D.; Tiwari, M.; Shrivastava, A. K.; Ghorpade, R.; Bapna, S.; Kaushik, M. P. Eur. J. Med. Chem. 2010, 45, 430–438. doi:10.1016/j.ejmech.2009.10.023 |
| 49. | Boersch, C.; Merkul, E.; Müller, T. J. J. Angew. Chem., Int. Ed. 2011, 50, 10448–10452. doi:10.1002/anie.201103296 |
| 18. | Bansal, E.; Srivastava, V. K.; Kumar, A. Eur. J. Med. Chem. 2001, 36, 81–92. doi:10.1016/s0223-5234(00)01179-x |
| 22. | Ciupa, A.; De Bank, P. A.; Mahon, M. F.; Wood, P. J.; Caggiano, L. Med. Chem. Commun. 2013, 4, 956–961. doi:10.1039/c3md00077j |
| 17. | Holla, B. S.; Akberali, P. M.; Shivananda, M. K. Farmaco 2000, 55, 256–263. doi:10.1016/s0014-827x(00)00030-6 |
| 53. | CCDC 1902138 (5a), CCDC 1906570 (5r), and CCDC 1906571 (6a) contain the supplementary crystallographic data (excluding structure factors) for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre. |
| 14. | Dorlars, A.; Schellhammer, C.-W.; Schroeder, J. Angew. Chem., Int. Ed. Engl. 1975, 14, 665–679. doi:10.1002/anie.197506651 |
| 15. | Catalan, J.; Fabero, F.; Claramunt, R. M.; Santa Maria, M. D.; de la Concepcion Foces-Foces, M.; Hernandez Cano, F.; Martinez-Ripoll, M.; Elguero, J.; Sastre, R. J. Am. Chem. Soc. 1992, 114, 5039–5048. doi:10.1021/ja00039a014 |
| 16. | Yang, Z.; Zhang, K.; Gong, F.; Li, S.; Chen, J.; Ma, J. S.; Sobenina, L. N.; Mikhaleva, A. I.; Trofimov, B. A.; Yang, G. J. Photochem. Photobiol., A 2011, 217, 29–34. doi:10.1016/j.jphotochem.2010.09.012 |
| 20. | Rajendra Prasad, Y.; Lakshmana Rao, A.; Prasoona, L.; Murali, K.; Ravi Kumar, P. Bioorg. Med. Chem. Lett. 2005, 15, 5030–5034. doi:10.1016/j.bmcl.2005.08.040 |
| 53. | CCDC 1902138 (5a), CCDC 1906570 (5r), and CCDC 1906571 (6a) contain the supplementary crystallographic data (excluding structure factors) for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre. |
| 3. | Stanovnik, B.; Svete, J. Product Class 1: Pyrazoles. In Category 2, Hetarenes and Related Ring Systems; Neier, R.; Bellus, D., Eds.; Science of Synthesis; Georg Thieme Verlag: Stuttgart, New York, 2002; pp 15–225. doi:10.1055/sos-sd-012-00002 |
| 4. | Elguero, J.; Silva, A. M. S.; Tomé, A. C. Five-Membered Heterocycles: 1,2-Azoles. Part 1. Pyrazoles. In Modern Heterocyclic Chemistry; Alvarez-Builla, J.; Vaquero, J. J.; Barluenga, J., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp 635–725. doi:10.1002/9783527637737.ch8 |
| 5. | Eicher, T.; Hauptmann, S. The Chemistry of Heterocycles, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2003; pp 186–189. doi:10.1002/352760183x |
| 29. | Kumar Dey, D.; Lycka, A.; Mitra, S.; Rosair, G. M. J. Organomet. Chem. 2004, 689, 88–95. doi:10.1016/j.jorganchem.2003.09.035 |
| 30. | de Sousa, G. F.; Garcia, E.; Gatto, C. C.; Resck, I. S.; Deflon, V. M.; Ardisson, J. D. J. Mol. Struct. 2010, 981, 46–53. doi:10.1016/j.molstruc.2010.07.023 |
| 31. | Someya, C. I.; Inoue, S.; Irran, E.; Krackl, S.; Enthaler, S. Eur. J. Inorg. Chem. 2011, 2691–2697. doi:10.1002/ejic.201100248 |
| 48. | Merkul, E.; Dohe, J.; Gers, C.; Rominger, F.; Müller, T. J. J. Angew. Chem., Int. Ed. 2011, 50, 2966–2969. doi:10.1002/anie.201007194 |
| 50. | Merkt, F. K.; Höwedes, S. P.; Gers-Panther, C. F.; Gruber, I.; Janiak, C.; Müller, T. J. J. Chem. – Eur. J. 2018, 24, 8114–8125. doi:10.1002/chem.201800079 |
| 51. | Gers-Panther, C. F.; Fischer, H.; Nordmann, J.; Seiler, T.; Behnke, T.; Würth, C.; Frank, W.; Resch-Genger, U.; Müller, T. J. J. J. Org. Chem. 2017, 82, 567–578. doi:10.1021/acs.joc.6b02581 |
| 52. | Gers, C. F.; Nordmann, J.; Kumru, C.; Frank, W.; Müller, T. J. J. J. Org. Chem. 2014, 79, 3296–3310. doi:10.1021/jo4025978 |
| 48. | Merkul, E.; Dohe, J.; Gers, C.; Rominger, F.; Müller, T. J. J. Angew. Chem., Int. Ed. 2011, 50, 2966–2969. doi:10.1002/anie.201007194 |
| 49. | Boersch, C.; Merkul, E.; Müller, T. J. J. Angew. Chem., Int. Ed. 2011, 50, 10448–10452. doi:10.1002/anie.201103296 |
| 48. | Merkul, E.; Dohe, J.; Gers, C.; Rominger, F.; Müller, T. J. J. Angew. Chem., Int. Ed. 2011, 50, 2966–2969. doi:10.1002/anie.201007194 |
| 49. | Boersch, C.; Merkul, E.; Müller, T. J. J. Angew. Chem., Int. Ed. 2011, 50, 10448–10452. doi:10.1002/anie.201103296 |
| 41. | Lessing, T.; Müller, T. J. J. Appl. Sci. 2015, 5, 1803–1836. doi:10.3390/app5041803 |
| 42. | D'Souza, D. M.; Müller, T. J. J. Chem. Soc. Rev. 2007, 36, 1095–1108. doi:10.1039/b608235c |
| 43. | Müller, T. J. J. Drug Discovery Today: Technol. 2018, 29, 19–26. doi:10.1016/j.ddtec.2018.06.003 |
| 44. | Gers-Panther, C. F.; Müller, T. J. J. Multicomponent Synthesis of Heterocycles Initiated by Catalytic Generation of Ynones and Ynediones. In Advances in Heterocyclic Chemistry; Scriven, E. F. V.; Ramsden, C. A., Eds.; Academic Press, 2016; Vol. 120, pp 67–98. doi:10.1016/bs.aihch.2016.04.007 |
| 45. | Müller, T. J. J. Top. Heterocycl. Chem. 2010, 25, 25–94. doi:10.1007/7081_2010_43 |
| 46. | Willy, B.; Müller, T. J. J. Curr. Org. Chem. 2009, 13, 1777–1790. doi:10.2174/138527209789630479 |
| 47. | Willy, B.; Müller, T. J. J. ARKIVOC 2008, No. i, 195–208. doi:10.3998/ark.5550190.0009.107 |
| 24. | Machado, P.; Rosa, F. A.; Rossatto, M.; da S. Sant'Anna, G.; Sauzem, P. D.; Siqueira da Silva, R. M.; Rubin, M. A.; Ferreira, J.; Bonacorso, H. G.; Zanatta, N.; Martins, M. A. P. ARKIVOC 2007, No. xvi, 281–297. doi:10.3998/ark.5550190.0008.g28 |
| 25. | Sauzem, P. D.; Machado, P.; Rubin, M. A.; da S. Sant'Anna, G.; Faber, H. B.; de Souza, A. H.; Mello, C. F.; Beck, P.; Burrow, R. A.; Bonacorso, H. G.; Zanatta, N.; Martins, M. A. P. Eur. J. Med. Chem. 2008, 43, 1237–1247. doi:10.1016/j.ejmech.2007.07.018 |
| 26. | Holla, B. S.; Udupa, K. V.; Sridhar, K. R. Bull. Chem. Soc. Jpn. 1989, 62, 3409–3411. doi:10.1246/bcsj.62.3409 |
| 32. | Sevenard, D. V.; Khomutov, O. G.; Kodess, M. I.; Pashkevich, K. I.; Loop, I.; Lork, E.; Röschenthaler, G.-V. Can. J. Chem. 2001, 79, 183–194. doi:10.1139/v01-003 |
| 33. | Zelenin, K. N.; Tugusheva, A. R.; Yakimovitch, S. I.; Alekseev, V. V.; Zerova, E. V. Chem. Heterocycl. Compd. 2002, 38, 668–676. doi:10.1023/a:1019909117505 |
| 34. | Zelenin, K. N.; Alekseyev, V. V.; Tygysheva, A. R.; Yakimovitch, S. I. Tetrahedron 1995, 51, 11251–11256. doi:10.1016/0040-4020(95)00672-u |
| 35. | Rateb, L.; Azmy, B.; Nashed, M. A.; Iskander, M. F. Z. Naturforsch., B: Anorg. Chem., Org. Chem. 1978, 33, 1527–1534. doi:10.1515/znb-1978-1233 |
| 36. | Bonacorso, H. G.; Wiethan, C. W.; Porte, L. M. F.; Moraes, M. C.; Navarini, J.; Belo, C. R.; Luz, F. M.; Zanatta, N.; Martins, M. A. P. ARKIVOC 2013, No. iv, 291–305. doi:10.3998/ark.5550190.p008.164 |
| 37. | Martins, M. A. P.; Moreira, D. N.; Frizzo, C. P.; Longhi, K.; Zanatta, N.; Bonacorso, H. G. J. Braz. Chem. Soc. 2008, 19, 1361–1368. doi:10.1590/s0103-50532008000700019 |
| 38. | Moreira, D. N.; Frizzo, C. P.; Longhi, K.; Zanatta, N.; Bonacorso, H. G.; Martins, M. A. P. Monatsh. Chem. 2008, 139, 1049–1054. doi:10.1007/s00706-008-0874-8 |
| 39. | Martins, M. A. P.; Beck, P.; Machado, P.; Brondani, S.; Moura, S.; Zanatta, N.; Bonacorso, H. G.; Flores, A. F. C. J. Braz. Chem. Soc. 2006, 17, 408–411. doi:10.1590/s0103-50532006000200027 |
| 40. | Buriol, L.; Frizzo, C. P.; Marzari, M. R. B.; Moreira, D. N.; Prola, L. D. T.; Zanatta, N.; Bonacorso, H. G.; Martins, M. A. P. J. Braz. Chem. Soc. 2010, 21, 1037–1044. doi:10.1590/s0103-50532010000600012 |
© 2019 Görgen et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)