Abstract
A systematic comparison of lipophilicity modulations upon fluorination of isopropyl, cyclopropyl and 3-oxetanyl substituents, at a single carbon atom, is provided using directly comparable, and easily accessible model compounds. In addition, comparison with relevant linear chain derivatives is provided, as well as lipophilicity changes occurring upon chain extension of acyclic precursors to give cyclopropyl containing compounds. For the compounds investigated, fluorination of the isopropyl substituent led to larger lipophilicity modulation compared to fluorination of the cyclopropyl substituent.

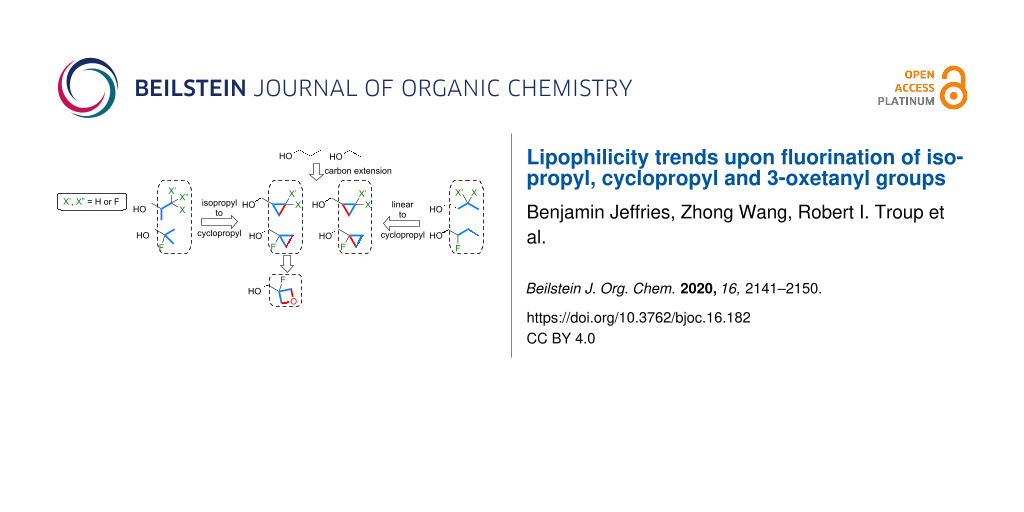
Graphical Abstract
Introduction
The introduction of small alkyl groups onto bioactive compounds as space filling groups is a common strategy in the drug optimization process. It is typically employed to ensure proper fitting of the part(s) of a ligand that interact with a receptor [1,2]. Apart from volume considerations, shape complementarity is regarded as important as well [3-5]. Hence, the modification of existing appendages is also commonly employed. For example, an isopropyl and a trifluoromethyl group have very similar volumes, but a very different shape [6].
However, even the introduction of relatively small methyl groups can impart profound consequences (“the magic methyl effect”) [7-9] on the activity and conformational profile, which can be very beneficial if this promotes the population of the desired bioactive conformation(s). The introduction or extension of alkyl groups generally leads to an increase in lipophilicity, which is more often than not undesired. Hence, this has led to an interest in how to add volume without adding lipophilicity, i.e., how to extend a carbon chain without a concomitant increase in logP.
A much-used isosteric replacement is the change of an isopropyl group, or more commonly an α,α-disubstituted geminal dimethyl group [10], into a cyclopropyl group [11,12]. Apart from a slight shape/volume alteration caused by the significant change in bond angles and the deletion of two hydrogen atoms, this bioisosteric modification also usually results in improved metabolic stability, increased rigidity and, due to its electron-withdrawing nature, pKa modulation of adjacent acid/base groups. Importantly, it also leads to a lipophilicity reduction, as illustrated by the Hansch π-values of isopropyl (1.53) [13] and cyclopropyl (1.14) [14]. This compares to π = 1.98 for a tert-butyl group [13]. However, exceptions are known, as illustrated for 1 and 2 (Figure 1) [15]. In this case, the expected lipophilicity reducing effect is offset by the electron-withdrawing effect on the nitrogen (ΔlogP +0.34). The reduction in amine basicity also shifts the distribution of free base and protonated species, resulting in a higher proportion of free base in solution at pH 7.4, which leads to a large logD7.4 increase (ΔlogD7.4 +0.73).
![[1860-5397-16-182-1]](/bjoc/content/figures/1860-5397-16-182-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of lipophilicity modulation for geminal dimethyl to cyclopropyl and oxetane modifications (measured experimentally via shake-flask method).
Figure 1: Examples of lipophilicity modulation for geminal dimethyl to cyclopropyl and oxetane modifications ...
The modification of a geminal dimethyl moiety to an oxetanyl group has also proven to be a useful biomimetic replacement leading to a significant logP decrease (compare 3 with 4) [16-18]. However, when an amino group is located in the α-position (5), the introduction of the oxetanyl group leading to 6 induces a significant pKa(H) decrease. While the reduction in logP is still observed, the larger proportion of unprotonated substrate leads to a logD7.4 increase.
Aliphatic fluorination can also be employed to decrease lipophilicities [6,19-22]. However, there is comparatively less precedence of lipophilicity comparisons between fluorinated isopropyl groups, cyclopropanes and oxetanes. Examples of bioactive compounds are given in Figure 2.
![[1860-5397-16-182-2]](/bjoc/content/figures/1860-5397-16-182-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Lipophilicity modulation examples involving fluorinated cyclopropane derivatives (measured experimentally via shake-flask method).
Figure 2: Lipophilicity modulation examples involving fluorinated cyclopropane derivatives (measured experime...
During the optimization of the LpxC inhibitor 7a [23], it was shown that fluorination to give analogue 7b reduced the lipophilicity. Interestingly, while the nonfluorinated 7a and 7c diastereomers have different logD7.4 values, this is not the case for their fluorinated analogues 7b and 7d. Oxazine derivative 8 featured in the development of centrally active β-secretase (BACE1) inhibitors [24]. Although the logD7.4 of the unsubstituted cyclopropyl containing analogue was not provided, it can be seen that two-carbon extension of 8a, and one-carbon extension of 8b,c to give the cyclopropyl derivative 8e, cause a lipophilicity increase. Conversely, changing a CF2Me for a cPrF group (8d to 8e) gives a lipophilicity reduction.
Finally, a number of model compound series have been reported (Figure 3). In the isopropanol series A [22], monofluorination led to a 0.4 logP decrease, which is further extended upon a second monofluorination at the other methyl group (compare A1 with A2, A3). With one (A4) and two (A5) trifluoromethyl groups, large increases in lipophilicity are observed. An interesting series (B) of phenylcyclopropanes was reported by O’Hagan [25,26]. All-cis vicinal trifluorination as in B2 led to a significant reduction in lipophilicity compared to the nonfluorinated B1, which was explained by the facially polar motif caused by the C–F dipoles present on the same side of the ring. Consequently, the diastereomer B4 possessed a higher lipophilicity, although the lipophilicity of the vicinally difluorinated diastereomers B3 and B5 was identical, and also the same as the trifluorinated B4. Geminal difluorination caused an increase in lipophilicity compared to vicinal fluorination (compare B6 with B3 and B5, and B7 with B4 and B2). Interestingly, in analogy with the same lipophilicity of B3–B4, B6 and B7 also were equivalent in lipophilicity. Finally, the 3-fluorinated oxetanyl derivative C2 has a lower logD value than the nonfluorinated parent C1 [16,17].
![[1860-5397-16-182-3]](/bjoc/content/figures/1860-5397-16-182-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Lipophilicity changes upon fluorination of isopropyl, cyclopropane and oxetane rings (Series A, C: measured experimentally via shake-flask method; series B: measured experimentally by reversed-phase HPLC).
Figure 3: Lipophilicity changes upon fluorination of isopropyl, cyclopropane and oxetane rings (Series A, C: ...
In this contribution, we describe a systematic study on the lipophilicity modulations of the isopropyl, cyclopropyl and 3-oxetanyl groups and their various possible analogues, featuring fluorination at a single carbon atom (Figure 4), using the directly comparable isobutanol, cyclopropylmethanol and 3-oxetanylmethanol scaffolds. All compounds are either commercially available or are easily synthesized from available fluorinated building blocks. In addition, comparison of lipophilicities of relevant linear butanol derivatives is discussed, as well as that of the introduction of an isopropyl/cyclopropyl/oxetanyl group from 1-propanol and ethanol, which represents carbon extensions (volume increase).
![[1860-5397-16-182-4]](/bjoc/content/figures/1860-5397-16-182-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Lipophilicity modulations discussed in this contribution.
Figure 4: Lipophilicity modulations discussed in this contribution.
Results and Discussion
Lipophilicity data and discussion
The lipophilicities of the nonfluorinated E1 and F1 have not been reported, and have been estimated by a range of calculation methods as described in Supporting Information File 1 (section 2). The average logP value of the data from those estimation approaches for E1 was determined to be 0.24 ± 0.03, and for F1 −0.80 ± 0.23. The experimental lipophilicity values of the fluorinated isobutanol, cyclopropylmethanol and 3-oxetanylmethanol derivatives are given in Figure 5. They were measured by a 19F NMR-based method developed by our group [22,27], which is suitable for measuring the octanol/water partition coefficients P of (fluorinated) non-UV active substrates.
![[1860-5397-16-182-5]](/bjoc/content/figures/1860-5397-16-182-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Distribution of the experimental lipophilicity values of series D, E and F (* denotes an estimated value, see Supporting Information File 1).
Figure 5: Distribution of the experimental lipophilicity values of series D, E and F (* denotes an estimated ...
As expected, monofluorination decreases the lipophilicity compared to the nonfluorinated parent D1, with a much larger decrease for the β-fluorohydrin D2 than for the γ-fluorohydrin D3. Interestingly, for the linear propanol, β-fluorination and γ-fluorination lead to a similar logP decrease (−0.29 vs −0.26 for 2- and 3-fluoropropanol) [22], while for the higher alkanols, β-fluorination leads to more lipophilic compounds than γ-fluorination [28]. The higher logP of β-fluorinated alcohols has been explained both by the occurrence of conformations with opposing dipole moments of the C–F and C–O bonds, as well as the reduction of polarizability of the oxygen lone pairs due to the fluorine electronegativity. However, while D2 is a β-fluorohydrin, its fluorine is substituted at a tertiary position, with the C–F bond able to polarize the six C–H bonds of the methyl groups, which has a lipophilicity lowering effect. The electron-withdrawing effect of the fluorine is evident from the 1H NMR chemical shift values of the CH3 groups in D1 (0.91 ppm) [29], D3 (0.98 ppm), and D2 (1.37 ppm) [30].
Compared to the monofluorinated D3, difluorination (D4) and trifluorination (D5) at the same carbon atom increases lipophilicity. There is a notable difference in lipohilicity between D4 and D5. Aliphatic compounds containing a CF2H group are frequently less lipophilic than to their nonfluorinated equivalent [20,31] (except in presence of a vicinal C–O bond [22,32]), which is what is found here for D4. The CF3-containing D5 is more lipophilic than the parent D1, and these values are consistent with the trends seen for 1-propanol [22].
For cyclopropylmethanol E1, β-fluorination (E2) leads to a smaller lipophilicity decrease compared to that seen for D1→D2, leading to a logP value in between those of the two γ-fluorinated diastereomers E3 and E4. The decrease seen for E2 is in accord with the observations for the β-fluorinated analogues of 7a and 7c (see above, Figure 2). The cis-isomer E3 has a slightly lower lipophilicity than the trans-isomer E4. The lower lipophilicity decrease upon monofluorination in series E compared to series F could be related to the hybridization state of cyclopropyl carbon atoms. However, β-fluorination of the oxetanyl derivative F1 led to a lipophilicity increase. With the caveat that the value for F1 is not a measured value and the difference is well within the standard deviation (0.23 logP units), this increase is in contrast to the result observed for C1 (see above, Figure 3).
As discussed in the Introduction, converting acyclic alkanes to cyclopropane equivalents is a frequently used tactic in medicinal chemistry. The pairwise comparison of the isopropyl with the cyclopropyl substructures is best discussed via Figure 6, with the bold red bond emphasizing the structural change (cf D1 vs E1). For completion, the change observed from the corresponding linear 1-butanol equivalents with equidistant alcohol and fluorine groups, by connecting C2 and C4 as shown (cf G1 to E1), is also considered (see Supporting Information File 1, Figure S1 for a summary figure showing changes in logP between linear alkanols and cyclopropylmethanols).
![[1860-5397-16-182-6]](/bjoc/content/figures/1860-5397-16-182-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Comparison of lipophilicities between the linear alkyl, isopropyl, cyclopropyl, and 3-oxetanyl substituents by fluorination level (exchange two C–H bonds by a C–C bond or C–O–C bonds, same carbon count).
Figure 6: Comparison of lipophilicities between the linear alkyl, isopropyl, cyclopropyl, and 3-oxetanyl subs...
Firstly, the isobutanol (D) and their corresponding 1-butanol (G) series are compared. It is known that branching causes a reduction in logP, which has been explained by a less energetically unfavorable contact with water due to the resulting reduction in hydrophobic surface [33] (compare D1 with G1, Δ 0.12). This difference is similar upon β-fluorination (D2 vs G2, Δ 0.16) with a slightly greater magnitude which, as discussed above, is potentially due to the polarization of C–H bonds by fluorine. Interestingly, for γ-mono- and γ,γ-difluorination the linear butanol derivatives G3 and G4 are now less lipophilic than the branched D3 and D4, respectively. A possible explanation is that branching in D3 and D4 results in shielding of the polar C–F bonds preventing dipolar interactions.
Regarding the conversion of the iso- and n-butyl chains to the cyclopropylmethyl arrangement, as expected a significant lipophilicity decrease is observed for the nonfluorinated derivatives (compare D1, G1 with E1), with another large decrease when converting the cyclopropyl group to an oxetanyl moiety (compare E1 with F1). A much lower lipophilicity decrease is seen when converting the acyclic β-fluorinated compounds D2 and G2 to the cyclopropyl analogue E2, but an appreciable logP decrease is still seen going from E2 to the oxetanyl derivative F2. With γ-fluorination, the logP decreases are similarly small, both for monofluorination (compare D3, G3 with E3, E4) and for geminal difluorination (compare D4, G4 with E5). Finally, converting the linear 4,4-difluorobutan-1-ol (G5) to the corresponding difluorinated cyclopropylmethanol E5 only results in a minor lipophilicity decrease. Nevertheless, in all cases, for the same fluorination motif, the cyclopropane derivatives have a lower lipophilicity compared to their acyclic equivalents.
It is also useful to compare lipophilicities of acyclic and cyclopropane derivatives in which the isosterism represents conversion of a C–H and C–F bond into a C–C bond (Figure 7), hence transforming an acyclic to a cyclic group. Some interesting trends are identified. When the chain ring is closed with the loss of a C–F that was part of a single fluorine motif (Figure 7A), only very minimal lipophilicity differences were then observed. For 4-fluorobutan-1-ol (G6), and 3,4-difluorobutan-1-ol (G7), which have the lowest lipophilicities of the linear butanol series measured so far [28], the corresponding cyclopropyl isosteres E1 and E4 have a slightly higher lipophilicity, while E3 has the same lipophilicity as G7. The lipophilicity-reducing power of the vicinal 1,2-difluoromotif in G7 is well-described [34,35], but at least for series D, the motif present in E3 is another efficient candidate when a logP reduction operation is in order. In contrast, other such ring closures (e.g., starting from G2, D3, and G8) lead to a minute logP decrease.
![[1860-5397-16-182-7]](/bjoc/content/figures/1860-5397-16-182-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Comparison of lipophilicities between isopropyl and cyclopropyl substituents grouped by exchange of C–H/C–F bonds by a C–C bond (same carbon count). Organised by the exchanged fluorine being the only fluorine substituent on the carbon involved (A) or as part of a CF2 (B) or CF3 (C) group.
Figure 7: Comparison of lipophilicities between isopropyl and cyclopropyl substituents grouped by exchange of...
The situation is different when the “cyclisation” operation is achieved from geminal motifs (Figure 7B). Significant logP reductions are observed, especially from the internal geminal fluorination motif as in G9 to E2 (0.72 logP units). Starting from a trifluoromethyl group (Figure 7C), similar logP reductions are achieved (e.g., D5→E5 and G10→E5) which interestingly, are of the same magnitude as for the nonfluorinated derivatives (compare D1, G1 to E1, Figure 6). It is worth noting that “cyclisation” to give cyclopropanol derivatives substituted with a fluorinated methyl group (not shown) can also be considered. Marketed drugs with such substructures include Voxilaprevir and Glecaprevir.
Finally, it is useful to consider the lipophilicity changes upon extending a propyl or ethyl chain to a cyclopropylmethyl moiety, which represents a one-carbon and a two-carbon extension, respectively. It is known that adding a spiro-cyclopropyl moiety onto a methylene group of an aliphatic chain, which represents a two-carbon extension, can lead to a lipophilicity reduction [16,17].
The one-carbon extension from H1 to give E1 results in a small lipophilicity decrease (Figure 8), which is slightly extended with further β-fluorination (E2). In contrast, the one-carbon introduction of a cyclopropyl group in H2 to give E2 leads to a lipophilicity increase. Replacing the fluorine substituent by a cyclopropyl carbon (compare H2 with E1) also leads to a lipophilicity increase, but not when starting from a geminal difluoro motif (compare C4 with E2).
![[1860-5397-16-182-8]](/bjoc/content/figures/1860-5397-16-182-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
The two-carbon extension from I1 to the acyclic derivatives D1 and G1 leads to a large lipophilicity increase, which is much smaller for a two-carbon cyclopropyl extension (E1). This lipophilicity increase is somewhat attenuated with concomitant β-fluorination (E2) and γ-fluorination (E3). Interestingly, a two-carbon extension of I1 with concomitant vicinal difluorination (G7 [28]) or fluorinated cyclopropyl (E3) leads to a much smaller increase in logP compared to two-carbon extension as a cyclopropyl (E1).
Computational results
A number of open-access fragment-based calculation methods, as well as the internal AstraZeneca method, were used to obtain clogP values of the fluorinated butanol, isobutanol, cyclopropylmethanol and 3-oxetanylmethanol derivatives mentioned above (Supporting Information File 1, Table S6). For each method, the correlations with the experimental data were obtained (Supporting Information File 1, Figures S7–S11). Three methods gave excellent correlations (with r2 between 0.86 and 0.91, Supporting Information File 1, Table S7), but other methods gave only low correlations (r2 = 0.36, 0.49). This is perhaps due to the training sets used, but it clearly demonstrates that even for a set of relatively simple compounds, fragment-based clogP calculations are not guaranteed to give reliable lipophilicity data.
Next, theoretical lipophilicities were obtained using DFT calculations, based on the notion that the partition coefficient of a given solute between two phases relates to the difference in Gibbs energy of the free ligand conformations of this solute in the respective solvents. Quantum chemistry calculations of Gibbs energies in octanol and water thus provide theoretical estimations of lipophilicities. Such estimations require systematic conformational analyses and geometry optimizations. Ho and co-worker showed that the use of the lowest-energy conformations as calculated in the respective solvents gave more accurate lipophilicities than the use of the lowest energy conformations in the gas phase [36]. Somewhat surprisingly, implicit solvent models proved superior than explicit models, despite their shorter calculation times, with the SMD implicit solvent model being superior compared to the IEF-PCM and C-PCM models [36,37]. However, the implicit octanol solvent model does not take into account the large amount of water present in saturated octanol [36].
We have previously used the SMD implicit solvation model with the MN15 functional in theoretical lipophilicity predictions of linear fluorohydrins [27,28], which gave an excellent correlation with the experimental values (r2 = 0.957), although the absolute values were much higher (a slope of >2 was obtained).
Unfortunately, the theoretical lipophilicity values computed in the present work using the same methodology for series D, E, and F, as well as for the linear butanols G1–G4, G7, G8, pictured in Supporting Information File 1, Figure 9 and given in Supporting Information File 1, Table S11, did not provide any useful approximation in terms of rank order or absolute magnitude of effect.
![[1860-5397-16-182-9]](/bjoc/content/figures/1860-5397-16-182-9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Experimental and theoretical (in blue) values (calculated at the MN15/aug-cc-pVTZ//MN15/cc-pVTZ level of theory). Values for G1, G4, G7, G8 taken from the literature [28]. The distance between the lines relate to the relative lipophilicity differences. Experimental and theoretical data sets normalized towards the parent compound.
Figure 9: Experimental and theoretical (in blue) values (calculated at the MN15/aug-cc-pVTZ//MN15/cc-pVTZ lev...
For series D, E, G, the DFT-logP values cluster together with minimal lipophilicity differences, and within mean absolute error limits (estimated to be around 0.8 logP units), making detailed considerations meaningless. This was a surprise, as series D and E are relatively rigid, which simplifies conformational analysis. The remarkably similar DFT-logP values suggest that the influence of the fluorination is underrepresented in the calculations. The following observations are noteworthy. The trifluorinated D5 was calculated to have the largest logP value, in accord with the experiment. The geminally difluorinated derivatives are calculated to be more lipophilic than the monofluorinated derivatives, except in series G: G4 was calculated to be less lipophilic than G2. The E3/E4 diastereomers were calculated to have essentially the same lipophilicities.
The correlation with the experimental values shows a coefficient of determination value of 0.776 (Figure 10A), which is worse than many of the investigated fragment-based clogP methods (see above), for which such clustering is not observed. When these values are combined with the full available set of fluorohydrins calculated with the MN15 functional (Figure 10B), the correlation is now reduced from r2 = 0.957 to 0.860.
![[1860-5397-16-182-10]](/bjoc/content/figures/1860-5397-16-182-10.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: Correlation of the DFT-calculated lipophilicities with the experimental values. A) fluorinated series D, E, F, G2–4, G7, G8, B) Compilation with other fluorohydrins (taken from reference [28]).
Figure 10: Correlation of the DFT-calculated lipophilicities with the experimental values. A) fluorinated seri...
Interestingly, the theoretical and experimental values, while much different in absolute value and in relative difference, correspond almost perfectly regarding lipophilicity order when arranged by motif (Figure 11). As a point of interest, the calculations correctly estimate a lower lipophilicity for the branched D1 and D2 vs G1 and G2, but cannot significantly distinguish between the linear and branched γ-mono- and difluorinated G3/D3 and G4/D4. Cyclopropyl structures are always less lipophilic than their corresponding acyclic counterparts.
![[1860-5397-16-182-11]](/bjoc/content/figures/1860-5397-16-182-11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 11: Experimental (in black) and theoretical (in blue) values (calculated at the MN15/aug-cc-pVTZ//MN15/cc-pVTZ level of theory) of all compounds, organized by motif.
Figure 11: Experimental (in black) and theoretical (in blue) values (calculated at the MN15/aug-cc-pVTZ//MN15/...
Conclusion
Monofluorination of isobutanol significantly decreased the lipophilicity, with β-fluorination leading to a lower logP value than γ-fluorination. As expected, geminal difluorination also decreased the logP, but to a lesser extent compared to monofluorination. For the corresponding cyclopropyl derivative, cyclopropylmethanol, monofluorination at the β or γ position only led to a minor decrease in logP, and no logP difference is observed with the geminal difluorinated analogue. The typically observed lower lipophilicities of branched isomers vs their linear isomers are also observed for β-fluorohydrin containing compounds, but not when γ-mono- or difluorinated. Equally, converting (acyclic) alkyl groups to the corresponding cyclopropane equivalents (C–H/C–H→C–C) typically leads to a lipophilicity decrease, which remains the case when fluorination is present. For a C–F/C–H→C–C ‘cyclisation’, only minimal lipophilicity differences are observed (in either direction) when the C–F moiety was part on a monofluorinated motif. However, when the C–F moiety was part of a geminal (CF2 or CF3) motif, a large logP reduction is observed.
Fragment based clogP methods show great variation in their ability to estimate the lipophilicities of the small alkanol compounds described herein. To our surprise, theoretical lipophilicity predictions using the MN15 functional with the SMD implicit solvation model also performed poorly, despite the fact that most of the substrates are small and relatively rigid compounds. It was found that for a given series, the DFT-logP values are clustered together, indicating the influence of the fluorination is underrepresented in the calculations. This clearly indicates that further research towards theoretical lipophilicity prediction based in relative Gibbs energies in water and (wet) octanol is required.
Supporting Information
| Supporting Information File 1: Synthesis, characterisation and copies of spectra of the novel compounds, details of the calculations, logP determinations of the nonfluorinated parents E1 and F1, and experimental measurements of the lipophilicities of the fluorinated derivatives. | ||
| Format: PDF | Size: 2.3 MB | Download |
References
-
Persch, E.; Dumele, O.; Diederich, F. Angew. Chem., Int. Ed. 2015, 54, 3290–3327. doi:10.1002/anie.201408487
Return to citation in text: [1] -
Mecozzi, S.; Rebek, J., Jr. Chem. – Eur. J. 1998, 4, 1016–1022. doi:10.1002/(sici)1521-3765(19980615)4:6<1016::aid-chem1016>3.0.co;2-b
Return to citation in text: [1] -
Bissantz, C.; Kuhn, B.; Stahl, M. J. Med. Chem. 2010, 53, 5061–5084. doi:10.1021/jm100112j
Return to citation in text: [1] -
Zauhar, R. J.; Moyna, G.; Tian, L.; Li, Z.; Welsh, W. J. J. Med. Chem. 2003, 46, 5674–5690. doi:10.1021/jm030242k
Return to citation in text: [1] -
Kumar, A.; Zhang, K. Y. J. Front. Chem. (Lausanne, Switz.) 2018, 6, 315. doi:10.3389/fchem.2018.00315
Return to citation in text: [1] -
Smart, B. E. J. Fluorine Chem. 2001, 109, 3–11. doi:10.1016/s0022-1139(01)00375-x
Return to citation in text: [1] [2] -
Schönherr, H.; Cernak, T. Angew. Chem., Int. Ed. 2013, 52, 12256–12267. doi:10.1002/anie.201303207
Return to citation in text: [1] -
Kuntz, K. W.; Campbell, J. E.; Keilhack, H.; Pollock, R. M.; Knutson, S. K.; Porter-Scott, M.; Richon, V. M.; Sneeringer, C. J.; Wigle, T. J.; Allain, C. J.; Majer, C. R.; Moyer, M. P.; Copeland, R. A.; Chesworth, R. J. Med. Chem. 2016, 59, 1556–1564. doi:10.1021/acs.jmedchem.5b01501
Return to citation in text: [1] -
Barreiro, E. J.; Kümmerle, A. E.; Fraga, C. A. M. Chem. Rev. 2011, 111, 5215–5246. doi:10.1021/cr200060g
Return to citation in text: [1] -
Talele, T. T. J. Med. Chem. 2018, 61, 2166–2210. doi:10.1021/acs.jmedchem.7b00315
Return to citation in text: [1] -
Talele, T. T. J. Med. Chem. 2016, 59, 8712–8756. doi:10.1021/acs.jmedchem.6b00472
Return to citation in text: [1] -
Novakov, I. A.; Babushkin, A. S.; Yablokov, A. S.; Nawrozkij, M. B.; Vostrikova, O. V.; Shejkin, D. S.; Mkrtchyan, A. S.; Balakin, K. V. Russ. Chem. Bull. 2018, 67, 395–418. doi:10.1007/s11172-018-2087-6
Return to citation in text: [1] -
Hansch, C.; Leo, A.; Unger, S. H.; Kim, K. H.; Nikaitani, D.; Lien, E. J. J. Med. Chem. 1973, 16, 1207–1216. doi:10.1021/jm00269a003
Return to citation in text: [1] [2] -
Skagerberg, B.; Bonelli, D.; Clementi, S.; Cruciani, G.; Ebert, C. Quant. Struct.-Act. Relat. 1989, 8, 32–38. doi:10.1002/qsar.19890080105
Return to citation in text: [1] -
Letavic, M. A.; Aluisio, L.; Apodaca, R.; Bajpai, M.; Barbier, A. J.; Bonneville, A.; Bonaventure, P.; Carruthers, N. I.; Dugovic, C.; Fraser, I. C.; Kramer, M. L.; Lord, B.; Lovenberg, T. W.; Li, L. Y.; Ly, K. S.; Mcallister, H.; Mani, N. S.; Morton, K. L.; Ndifor, A.; Nepomuceno, S. D.; Pandit, C. R.; Sands, S. B.; Shah, C. R.; Shelton, J. E.; Snook, S. S.; Swanson, D. M.; Xiao, W. ACS Med. Chem. Lett. 2015, 6, 450–454. doi:10.1021/ml5005156
Return to citation in text: [1] -
Wuitschik, G.; Carreira, E. M.; Wagner, B.; Fischer, H.; Parrilla, I.; Schuler, F.; Rogers-Evans, M.; Müller, K. J. Med. Chem. 2010, 53, 3227–3246. doi:10.1021/jm9018788
Return to citation in text: [1] [2] [3] -
Wuitschik, G.; Rogers-Evans, M.; Müller, K.; Fischer, H.; Wagner, B.; Schuler, F.; Polonchuk, L.; Carreira, E. M. Angew. Chem., Int. Ed. 2006, 45, 7736–7739. doi:10.1002/anie.200602343
Return to citation in text: [1] [2] [3] -
Bull, J. A.; Croft, R. A.; Davis, O. A.; Doran, R.; Morgan, K. F. Chem. Rev. 2016, 116, 12150–12233. doi:10.1021/acs.chemrev.6b00274
Return to citation in text: [1] -
Böhm, H.-J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. ChemBioChem 2004, 5, 637–643. doi:10.1002/cbic.200301023
Return to citation in text: [1] -
Huchet, Q. A.; Kuhn, B.; Wagner, B.; Fischer, H.; Kansy, M.; Zimmerli, D.; Carreira, E. M.; Müller, K. J. Fluorine Chem. 2013, 152, 119–128. doi:10.1016/j.jfluchem.2013.02.023
Return to citation in text: [1] [2] -
Müller, K. Fluorination patterns in small alkyl groups: their impact on properties relevant to drug discovery. In Fluorine in Life Sciences: Pharmaceuticals, Medicinal Diagnostics, and Agrochemicals; Haufe, G.; Leroux, F., Eds.; Academic Press: London, U.K., 2018; pp 91–140. doi:10.1016/b978-0-12-812733-9.00002-7
Return to citation in text: [1] -
Linclau, B.; Wang, Z.; Compain, G.; Paumelle, V.; Fontenelle, C. Q.; Wells, N.; Weymouth-Wilson, A. Angew. Chem., Int. Ed. 2016, 55, 674–678. doi:10.1002/anie.201509460
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Panchaud, P.; Surivet, J.-P.; Diethelm, S.; Blumstein, A.-C.; Gauvin, J.-C.; Jacob, L.; Masse, F.; Mathieu, G.; Mirre, A.; Schmitt, C.; Enderlin-Paput, M.; Lange, R.; Gnerre, C.; Seeland, S.; Herrmann, C.; Locher, H. H.; Seiler, P.; Ritz, D.; Rueedi, G. J. Med. Chem. 2020, 63, 88–102. doi:10.1021/acs.jmedchem.9b01605
Return to citation in text: [1] -
Nakahara, K.; Fuchino, K.; Komano, K.; Asada, N.; Tadano, G.; Hasegawa, T.; Yamamoto, T.; Sako, Y.; Ogawa, M.; Unemura, C.; Hosono, M.; Ito, H.; Sakaguchi, G.; Ando, S.; Ohnishi, S.; Kido, Y.; Fukushima, T.; Dhuyvetter, D.; Borghys, H.; Gijsen, H. J. M.; Yamano, Y.; Iso, Y.; Kusakabe, K.-i. J. Med. Chem. 2018, 61, 5525–5546. doi:10.1021/acs.jmedchem.8b00011
Return to citation in text: [1] -
Fang, Z.; Cordes, D. B.; Slawin, A. M. Z.; O'Hagan, D. Chem. Commun. 2019, 55, 10539–10542. doi:10.1039/c9cc05749h
Return to citation in text: [1] -
Thomson, C. J.; Zhang, Q.; Al-Maharik, N.; Bühl, M.; Cordes, D. B.; Slawin, A. M. Z.; O’Hagan, D. Chem. Commun. 2018, 54, 8415–8418. doi:10.1039/c8cc04964e
Return to citation in text: [1] -
Wang, Z.; Jeffries, B. F.; Felstead, H. R.; Wells, N. J.; Chiarparin, E.; Linclau, B. J. Visualized Exp. 2019, No. 143, e58567. doi:10.3791/58567
Return to citation in text: [1] [2] -
Jeffries, B.; Wang, Z.; Felstead, H. R.; Le Questel, J.-Y.; Scott, J. S.; Chiarparin, E.; Graton, J.; Linclau, B. J. Med. Chem. 2020, 63, 1002–1031. doi:10.1021/acs.jmedchem.9b01172
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Siebum, A. H. G.; Woo, W. S.; Lugtenburg, J. Eur. J. Org. Chem. 2003, 4664–4678. doi:10.1002/ejoc.200300410
Return to citation in text: [1] -
Scott, J. S.; Bailey, A.; Buttar, D.; Carbajo, R. J.; Curwen, J.; Davey, P. R. J.; Davies, R. D. M.; Degorce, S. L.; Donald, C.; Gangl, E.; Greenwood, R.; Groombridge, S. D.; Johnson, T.; Lamont, S.; Lawson, M.; Lister, A.; Morrow, C. J.; Moss, T. A.; Pink, J. H.; Polanski, R. J. Med. Chem. 2019, 62, 1593–1608. doi:10.1021/acs.jmedchem.8b01837
Return to citation in text: [1] -
Zafrani, Y.; Sod-Moriah, G.; Yeffet, D.; Berliner, A.; Amir, D.; Marciano, D.; Elias, S.; Katalan, S.; Ashkenazi, N.; Madmon, M.; Gershonov, E.; Saphier, S. J. Med. Chem. 2019, 62, 5628–5637. doi:10.1021/acs.jmedchem.9b00604
Return to citation in text: [1] -
Kubyshkin, V.; Budisa, N. Beilstein J. Org. Chem. 2017, 13, 2442–2457. doi:10.3762/bjoc.13.241
Return to citation in text: [1] -
Bazzini, P.; Wermuth, C. G. Substituents and Functions: Qualitative and Quantitative Aspects of Structure–Activity Relationships. In The Practice of Medicinal Chemistry, 3rd ed.; Wermuth, C. G., Ed.; Academic Press: New York, NY, USA, 2008; pp 429–463. doi:10.1016/b978-0-12-374194-3.00020-2
Return to citation in text: [1] -
Huchet, Q. A.; Kuhn, B.; Wagner, B.; Kratochwil, N. A.; Fischer, H.; Kansy, M.; Zimmerli, D.; Carreira, E. M.; Müller, K. J. Med. Chem. 2015, 58, 9041–9060. doi:10.1021/acs.jmedchem.5b01455
Return to citation in text: [1] -
Erdeljac, N.; Kehr, G.; Ahlqvist, M.; Knerr, L.; Gilmour, R. Chem. Commun. 2018, 54, 12002–12005. doi:10.1039/c8cc05643a
Return to citation in text: [1] -
Kundi, V.; Ho, J. J. Phys. Chem. B 2019, 123, 6810–6822. doi:10.1021/acs.jpcb.9b04061
Return to citation in text: [1] [2] [3] -
Michalík, M.; Lukeš, V. Acta Chim. Slovaca 2016, 9, 89–94. doi:10.1515/acs-2016-0015
Return to citation in text: [1]
| 34. | Huchet, Q. A.; Kuhn, B.; Wagner, B.; Kratochwil, N. A.; Fischer, H.; Kansy, M.; Zimmerli, D.; Carreira, E. M.; Müller, K. J. Med. Chem. 2015, 58, 9041–9060. doi:10.1021/acs.jmedchem.5b01455 |
| 35. | Erdeljac, N.; Kehr, G.; Ahlqvist, M.; Knerr, L.; Gilmour, R. Chem. Commun. 2018, 54, 12002–12005. doi:10.1039/c8cc05643a |
| 16. | Wuitschik, G.; Carreira, E. M.; Wagner, B.; Fischer, H.; Parrilla, I.; Schuler, F.; Rogers-Evans, M.; Müller, K. J. Med. Chem. 2010, 53, 3227–3246. doi:10.1021/jm9018788 |
| 17. | Wuitschik, G.; Rogers-Evans, M.; Müller, K.; Fischer, H.; Wagner, B.; Schuler, F.; Polonchuk, L.; Carreira, E. M. Angew. Chem., Int. Ed. 2006, 45, 7736–7739. doi:10.1002/anie.200602343 |
| 28. | Jeffries, B.; Wang, Z.; Felstead, H. R.; Le Questel, J.-Y.; Scott, J. S.; Chiarparin, E.; Graton, J.; Linclau, B. J. Med. Chem. 2020, 63, 1002–1031. doi:10.1021/acs.jmedchem.9b01172 |
| 1. | Persch, E.; Dumele, O.; Diederich, F. Angew. Chem., Int. Ed. 2015, 54, 3290–3327. doi:10.1002/anie.201408487 |
| 2. | Mecozzi, S.; Rebek, J., Jr. Chem. – Eur. J. 1998, 4, 1016–1022. doi:10.1002/(sici)1521-3765(19980615)4:6<1016::aid-chem1016>3.0.co;2-b |
| 10. | Talele, T. T. J. Med. Chem. 2018, 61, 2166–2210. doi:10.1021/acs.jmedchem.7b00315 |
| 22. | Linclau, B.; Wang, Z.; Compain, G.; Paumelle, V.; Fontenelle, C. Q.; Wells, N.; Weymouth-Wilson, A. Angew. Chem., Int. Ed. 2016, 55, 674–678. doi:10.1002/anie.201509460 |
| 7. | Schönherr, H.; Cernak, T. Angew. Chem., Int. Ed. 2013, 52, 12256–12267. doi:10.1002/anie.201303207 |
| 8. | Kuntz, K. W.; Campbell, J. E.; Keilhack, H.; Pollock, R. M.; Knutson, S. K.; Porter-Scott, M.; Richon, V. M.; Sneeringer, C. J.; Wigle, T. J.; Allain, C. J.; Majer, C. R.; Moyer, M. P.; Copeland, R. A.; Chesworth, R. J. Med. Chem. 2016, 59, 1556–1564. doi:10.1021/acs.jmedchem.5b01501 |
| 9. | Barreiro, E. J.; Kümmerle, A. E.; Fraga, C. A. M. Chem. Rev. 2011, 111, 5215–5246. doi:10.1021/cr200060g |
| 25. | Fang, Z.; Cordes, D. B.; Slawin, A. M. Z.; O'Hagan, D. Chem. Commun. 2019, 55, 10539–10542. doi:10.1039/c9cc05749h |
| 26. | Thomson, C. J.; Zhang, Q.; Al-Maharik, N.; Bühl, M.; Cordes, D. B.; Slawin, A. M. Z.; O’Hagan, D. Chem. Commun. 2018, 54, 8415–8418. doi:10.1039/c8cc04964e |
| 6. | Smart, B. E. J. Fluorine Chem. 2001, 109, 3–11. doi:10.1016/s0022-1139(01)00375-x |
| 23. | Panchaud, P.; Surivet, J.-P.; Diethelm, S.; Blumstein, A.-C.; Gauvin, J.-C.; Jacob, L.; Masse, F.; Mathieu, G.; Mirre, A.; Schmitt, C.; Enderlin-Paput, M.; Lange, R.; Gnerre, C.; Seeland, S.; Herrmann, C.; Locher, H. H.; Seiler, P.; Ritz, D.; Rueedi, G. J. Med. Chem. 2020, 63, 88–102. doi:10.1021/acs.jmedchem.9b01605 |
| 28. | Jeffries, B.; Wang, Z.; Felstead, H. R.; Le Questel, J.-Y.; Scott, J. S.; Chiarparin, E.; Graton, J.; Linclau, B. J. Med. Chem. 2020, 63, 1002–1031. doi:10.1021/acs.jmedchem.9b01172 |
| 3. | Bissantz, C.; Kuhn, B.; Stahl, M. J. Med. Chem. 2010, 53, 5061–5084. doi:10.1021/jm100112j |
| 4. | Zauhar, R. J.; Moyna, G.; Tian, L.; Li, Z.; Welsh, W. J. J. Med. Chem. 2003, 46, 5674–5690. doi:10.1021/jm030242k |
| 5. | Kumar, A.; Zhang, K. Y. J. Front. Chem. (Lausanne, Switz.) 2018, 6, 315. doi:10.3389/fchem.2018.00315 |
| 24. | Nakahara, K.; Fuchino, K.; Komano, K.; Asada, N.; Tadano, G.; Hasegawa, T.; Yamamoto, T.; Sako, Y.; Ogawa, M.; Unemura, C.; Hosono, M.; Ito, H.; Sakaguchi, G.; Ando, S.; Ohnishi, S.; Kido, Y.; Fukushima, T.; Dhuyvetter, D.; Borghys, H.; Gijsen, H. J. M.; Yamano, Y.; Iso, Y.; Kusakabe, K.-i. J. Med. Chem. 2018, 61, 5525–5546. doi:10.1021/acs.jmedchem.8b00011 |
| 28. | Jeffries, B.; Wang, Z.; Felstead, H. R.; Le Questel, J.-Y.; Scott, J. S.; Chiarparin, E.; Graton, J.; Linclau, B. J. Med. Chem. 2020, 63, 1002–1031. doi:10.1021/acs.jmedchem.9b01172 |
| 13. | Hansch, C.; Leo, A.; Unger, S. H.; Kim, K. H.; Nikaitani, D.; Lien, E. J. J. Med. Chem. 1973, 16, 1207–1216. doi:10.1021/jm00269a003 |
| 16. | Wuitschik, G.; Carreira, E. M.; Wagner, B.; Fischer, H.; Parrilla, I.; Schuler, F.; Rogers-Evans, M.; Müller, K. J. Med. Chem. 2010, 53, 3227–3246. doi:10.1021/jm9018788 |
| 17. | Wuitschik, G.; Rogers-Evans, M.; Müller, K.; Fischer, H.; Wagner, B.; Schuler, F.; Polonchuk, L.; Carreira, E. M. Angew. Chem., Int. Ed. 2006, 45, 7736–7739. doi:10.1002/anie.200602343 |
| 18. | Bull, J. A.; Croft, R. A.; Davis, O. A.; Doran, R.; Morgan, K. F. Chem. Rev. 2016, 116, 12150–12233. doi:10.1021/acs.chemrev.6b00274 |
| 36. | Kundi, V.; Ho, J. J. Phys. Chem. B 2019, 123, 6810–6822. doi:10.1021/acs.jpcb.9b04061 |
| 14. | Skagerberg, B.; Bonelli, D.; Clementi, S.; Cruciani, G.; Ebert, C. Quant. Struct.-Act. Relat. 1989, 8, 32–38. doi:10.1002/qsar.19890080105 |
| 6. | Smart, B. E. J. Fluorine Chem. 2001, 109, 3–11. doi:10.1016/s0022-1139(01)00375-x |
| 19. | Böhm, H.-J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. ChemBioChem 2004, 5, 637–643. doi:10.1002/cbic.200301023 |
| 20. | Huchet, Q. A.; Kuhn, B.; Wagner, B.; Fischer, H.; Kansy, M.; Zimmerli, D.; Carreira, E. M.; Müller, K. J. Fluorine Chem. 2013, 152, 119–128. doi:10.1016/j.jfluchem.2013.02.023 |
| 21. | Müller, K. Fluorination patterns in small alkyl groups: their impact on properties relevant to drug discovery. In Fluorine in Life Sciences: Pharmaceuticals, Medicinal Diagnostics, and Agrochemicals; Haufe, G.; Leroux, F., Eds.; Academic Press: London, U.K., 2018; pp 91–140. doi:10.1016/b978-0-12-812733-9.00002-7 |
| 22. | Linclau, B.; Wang, Z.; Compain, G.; Paumelle, V.; Fontenelle, C. Q.; Wells, N.; Weymouth-Wilson, A. Angew. Chem., Int. Ed. 2016, 55, 674–678. doi:10.1002/anie.201509460 |
| 27. | Wang, Z.; Jeffries, B. F.; Felstead, H. R.; Wells, N. J.; Chiarparin, E.; Linclau, B. J. Visualized Exp. 2019, No. 143, e58567. doi:10.3791/58567 |
| 28. | Jeffries, B.; Wang, Z.; Felstead, H. R.; Le Questel, J.-Y.; Scott, J. S.; Chiarparin, E.; Graton, J.; Linclau, B. J. Med. Chem. 2020, 63, 1002–1031. doi:10.1021/acs.jmedchem.9b01172 |
| 13. | Hansch, C.; Leo, A.; Unger, S. H.; Kim, K. H.; Nikaitani, D.; Lien, E. J. J. Med. Chem. 1973, 16, 1207–1216. doi:10.1021/jm00269a003 |
| 36. | Kundi, V.; Ho, J. J. Phys. Chem. B 2019, 123, 6810–6822. doi:10.1021/acs.jpcb.9b04061 |
| 11. | Talele, T. T. J. Med. Chem. 2016, 59, 8712–8756. doi:10.1021/acs.jmedchem.6b00472 |
| 12. | Novakov, I. A.; Babushkin, A. S.; Yablokov, A. S.; Nawrozkij, M. B.; Vostrikova, O. V.; Shejkin, D. S.; Mkrtchyan, A. S.; Balakin, K. V. Russ. Chem. Bull. 2018, 67, 395–418. doi:10.1007/s11172-018-2087-6 |
| 15. | Letavic, M. A.; Aluisio, L.; Apodaca, R.; Bajpai, M.; Barbier, A. J.; Bonneville, A.; Bonaventure, P.; Carruthers, N. I.; Dugovic, C.; Fraser, I. C.; Kramer, M. L.; Lord, B.; Lovenberg, T. W.; Li, L. Y.; Ly, K. S.; Mcallister, H.; Mani, N. S.; Morton, K. L.; Ndifor, A.; Nepomuceno, S. D.; Pandit, C. R.; Sands, S. B.; Shah, C. R.; Shelton, J. E.; Snook, S. S.; Swanson, D. M.; Xiao, W. ACS Med. Chem. Lett. 2015, 6, 450–454. doi:10.1021/ml5005156 |
| 36. | Kundi, V.; Ho, J. J. Phys. Chem. B 2019, 123, 6810–6822. doi:10.1021/acs.jpcb.9b04061 |
| 37. | Michalík, M.; Lukeš, V. Acta Chim. Slovaca 2016, 9, 89–94. doi:10.1515/acs-2016-0015 |
| 22. | Linclau, B.; Wang, Z.; Compain, G.; Paumelle, V.; Fontenelle, C. Q.; Wells, N.; Weymouth-Wilson, A. Angew. Chem., Int. Ed. 2016, 55, 674–678. doi:10.1002/anie.201509460 |
| 16. | Wuitschik, G.; Carreira, E. M.; Wagner, B.; Fischer, H.; Parrilla, I.; Schuler, F.; Rogers-Evans, M.; Müller, K. J. Med. Chem. 2010, 53, 3227–3246. doi:10.1021/jm9018788 |
| 17. | Wuitschik, G.; Rogers-Evans, M.; Müller, K.; Fischer, H.; Wagner, B.; Schuler, F.; Polonchuk, L.; Carreira, E. M. Angew. Chem., Int. Ed. 2006, 45, 7736–7739. doi:10.1002/anie.200602343 |
| 22. | Linclau, B.; Wang, Z.; Compain, G.; Paumelle, V.; Fontenelle, C. Q.; Wells, N.; Weymouth-Wilson, A. Angew. Chem., Int. Ed. 2016, 55, 674–678. doi:10.1002/anie.201509460 |
| 27. | Wang, Z.; Jeffries, B. F.; Felstead, H. R.; Wells, N. J.; Chiarparin, E.; Linclau, B. J. Visualized Exp. 2019, No. 143, e58567. doi:10.3791/58567 |
| 33. | Bazzini, P.; Wermuth, C. G. Substituents and Functions: Qualitative and Quantitative Aspects of Structure–Activity Relationships. In The Practice of Medicinal Chemistry, 3rd ed.; Wermuth, C. G., Ed.; Academic Press: New York, NY, USA, 2008; pp 429–463. doi:10.1016/b978-0-12-374194-3.00020-2 |
| 28. | Jeffries, B.; Wang, Z.; Felstead, H. R.; Le Questel, J.-Y.; Scott, J. S.; Chiarparin, E.; Graton, J.; Linclau, B. J. Med. Chem. 2020, 63, 1002–1031. doi:10.1021/acs.jmedchem.9b01172 |
| 22. | Linclau, B.; Wang, Z.; Compain, G.; Paumelle, V.; Fontenelle, C. Q.; Wells, N.; Weymouth-Wilson, A. Angew. Chem., Int. Ed. 2016, 55, 674–678. doi:10.1002/anie.201509460 |
| 32. | Kubyshkin, V.; Budisa, N. Beilstein J. Org. Chem. 2017, 13, 2442–2457. doi:10.3762/bjoc.13.241 |
| 22. | Linclau, B.; Wang, Z.; Compain, G.; Paumelle, V.; Fontenelle, C. Q.; Wells, N.; Weymouth-Wilson, A. Angew. Chem., Int. Ed. 2016, 55, 674–678. doi:10.1002/anie.201509460 |
| 30. | Scott, J. S.; Bailey, A.; Buttar, D.; Carbajo, R. J.; Curwen, J.; Davey, P. R. J.; Davies, R. D. M.; Degorce, S. L.; Donald, C.; Gangl, E.; Greenwood, R.; Groombridge, S. D.; Johnson, T.; Lamont, S.; Lawson, M.; Lister, A.; Morrow, C. J.; Moss, T. A.; Pink, J. H.; Polanski, R. J. Med. Chem. 2019, 62, 1593–1608. doi:10.1021/acs.jmedchem.8b01837 |
| 20. | Huchet, Q. A.; Kuhn, B.; Wagner, B.; Fischer, H.; Kansy, M.; Zimmerli, D.; Carreira, E. M.; Müller, K. J. Fluorine Chem. 2013, 152, 119–128. doi:10.1016/j.jfluchem.2013.02.023 |
| 31. | Zafrani, Y.; Sod-Moriah, G.; Yeffet, D.; Berliner, A.; Amir, D.; Marciano, D.; Elias, S.; Katalan, S.; Ashkenazi, N.; Madmon, M.; Gershonov, E.; Saphier, S. J. Med. Chem. 2019, 62, 5628–5637. doi:10.1021/acs.jmedchem.9b00604 |
| 28. | Jeffries, B.; Wang, Z.; Felstead, H. R.; Le Questel, J.-Y.; Scott, J. S.; Chiarparin, E.; Graton, J.; Linclau, B. J. Med. Chem. 2020, 63, 1002–1031. doi:10.1021/acs.jmedchem.9b01172 |
| 29. | Siebum, A. H. G.; Woo, W. S.; Lugtenburg, J. Eur. J. Org. Chem. 2003, 4664–4678. doi:10.1002/ejoc.200300410 |
© 2020 Jeffries et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)