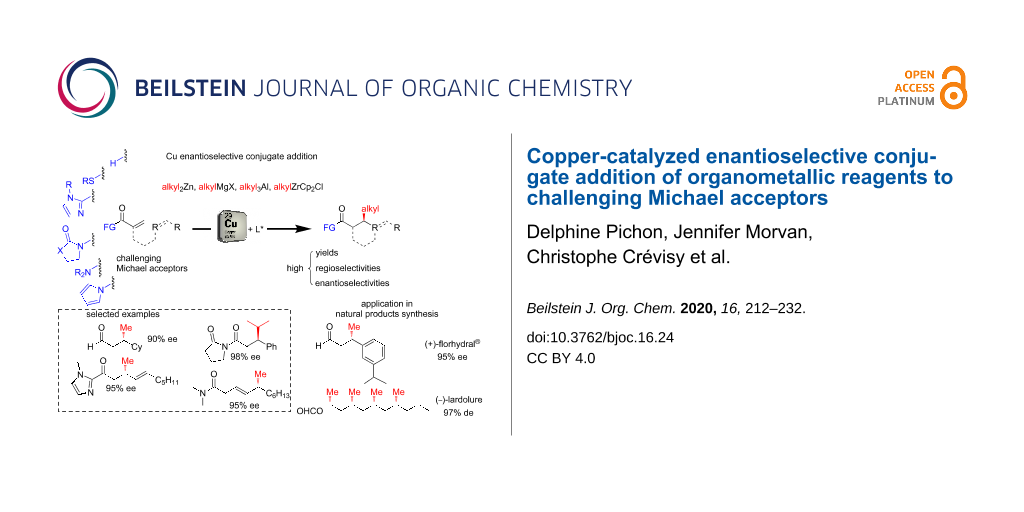
Abstract
The copper-catalyzed enantioselective conjugate addition (ECA) of organometallic nucleophiles to electron-deficient alkenes (Michael acceptors) represents an efficient and attractive methodology for providing a wide range of relevant chiral molecules. In order to increase the attractiveness of this useful catalytic transformation, some Michael acceptors bearing challenging electron-deficient functions (i.e., aldehydes, thioesters, acylimidazoles, N-acyloxazolidinones, N-acylpyrrolidinones, amides, N-acylpyrroles) were recently investigated. Remarkably, only a few chiral copper-based catalytic systems have successfully achieved the conjugate addition of different organometallic reagents to these challenging Michael acceptors, with excellent regio- and enantioselectivity. Furthermore, thanks to their easy derivatization, the resulting chiral conjugated products could be converted into various natural products. The aim of this tutorial review is to summarize recent advances accomplished in this stimulating field.

Graphical Abstract
Introduction
Generating high molecular complexity and controlling multiple stereogenic centers in a minimum number of steps is nowadays one of the most important challenges in organic chemistry for the synthesis of complex chiral molecules. The transition metal (TM)-catalyzed enantioselective conjugate addition (ECA) of nucleophiles to electron-deficient alkenes (Michael acceptors) is one of the most relevant and versatile methods to achieve this goal [1-4]. Among the plethora of metals studied, copper-based catalytic systems proved to be highly efficient for the conjugate addition of various organometallic reagents, such as diorganozinc, triorganoaluminium, and Grignard reagents to Michael acceptors. In that respect, since the pioneering example reported by Alexakis and co-workers in 1993 [5], a wide range of cyclic and acyclic electron-deficient alkenes, such as α,β-unsaturated ketones, esters, nitriles, sulfones, or nitroolefines, was intensively studied, leading to the expected 1,4-products in excellent yields and remarkable enantioselectivities. More recently, tremendous breakthroughs were achieved in this field, notably by the formation of all-carbon quaternary chiral centers [6] and the challenging 1,6-, 1,8-, or 1,10-selective addition to cyclic or aliphatic polyenic substrates [7-9]. Furthermore, Cu ECA transformations were also successfully applied to the synthesis of natural products [10]. Nevertheless, it is worth to underline that the choice of the electron-withdrawing group (EWG) on the Michael substrates appears not so simple. First, the hardness of the involved organometallic reagents has to be considered in order to overcome or limit undesirable side reactions. Although the main role of copper is to form a transient organocuprate reactive species with the hard nucleophiles to avoid the formation of the nondesired 1,2-product, some Grignard or aluminium reagents remain too reactive and incompatible with some electron-withdrawing functions. In contrast, some organometallic reagents, such as dimethylzinc, are poorly reactive and require a higher electrophilicity of the Michael acceptors to provide the desired conjugated products. Second, in order to be attractive for the total synthesis of relevant molecules, the involved EWG should preferably allow readily applicable and practicable postfunctionalizations [10].
Among the plethora of studied Michael acceptors, α,β-unsaturated aldehydes, thioesters, acylimidazoles, N-acyloxazolidinones, N-acylpyrrolidinones, amides, and N-acylpyrroles have been scarcely investigated in Cu ECA despite their usefulness for postfunctionalizations. This tutorial review aims to describe the early examples and recent advances in copper-catalyzed asymmetric conjugate additions of dialkylzinc, Grignard, or trialkylaluminium reagents toward those challenging substrates and their fruitful application in the total synthesis of natural products.
Review
Enantioselective conjugate addition to challenging Michael acceptors
Copper-catalyzed ECA to α,β-unsaturated aldehydes
Nowadays, β-substituted enals represent probably the most challenging Michael acceptors in the copper-catalyzed ECA of organometallic reagents [11-13]. This challenge is reinforced by the fact that the resulting chiral β-functionalized aldehydes are considered as an important motif that is ubiquitous in numerous natural molecules. However, as depicted in Scheme 1, due to their stronger reactivity than that of usual esters or ketones, a competitive 1,2-addition to the carbonyl function of enals could occur, leading to the corresponding alcohol as a byproduct. Moreover, even if the 1,4-addition is favored, thanks to the copper/ligand catalytic species, the resulting metallic enolate intermediate can also react with the starting material to form the aldol byproduct, significantly altering the yield of the expected 1,4-product (Scheme 1).
![[1860-5397-16-24-i1]](/bjoc/content/inline/1860-5397-16-24-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Competitive side reactions in the Cu ECA of organometallic reagents to α,β-unsaturated aldehydes.
Scheme 1: Competitive side reactions in the Cu ECA of organometallic reagents to α,β-unsaturated aldehydes.
The first successful copper-catalyzed ECA to α,β-unsaturated aldehydes with organozinc and Grignard reagents was reported by Alexakis and co-workers in 2010 [14]. After screening various chiral phosphine-based ligands, the combinations of either phosphoramidite L1 with Cu(OTf)2, or (R)-BINAP (L2) with copper thiophenecarboxylate (CuTC) appeared to be the most efficient for the addition of Et2Zn to a variety of cyclic and acyclic aldehydes 1. High 1,4-regioselectivities and promising stereoselectivities ranging from 27 to 90% ee were achieved (Scheme 2a). It is noteworthy that the addition of dimethylzinc was also successfully achieved, as the desired 1,4-methylated products were exclusively formed in moderate to good yields, with ee values of up to 76%. When the conjugate addition was performed with Grignard reagents, significant amounts of 1,2-products and enols were formed, despite the use of cryogenic conditions. (R)-BINAP (L2) gave the best regio- and enantioselectivity, with 62% of the 1,4-product and 89% ee with EtMgBr (Scheme 2b). To overcome the low regioselectivity, the authors took into account previous works showing that the 1,4-regioselectivity in the addition of cuprates to enals could be improved in the presence of a slight excess of TMSCl [15-19]. Indeed, using TMSCl in combination with (R)-TolBINAP (L3), a promising 85% regioselectivity was observed, without altering the enantioselectivity (90% ee), whereas only 32% of the desired 1,4- product was obtained without TMSCl [20]. With those optimized conditions, various enals and Grignard reagents were screened. Nevertheless, despite the presence of TMSCl, the 1,4:1,2 ratio varied significantly (from 85:15 to 10:90), while the level of enantioselectivity remained relatively good, reaching up to 90%.
![[1860-5397-16-24-i2]](/bjoc/content/inline/1860-5397-16-24-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Cu-catalyzed ECA of α,β-unsaturated aldehydes with phosphoramidite- (a) and phosphine-based ligands (b).
Scheme 2: Cu-catalyzed ECA of α,β-unsaturated aldehydes with phosphoramidite- (a) and phosphine-based ligands...
Following this, Alexakis and Quintard invented an efficient stepwise one-pot copper-catalyzed asymmetric ECA/organocatalyzed α-substitution of enals [21]. By using (R)-BINAP (L2)/CuTC in combination with chiral prolinol derivatives L4–6 as organocatalysts, various α,β-functionalized aldehydes were synthesized in good isolated yields (57–74%) and remarkable enantioselectivity (99%) from diethylzinc or dimethylzinc as nucleophiles and vinyl sulfones as electrophiles (Scheme 3). Of note, both the anti- and the syn-product could be predominantly formed (with a anti:syn ratio from 83:17 to 15:85), and no diastereocontrol occurred in the absence of the organocatalyst. Interestingly, this simple protocol was successfully applied to the enantioselective synthesis of valnoctamide, a commercialized mild tranquilizer. Finally, this methodology was extended to the sequential Michael/halogenation reaction using NFSI or NCS as electrophiles, with similar efficiency.
![[1860-5397-16-24-i3]](/bjoc/content/inline/1860-5397-16-24-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: One-pot Cu-catalyzed ECA/organocatalyzed α-substitution of enals.
Scheme 3: One-pot Cu-catalyzed ECA/organocatalyzed α-substitution of enals.
Similarly, a cocatalyzed enantioselective β-functionalization of enals was developed by Córdova, Ibrahem, and co-workers in 2011 (Scheme 4) [22]. By mixing high catalytic loadings of Cu(OTf)2, PPh3, and TMS-protected diarylprolinol L4, the conjugate addition of Et2Zn or Me2Zn to various β-substituted enals proved to be highly enantioselective (ee up to 96%), but moderate to good 1,4:1,2 ratios were obtained (51:49 to 97:3). Of note, chiral phosphines were also screened, but without any improvement of selectivity. Furthermore, this methodology was then efficiently applied to the total synthesis of several bisabolane sesquiterpenes, which exhibited anticancer and antimicrobial activities or are employed as ingredients in perfumes and cosmetics (Scheme 4).
![[1860-5397-16-24-i4]](/bjoc/content/inline/1860-5397-16-24-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Combination of copper and amino catalysis for enantioselective β-functionalizations of enals.
Scheme 4: Combination of copper and amino catalysis for enantioselective β-functionalizations of enals.
The last report on ECAs of enals [23] was disclosed in 2016 by Alexakis and co-workers [24]. They achieved to develop three sets of optimized conditions for the CuTC-catalyzed conjugate addition of diorganozinc compounds, Grignard, and triorganoaluminium reagents to α,β-unsaturated aldehydes (Scheme 5). With diethyl- and dimethylzinc, and in the presence of the most efficient chiral ligand (R)-H8-BINAP (L7), moderate to excellent regioselectivities (1,4:1,2 ratios up to 100:0) were observed, and the desired 1,4-products were formed with remarkable enantioselectivities (58 to 96% ee). With Grignard reagents, the best ee values (45 to 90%) were obtained with (R)-TolBINAP (L3), but despite the presence of TMSCl, the regioselectivities remained modest, with a highest 1,4:1,2 ratio of 85:15. At last, (R)-SEGPHOS (L8) promoted the conjugate addition of Me3Al to cinnamaldehyde, with a remarkable 96% ee and a moderate 1,4:1,2 ratio. However, albeit no trace of aldol byproduct was detected, the reaction was incomplete (66% conversion). The use of TMSCl improved the conversion to 88%, but this was detrimental to the enantiocontrol (8% ee). These methodologies were applied to the straightforward synthesis of valuable (R)-citronellal and (S)-Florhydral®, which were obtained with excellent enantioselectivities (87 and 96%, respectively). However, moderate to significant amounts of the 1,2-products were also formed.
![[1860-5397-16-24-i5]](/bjoc/content/inline/1860-5397-16-24-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Optimized conditions for the Cu ECAs of R2Zn, RMgBr, and AlMe3 with α,β-unsaturated aldehydes.
Scheme 5: Optimized conditions for the Cu ECAs of R2Zn, RMgBr, and AlMe3 with α,β-unsaturated aldehydes.
As highlighted by these pioneering works, the direct copper-catalyzed conjugate addition of organometallic reagents to α,β-unsaturated aldehydes still remains an important challenge. Albeit some promising excellent regioselectivities and high enantioselectivities were achieved, this was often limited to a few organometallic reagents/enal substrates, as mentioned above. In that respect, indirect pathways were developed as alternative strategies, involving electron-deficient functions that can subsequently easily be converted to aldehydes.
α,β-Unsaturated thioesters
In 2005, Feringa, Minnaard, and co-workers were the first to report the ECA of Grignard reagents to α,β-unsaturated thioesters [25]. Advantageously, the latter were also readily accessible but significantly more reactive than α,β-unsaturated esters. Indeed, the thioester fragments featured a reduced electron delocalization compared to oxoesters, which resulted in a higher reactivity in conjugate additions, even with the less reactive MeMgBr. As depicted in Scheme 6, excellent yields and remarkable enantioselectivities (up to 96%) were obtained in ECAs of linear aliphatic Grignard reagents (in particular MeMgBr) to a wide range of substrates, catalyzed by CuBr∙SMe2/(R,S)-Josiphos (L9).
![[1860-5397-16-24-i6]](/bjoc/content/inline/1860-5397-16-24-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: CuECA of Grignard reagents to α,β-unsaturated thioesters and their application in the asymmetric total synthesis of (–)-lardolure.
Scheme 6: CuECA of Grignard reagents to α,β-unsaturated thioesters and their application in the asymmetric to...
However, the catalytic system was poorly selective toward sterically hindered organomagnesium nucleophiles (15–25% ee). The synthetic versatility of the thioester function was illustrated in the synthesis of (−)-lardolure (26% overall yield over 12 steps) via a relevant diastereoselective and enantioselective iterative route, affording the highly desirable deoxypropionate moiety in high 97% de. The Josiphos (L9)/CuBr∙SMe2 catalytic system was also efficient to promote the ECAs of MeMgBr to the less reactive aromatic α,β-unsaturated thioesters (ee values up to >99%) [26]. In order to extend their methodology to less reactive bulky Grignard reagents and/or substrates, a catalytic system of wider application, involving (S)-TolBINAP (ent-L3)/CuI was developed by the same authors [26]. The expected 1,4-products were isolated in good yields and moderate to excellent enantioselectivities (up to 99% ee, Scheme 7), depending on the steric hindrance of the reagent. Unfortunately, the addition of PhMgBr remained unsuccessful. This powerful catalytic protocol was illustrated by Bates and Sridhar in the enantioselective total synthesis of (−)-mintlactone [27]. The key step, furnishing the β-methylated thioester 8j, was accomplished in a good yield of 84% and a high ee of 94% (Scheme 7).
![[1860-5397-16-24-i7]](/bjoc/content/inline/1860-5397-16-24-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Improved Cu ECA of Grignard reagents to α,β-unsaturated thioesters, and their application in the asymmetric total synthesis of (−)-mintlactone.
Scheme 7: Improved Cu ECA of Grignard reagents to α,β-unsaturated thioesters, and their application in the as...
In 2008, Feringa and Minnaard evaluated the ECA of Grignard reagents to γ-substituted α,β-unsaturated thioesters that could lead to vicinal (i.e., 1,2-relation) dialkyl arrays, a highly desirable moiety that is ubiquitous in a wide range of natural products [28]. As depicted in Scheme 8, TolBINAP (L3)/CuI, which appeared to be a better catalytic system than Josiphos (L9)/CuBr∙SMe2, afforded either the syn or anti 1,4 product 13 in good isolated yields and excellent diastereoselectivities and enantioselectivities (dr up to 99:1 and ee up to >99.5). The value of the protocol was successfully illustrated through the enantioselective total syntheses of (−)-lasiol and (+)-faranal, two useful natural pheromones.
![[1860-5397-16-24-i8]](/bjoc/content/inline/1860-5397-16-24-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Catalytic enantioselective synthesis of vicinal dialkyl arrays via Cu ECA of Grignard reagents to γ-substituted α,β-unsaturated thioesters.
Scheme 8: Catalytic enantioselective synthesis of vicinal dialkyl arrays via Cu ECA of Grignard reagents to γ...
Shortly after, Feringa, Minnaard, and co-workers demonstrated the efficiency of (S,R)-reversed Josiphos (L10) in the copper-catalyzed 1,6-ECA of MeMgBr to α,β,γ,δ-bisunsaturated thioesters [29,30]. The expected 1,6-products were selectively formed (the 1,6:1,4 ratio ranged from 85:15 to 99:1) in high yields (78–88%) and good enantioselectivities (82–89%, Scheme 9). It is worth to note that this protocol failed in the case of linear dienoates [29]. Interestingly, after a subsequent reconjugation step in the presence of DBU, the resulting enantioenriched γ-methylated α,β-unsaturated thioester 18a was subsequently reacted in a 1,4-ECA reaction catalyzed by Josiphos (L9)/CuBr·SMe2. Using both enantiomers of the chiral ligand, either anti- or syn-1,3-deoxypropionate units were produced in good yields and excellent enantioselectivities (85–92% ee). Furthermore, an iterative procedure was also performed leading to all-syn or anti/syn-5,7,9-stereotriads, with high yields and stereoselectivity. This methodology was also tested on linear polyenic thioesters [9]. The challenging 1,8- and 1,10-products 21a/b were obtained, but the stereoselectivity dropped when the distance between the reacting olefin and the ester function was increased (1,8-ECA 72% ee; 1,10-ECA 45% ee). However, the regioselectivity (59–86%) and yield (44–63%) remained decent.
![[1860-5397-16-24-i9]](/bjoc/content/inline/1860-5397-16-24-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: 1,6-Cu ECA of MeMgBr to α,β,γ,δ-bisunsaturated thioesters: an iterative approach to deoxypropionate units.
Scheme 9: 1,6-Cu ECA of MeMgBr to α,β,γ,δ-bisunsaturated thioesters: an iterative approach to deoxypropionate...
The efficiency of TolBINAP (L3)/CuI was also demonstrated in the ECA of Grignard reagents to the 4-chloro-α,β-unsaturated thioester 22 [31]. Interestingly, the presence of the internal chloro leaving group allowed a powerful tandem conjugate addition–enolate trapping that led to valuable trans-1-alkyl-2-substituted cyclopropanes (Scheme 10). Various Grignard reagents were used, affording the corresponding cyclopropanes in moderate to high yields (50–92%) and good to excellent ee values (70–96%), except for PhMgBr (26% ee).
![[1860-5397-16-24-i10]](/bjoc/content/inline/1860-5397-16-24-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Tandem Cu ECA/intramolecular enolate trapping involving 4-chloro-α,β-unsaturated thioester 22.
Scheme 10: Tandem Cu ECA/intramolecular enolate trapping involving 4-chloro-α,β-unsaturated thioester 22.
In 2010, Hall and Lee described a successful synthesis of enantioenriched boronate derivatives through catalytic ECA of Grignard reagents to 3-boronyl α,β-unsaturated thioesters (Scheme 11) [32]. By applying an L3/CuI catalytic protocol previously developed by Feringa and Minnaard, MeMgBr and a range of aromatic Grignard reagents were selectively introduced, leading to the expected 1,4-products in high yields (50–82%) and ee values (82–98%). Unfortunately, ortho-substituted aromatic or hindered alkenyl reagents led to the corresponding products without showing any enantioselectivity.
![[1860-5397-16-24-i11]](/bjoc/content/inline/1860-5397-16-24-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Cu ECA of Grignard reagents to 3-boronyl α,β-unsaturated thioesters.
Scheme 11: Cu ECA of Grignard reagents to 3-boronyl α,β-unsaturated thioesters.
Very recently, Fletcher and Gao reported the first copper-catalyzed ECA of alkylzirconium reagents to α,β-unsaturated thioesters [33]. Starting from diversely functionalized alkenes, the resulting hydrozirconated adducts were reacted with various β-substituted Michael acceptors in the presence of CuCl and the chiral phosphoramidite L11 (Scheme 12). Remarkably, the corresponding 1,4-products were isolated in moderate to good yields (around 70%) and up to 99% ee. The high versatility of the protocol was illustrated by the synthesis of commercially relevant fragrances (phenoxanol and hydroxycitronellal). Additionally, an efficient iterative route was also described, allowing to produce the highly functionalized deoxypropionate fragment 30 in good overall yields and excellent stereocontrol for all stereogenic centers (up to 98:2 dr).
![[1860-5397-16-24-i12]](/bjoc/content/inline/1860-5397-16-24-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Cu ECA of alkylzirconium reagents to α,β-unsaturated thioesters.
Scheme 12: Cu ECA of alkylzirconium reagents to α,β-unsaturated thioesters.
α,β-Unsaturated acylimidazoles
The pioneering and successful use of α,β-unsaturated acylimidazoles as Michael acceptors in enantioselective catalysis was reported by Evans and co-workers in 2005 [34]. The selected asymmetric transformation was the Friedel–Crafts 1,4-addition involving indole derivatives as nucleophiles, catalyzed by a scandium(III) triflate complex with chiral bis(oxazolinyl)pyridine ligands. As highlighted by Evans, the acylimidazole moiety constituted a privileged surrogate of esters, amides, ketones, and aldehydes. Indeed, this peculiar function, which was readily accessible from the corresponding aldehydes or Weinreb amides, could be efficiently converted into a wide range of carbonyl derivatives, as depicted in Scheme 13.
![[1860-5397-16-24-i13]](/bjoc/content/inline/1860-5397-16-24-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Conversion of acylimidazoles into aldehydes, ketones, acids, esters, amides, and amines.
Scheme 13: Conversion of acylimidazoles into aldehydes, ketones, acids, esters, amides, and amines.
The successful use of α,β-unsaturated acylimidazole in Cu ECAs using organometallic reagents has been introduced very recently. Pioneering works in this field were published in 2011 by Roelfes, Liskamp, and co-workers, with the 1,4-addition of dimethyl malonate to cinnamyl 2-acyl-1-methylimidazole (31). Unfortunately, in the presence of Cu(NO3)2 and the triazacyclophane-based ligand L12, the product was obtained in a good yield of 90%, but a poor enantioselectivity (24% ee) was observed (Scheme 14) [35].
![[1860-5397-16-24-i14]](/bjoc/content/inline/1860-5397-16-24-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Cu ECA of dimethyl malonate to α,β-unsaturated acylimidazole 31 with triazacyclophane-based ligand L12.
Scheme 14: Cu ECA of dimethyl malonate to α,β-unsaturated acylimidazole 31 with triazacyclophane-based ligand ...
In 2012, Sawamura and co-workers described the first highly enantioselective copper-catalyzed conjugate addition of alkyl boranes to α,β-unsaturated 2-acyl-1-methylbenzimidazoles 33 [36]. Based on a previous study dealing with the CuCl/IMes-catalyzed addition of various alkylated 9BBN derivatives [37], the authors screened a set of various chiral NHC precursors. The imidazolium compound L13 appeared to be the most efficient one, affording the desired 1,4 products in high yields (57 to 93%) and excellent ee values (up to 93%, Scheme 15). Advantageously, this methodology was highly tolerant towards a wide range of functional groups, whether on alkylboranes or on substrates.
![[1860-5397-16-24-i15]](/bjoc/content/inline/1860-5397-16-24-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Cu/L13-catalyzed ECA of alkylboranes to α,β-unsaturated acylimidazoles.
Scheme 15: Cu/L13-catalyzed ECA of alkylboranes to α,β-unsaturated acylimidazoles.
In 2015, Mauduit, Campagne, and co-workers set up a highly enantioselective 1,4-addition of dimethylzinc to a wide scope of α,β-unsaturated acylimidazoles 35 [38]. Among the various ligands screened in combination with copper(II) triflate, the hydroxyalkyl-chelating NHC precursor L14 proved to be the most efficient one, giving the 1,4 product with moderate to good yields (34–86%) and excellent enantioinduction (86 to 95% ee, Scheme 16). The methodology was successfully applied to various extended Michael acceptor systems (dienic or trienic acylimidazoles), leading preferably to the corresponding 1,4 products in moderate to good yields (28–85%) with remarkable regio- (>95%) and enantioselectivities (91–95% ee) [39]. Interestingly, DFT calculations supported the crucial role of the imidazole moiety towards the 1,4-addition (vs 1,6 or 1,8) [39].
![[1860-5397-16-24-i16]](/bjoc/content/inline/1860-5397-16-24-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Cu/hydroxyalkyl-NHC-catalyzed ECA of dimethylzinc to α,β-unsaturated acylimidazoles.
Scheme 16: Cu/hydroxyalkyl-NHC-catalyzed ECA of dimethylzinc to α,β-unsaturated acylimidazoles.
Thanks to the efficient post-transformation of the acylimidazole function, the synthetic potential of this methodology was illustrated in the synthesis of relevant molecules, such as a ionone derivative, (+)-ar-turmerone, and (+)-Florhydral®, which were formed in good yields, without alteration of their optical purity [38,39].
Moreover, an iterative Cu ECA process allowing the selective introduction of a second methyl stereogenic center was then explored to develop a straightforward access to 1,3-deoxypropionate units, a scaffold ubiquitous in numerous natural products (Scheme 17) [40]. Starting from enantioenriched β-methylated aldehyde 37, the regeneration of the α,β-unsaturated 2-acyl-1-methylimidazole moiety was performed in high yield and E/Z selectivity via a two-step protocol. The resulting Michael acceptor was then engaged in an ECA to afford the expected 1,3-dimethyl product in 69% yield and a good diastereomeric excess of 94% (Scheme 17). Following this iterative methodology, the synthesis of 3,5,7-all-syn- and anti,anti-stereotriads 40a/b were successfully achieved in high diastereomeric ratios (up to >95:5) and good overall yields from (R)- or (S)-citronellal.
![[1860-5397-16-24-i17]](/bjoc/content/inline/1860-5397-16-24-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Stereocontrolled synthesis of 3,5,7-all-syn and anti,anti-stereotriads via iterative Cu ECAs.
Scheme 17: Stereocontrolled synthesis of 3,5,7-all-syn and anti,anti-stereotriads via iterative Cu ECAs.
More recently, Mauduit, Campagne, and co-workers reported an efficient Cu/Taniaphos-catalyzed β-borylation of an α,β-unsaturated acylimidazole, leading to various enantioenriched β-hydroxy products after oxidation (up to >98% ee) [41]. Interestingly, following the aforementioned iterative ECA strategy, the postfunctionalized chiral acylimidazole 41 derived from (S)-citronellal was efficiently converted to highly desirable anti,syn- and anti,anti-3,5,7-(Me,OR,Me)-substituted products 42a/b, which were isolated in good yields and excellent diastereomeric ratios (up to >95:5, Scheme 18).
![[1860-5397-16-24-i18]](/bjoc/content/inline/1860-5397-16-24-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Stereocontrolled synthesis of anti,syn- and anti,anti-3,5,7-(Me,OR,Me) units via iterative Cu ECA/BCA.
Scheme 18: Stereocontrolled synthesis of anti,syn- and anti,anti-3,5,7-(Me,OR,Me) units via iterative Cu ECA/B...
α,β-Unsaturated N-acyloxazolidinone, N-acylpyrrolidinone, and amide derivatives
Similar to acylimidazole Michael acceptors, the first use of α,β-unsaturated N-acyloxazolidinones was also described in asymmetric Friedel–Crafts 1,4-additions catalyzed by chiral copper/bisoxazolidine Lewis acids [42-46]. Thanks to the easy post-transformation of the oxazolidine moiety, the resulting enantioenriched products (up to 99% ee) were successfully converted to relevant molecules, such as trans-whisky lactone [43].
In 2003, Hoveyda and Hird reported the first 1,4-addition of alkylmetal nucleophiles to α,β-unsaturated N-acyloxazolidinones (Scheme 19) [47]. The chiral triamidophosphane ligand L15a as a copper(I) triflate complex efficiently promoted the catalytic conjugate addition of dialkylzinc species to various N-acyloxazolidinone Michael acceptors, in most of the cases with high isolated yields (61 to 95%) and excellent enantioselectivities (up to >98%). Furthermore, the resulting enantioenriched β-alkylated N-acyloxazolidinones could be converted to various derivatives (aldehydes, ketones, Weinreb amides, or carboxylic acids) in good yields and without alteration of the ee values. In 2006, aminohydroxyphosphine L15b was used as a new designer ligand by Nakamura and co-workers for the addition of diethylzinc to crotonyl N-acyloxazolidinone [48]. The 1,4-product was also formed in high enantioselectivity (>98% ee) and high yield (91%).
![[1860-5397-16-24-i19]](/bjoc/content/inline/1860-5397-16-24-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Cu-catalyzed ECA of dialkylzinc reagents to α,β-unsaturated N-acyloxazolidinones.
Scheme 19: Cu-catalyzed ECA of dialkylzinc reagents to α,β-unsaturated N-acyloxazolidinones.
The same year, Pineschi et al. evaluated various α,β-unsaturated acyl derivatives for the copper/(R,S,S)-phosphoramidite L16-catalyzed addition of diethylzinc (Scheme 20) [49]. Although the Michael acceptor bearing the N-acyloxazolidinone moiety that was successfully used by Hoveyda (95% ee) gave a lower enantioselectivity (64%), a more satisfactory enantiocontrol was obtained with the substrate having a 2-pyrrolidinone fragment (87% ee). The scope was then extended to various α,β-unsaturated N-acyl-2-pyrrolidinones and dialkylzinc reagents, leading to the corresponding 1,4-products in low to good yields (7–88%), with good to excellent enantioinduction (60 to >99% ee). Trimethylaluminium reagents were also investigated, but unfortunately, only low ee values were observed (20–36% ee), whereas no reactivity was observed with α,β-unsaturated amides.
![[1860-5397-16-24-i20]](/bjoc/content/inline/1860-5397-16-24-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Cu/phosphoramidite L16-catalyzed ECA of dialkylzincs to α,β-unsaturated N-acyl-2-pyrrolidinones.
Scheme 20: Cu/phosphoramidite L16-catalyzed ECA of dialkylzincs to α,β-unsaturated N-acyl-2-pyrrolidinones.
Although Pineschi’s conditions were ineffective for amides [49], Harutyunyan and co-workers achieved an important breakthrough by reporting the first enantioselective alkylation of α,β-unsaturated amides [50]. Indeed, due to their poor reactivity compared to other Michael acceptors, catalytic asymmetric conjugate additions of organometallic reagents to N,N-dialkylenamides remained a real challenge. However, thanks to the synergistic action of the boron-based Lewis acid BF3∙Et2O, CuBr∙SMe2, and the chiral (R,S)-Josiphos ligand (L9), an efficient (yields up to 86%) and highly regio- and enantioselective (ee values up to 99%) protocol was developed for 1,4-additions of various Grignard reagents to a wide scope of substrates (Scheme 21). Notably, the introduction of methyl and functionalized alkyl groups was performed with remarkable stereoselectivity (97–99%). Furthermore, this catalytic system was easily upscalable (up to 10 g), and the chiral catalyst could be recycled without any loss of efficiency. Unfortunately, although a wide range of Grignard reagents led to excellent results, PhMgBr provided low conversion and the racemic 1,4-product. Additionally, amide substrates featuring a bis(para-methoxybenzyl) moiety could be converted into relevant β-alkyl-substituted chiral amines, ubiquitous in numerous pharmaceutical ingredients, such as 52, a direct precursor of a drug candidate. Moreover, tandem ECA/enolate trapping was also studied, providing the trans-cyclopentane product 56 as a single diastereoisomer (92% ee).
![[1860-5397-16-24-i21]](/bjoc/content/inline/1860-5397-16-24-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Cu/(R,S)-Josiphos (L9)-catalyzed ECA of Grignard reagents to α,β-unsaturated amides.
Scheme 21: Cu/(R,S)-Josiphos (L9)-catalyzed ECA of Grignard reagents to α,β-unsaturated amides.
In 2018, the same authors reported the 1,6- and 1,4-additions of various Grignard reagents to a wide scope of conjugated dienyl amides (Scheme 22) [51]. Interestingly, the authors observed that the regioselectivity was directed by the substituent in the δ-position of the substrate: dienic amides featuring linear or functionalized aliphatic substituents in the δ-position led predominantly to 1,6-products, whereas those featuring electron-rich and electron-poor aromatic, heteroaromatic, and branched aliphatic substituents in the δ-position afforded preferably the 1,4-products. Importantly, when the morpholine moiety was used as N-substituent, the addition of diethylzinc to the enamide afforded the 1,6-addition product with 78% isolated yield and 91% ee. It is worth to underline that the morpholine group could easily allow further postfunctionalizations. Furthermore, thanks to the highly 1,6-enantioselective additions of methylmagnesium bromide (95% ee), this methodology was applied to the synthesis of the natural product penicillenol A by oxidative cleavage of the resulting 1,6 product 59c, affording the key synthon 60 with a slight erosion of the optical purity (Scheme 22).
![[1860-5397-16-24-i22]](/bjoc/content/inline/1860-5397-16-24-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Cu/Josiphos (L9)-catalyzed ECA of Grignard reagents to polyunsaturated amides.
Scheme 22: Cu/Josiphos (L9)-catalyzed ECA of Grignard reagents to polyunsaturated amides.
α,β-Unsaturated N-acylpyrroles
α,β-Unsaturated N-acylpyrroles were also investigated as Michael acceptors in enantioselective conjugate additions using organometallic reagents. In 2013, Endo and Shibata described a catalytic system based on multinuclear copper/aluminium complexes and phenolphosphine-based ligands L17a/b and L18, allowing the selective 1,4-addition of trimethylaluminium to three α,β-unsaturated N-acylpyrroles with moderate to good yields (54 to 87%) and excellent enantioselectivities (94 to 97% ee, Scheme 23) [52]. This methodology was successfully applied to the synthesis of various natural molecules, such as (S)-Florhydral® and (S)-(+)-ar-turmerone or key intermediates in the synthesis of 8-deoxyanisatin and frondosin.
![[1860-5397-16-24-i23]](/bjoc/content/inline/1860-5397-16-24-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Cu-catalyzed ECA of trimethylaluminium to N-acylpyrrole derivatives.
Scheme 23: Cu-catalyzed ECA of trimethylaluminium to N-acylpyrrole derivatives.
Conclusion
The enantioselective Cu-catalyzed conjugate addition of organometallic reagents to Michael acceptors has been extensively studied for many decades and led to remarkable results. However, for a long time, some classes of Michael acceptors (α,β-unsaturated aldehydes, thioesters, acylimidazoles, N-acyloxazolidinone, N-acylpyrrolidinone, amides, N-acylpyrroles) have been neglected to varying degrees, probably owing to their particular reactivity, which led to less impressive results. Nevertheless, these substrates present a high potential in total synthesis, since the chiral products can be easily transformed into various natural compounds. For example, the aldehyde function, which is directly obtained from α,β-unsaturated aldehydes or is accessible through postderivatization of either acylimidazole or thioester functions, is present in many natural compounds and is also a key functional group for many synthetic strategies. Furthermore, some of the functional groups listed above can be converted into various other synthetically useful groups, such as ketones, esters, carboxylic acids, and (Weinreb) amide groups.
More recently, some works were reported, which showed that through a judicious selection of chiral ligands and a fine-tuning of the reactivities of both partners, interesting selectivities could be reached with these more challenging electron-deficient alkenes, including, in some cases, di- or trienic acceptors, which accordingly extends the scope and the synthetic applicability of the method. The potential of the methodology has been illustrated through the efficient conversion of some 1,4-products into various chiral natural products. In addition, iterative procedures leading to chiral 1,3,5-(Me,Me,Me) and 1,3,5-(Me,OH,Me) motifs in a stereocontrolled way were successfully applied from α,β-unsaturated acylimidazoles and α,β-unsaturated thioesters, and thus opening a new field for the total synthesis of natural products. We hope that the results collected in this review will encourage chemists on one hand to continue the search for improved procedures combining simple, easily accessible chiral ligands, lower catalytic loadings, high ee values and productivity, and a wide scope, and on the other hand, to include this highly promising methodology in many synthetic strategies.
References
-
Mauduit, M.; Baslé, O.; Clavier, H.; Crévisy, C.; Denicourt-Nowicki, A. Metal-Catalyzed Asymmetric Nucleophilic Addition to Electron-Deficient Alkenes. In Comprehensive Organic Synthesis, 2nd ed.; Molander, G. A.; Knochel, P., Eds.; Elsevier: Amsterdam, Netherlands, 2014; Vol. 4, pp 189–341. doi:10.1016/b978-0-08-097742-3.00406-7
Return to citation in text: [1] -
Alexakis, A.; Bäckvall, J. E.; Krause, N.; Pàmies, O.; Diéguez, M. Chem. Rev. 2008, 108, 2796–2823. doi:10.1021/cr0683515
Return to citation in text: [1] -
Harutyunyan, S. R.; den Hartog, T.; Geurts, K.; Minnaard, A. J.; Feringa, B. L. Chem. Rev. 2008, 108, 2824–2852. doi:10.1021/cr068424k
Return to citation in text: [1] -
Jerphagnon, T.; Pizzuti, M. G.; Minnaard, A. J.; Feringa, B. L. Chem. Soc. Rev. 2009, 38, 1039–1075. doi:10.1039/b816853a
Return to citation in text: [1] -
Alexakis, A.; Frutos, J.; Mangeney, P. Tetrahedron: Asymmetry 1993, 4, 2427–2430. doi:10.1016/s0957-4166(00)82216-8
Return to citation in text: [1] -
Martin, D.; Kehrli, S.; d'Augustin, M.; Clavier, H.; Mauduit, M.; Alexakis, A. J. Am. Chem. Soc. 2006, 128, 8416–8417. doi:10.1021/ja0629920
Return to citation in text: [1] -
Tissot, M.; Li, H.; Alexakis, A. Copper-catalyzed asymmetric conjugate addition and allylic substitution of organometallic reagents to extended multiple bond systems. In Copper catalyzed asymmetric synthesis; Alexakis, A.; Krause, N.; Woodward, S., Eds.; Wiley-VCH: Weinheim, Germany, 2014; pp 69–84. doi:10.1002/9783527664573.ch3
Return to citation in text: [1] -
Schmid, T. E.; Drissi-Amraoui, S.; Crévisy, C.; Baslé, O.; Mauduit, M. Beilstein J. Org. Chem. 2015, 11, 2418–2434. doi:10.3762/bjoc.11.263
Return to citation in text: [1] -
den Hartog, T.; Huang, Y.; Fañanás-Mastral, M.; Meuwese, A.; Rudolph, A.; Pérez, M.; Minnaard, A. J.; Feringa, B. L. ACS Catal. 2015, 5, 560–574. doi:10.1021/cs501297s
See for mechanistic considerations.
Return to citation in text: [1] [2] -
Calvo, B. C.; Buter, J.; Minnaard, A. J. Applications to the synthesis of natural products. In Copper-Catalyzed Asymmetric Synthesis; Alexakis, A.; Krause, N.; Woodward, S., Eds.; Wiley-VCH: Weinheim, Germany, 2014; pp 373–448. doi:10.1002/9783527664573.ch14
Return to citation in text: [1] [2] -
Jones, P.; Reddy, C. K.; Knochel, P. Tetrahedron 1998, 54, 1471–1490. doi:10.1016/s0040-4020(97)10382-9
See for previous results in copper-free conjugate addition of organometallic reagents to α,β-unsaturated aldehydes.
Return to citation in text: [1] -
Bräse, S.; Höfener, S. Angew. Chem., Int. Ed. 2005, 44, 7879–7881. doi:10.1002/anie.200501732
See for the first enantioselective copper-free conjugate addition of organometallic reagents to α,β-unsaturated aldehydes.
Return to citation in text: [1] -
Marshall, J. A.; Herold, M.; Eidam, H. S.; Eidam, P. Org. Lett. 2006, 8, 5505–5508. doi:10.1021/ol062154a
See for the first copper-catalyzed diastereoselective conjugate addition of organometallic reagents to α,β-unsaturated aldehydes.
Return to citation in text: [1] -
Palais, L.; Babel, L.; Quintard, A.; Belot, S.; Alexakis, A. Org. Lett. 2010, 12, 1988–1991. doi:10.1021/ol100441r
Return to citation in text: [1] -
Corey, E. J.; Boaz, N. W. Tetrahedron Lett. 1985, 26, 6015–6018. doi:10.1016/s0040-4039(00)95113-x
Return to citation in text: [1] -
Corey, E. J.; Boaz, N. W. Tetrahedron Lett. 1985, 26, 6019–6022. doi:10.1016/s0040-4039(00)95114-1
Return to citation in text: [1] -
Alexakis, A.; Berlan, J.; Besace, Y. Tetrahedron Lett. 1986, 27, 1047–1050. doi:10.1016/s0040-4039(86)80044-2
Return to citation in text: [1] -
Nakamura, E.; Matsuzawa, S.; Horiguchi, Y.; Kuwajima, I. Tetrahedron Lett. 1986, 27, 4029–4032. doi:10.1016/s0040-4039(00)84902-3
Return to citation in text: [1] -
Chuit, C.; Foulon, J. P.; Normant, J. F. Tetrahedron 1980, 36, 2305–2310. doi:10.1016/0040-4020(80)80126-8
Return to citation in text: [1] -
Gremaud, L.; Palais, L.; Alexakis, A. Chimia 2012, 66, 196–200. doi:10.2533/chimia.2012.196
Return to citation in text: [1] -
Quintard, A.; Alexakis, A. Adv. Synth. Catal. 2010, 352, 1856–1860. doi:10.1002/adsc.201000309
Return to citation in text: [1] -
Afewerki, S.; Breistein, P.; Pirttilä, K.; Deiana, L.; Dziedzic, P.; Ibrahem, I.; Córdova, A. Chem. – Eur. J. 2011, 17, 8784–8788. doi:10.1002/chem.201100756
Return to citation in text: [1] -
Fañanás-Mastral, M.; Feringa, B. L. J. Am. Chem. Soc. 2010, 132, 13152–13153. doi:10.1021/ja105585y
See for a relevant alternative copper catalytic process based on the in situ enantioselective allylic alkylation of α,β-unstaurated aldehydes leading to β-substituted aldehydes with high ee values (up to 94%).
Return to citation in text: [1] -
Goncalves-Contal, S.; Gremaud, L.; Palais, L.; Babel, L.; Alexakis, A. Synthesis 2016, 48, 3301–3308. doi:10.1055/s-0035-1562487
Return to citation in text: [1] -
Des Mazery, R.; Pullez, M.; López, F.; Harutyunyan, S. R.; Minnaard, A. J.; Feringa, B. L. J. Am. Chem. Soc. 2005, 127, 9966–9967. doi:10.1021/ja053020f
Return to citation in text: [1] -
Maciá Ruiz, B.; Geurts, K.; Fernández-Ibáñez, M. Á.; ter Horst, B.; Minnaard, A. J.; Feringa, B. L. Org. Lett. 2007, 9, 5123–5126. doi:10.1021/ol702425a
Return to citation in text: [1] [2] -
Bates, R. W.; Sridhar, S. J. Org. Chem. 2008, 73, 8104–8105. doi:10.1021/jo801433f
Return to citation in text: [1] -
van Zijl, A. W.; Szymanski, W.; López, F.; Minnaard, A. J.; Feringa, B. L. J. Org. Chem. 2008, 73, 6994–7002. doi:10.1021/jo8010649
Return to citation in text: [1] -
den Hartog, T.; Harutyunyan, S. R.; Font, D.; Minnaard, A. J.; Feringa, B. L. Angew. Chem., Int. Ed. 2008, 47, 398–401. doi:10.1002/anie.200703702
Return to citation in text: [1] [2] -
den Hartog, T.; Jan van Dijken, D.; Minnaard, A. J.; Feringa, B. L. Tetrahedron: Asymmetry 2010, 21, 1574–1584. doi:10.1016/j.tetasy.2010.04.057
Return to citation in text: [1] -
den Hartog, T.; Rudolph, A.; Maciá, B.; Minnaard, A. J.; Feringa, B. L. J. Am. Chem. Soc. 2010, 132, 14349–14351. doi:10.1021/ja105704m
Return to citation in text: [1] -
Lee, J. C. H.; Hall, D. G. J. Am. Chem. Soc. 2010, 132, 5544–5545. doi:10.1021/ja9104057
Return to citation in text: [1] -
Gao, Z.; Fletcher, S. P. Chem. Commun. 2017, 53, 10216–10219. doi:10.1039/c7cc05433e
Return to citation in text: [1] -
Evans, D. A.; Fandrick, K. R.; Song, H.-J. J. Am. Chem. Soc. 2005, 127, 8942–8943. doi:10.1021/ja052433d
Return to citation in text: [1] -
Bauke Albada, H.; Rosati, F.; Coquière, D.; Roelfes, G.; Liskamp, R. M. J. Eur. J. Org. Chem. 2011, 1714–1720. doi:10.1002/ejoc.201001522
Return to citation in text: [1] -
Yoshida, M.; Ohmiya, H.; Sawamura, M. J. Am. Chem. Soc. 2012, 134, 11896–11899. doi:10.1021/ja304481a
Return to citation in text: [1] -
Ohmiya, H.; Yoshida, M.; Sawamura, M. Org. Lett. 2011, 13, 482–485. doi:10.1021/ol102819k
Return to citation in text: [1] -
Drissi-Amraoui, S.; Morin, M. S. T.; Crévisy, C.; Baslé, O.; Marcia de Figueiredo, R.; Mauduit, M.; Campagne, J.-M. Angew. Chem., Int. Ed. 2015, 54, 11830–11834. doi:10.1002/anie.201506189
Return to citation in text: [1] [2] -
Drissi-Amraoui, S.; Schmid, T. E.; Lauberteaux, J.; Crévisy, C.; Baslé, O.; de Figueiredo, R. M.; Halbert, S.; Gérard, H.; Mauduit, M.; Campagne, J.-M. Adv. Synth. Catal. 2016, 358, 2519–2540. doi:10.1002/adsc.201600458
Return to citation in text: [1] [2] [3] -
ter Horst, B.; Feringa, B. L.; Minnaard, A. J. Chem. Commun. 2010, 46, 2535–2547. doi:10.1039/b926265b
See for a review on iterative ECA processes.
Return to citation in text: [1] -
Lauberteaux, J.; Crévisy, C.; Baslé, O.; de Figueiredo, R. M.; Mauduit, M.; Campagne, J.-M. Org. Lett. 2019, 21, 1872–1876. doi:10.1021/acs.orglett.9b00479
Return to citation in text: [1] -
Kitajima, H.; Katsuki, T. Synlett 1997, 568–570. doi:10.1055/s-1997-3235
Return to citation in text: [1] -
Nishikori, H.; Ito, K.; Katsuki, T. Tetrahedron: Asymmetry 1998, 9, 1165–1170. doi:10.1016/s0957-4166(98)00080-9
Return to citation in text: [1] [2] -
Evans, D. A.; Willis, M. C.; Johnston, J. N. Org. Lett. 1999, 1, 865–868. doi:10.1021/ol9901570
Return to citation in text: [1] -
Evans, D. A.; Rovis, T.; Johnson, J. S. Pure Appl. Chem. 1999, 71, 1407–1415. doi:10.1351/pac199971081407
Return to citation in text: [1] -
Evans, D. A.; Scheidt, K. A.; Johnston, J. N.; Willis, M. C. J. Am. Chem. Soc. 2001, 123, 4480–4491. doi:10.1021/ja010302g
Return to citation in text: [1] -
Hird, A. W.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 42, 1276–1279. doi:10.1002/anie.200390328
Return to citation in text: [1] -
Hajra, A.; Yoshikai, N.; Nakamura, E. Org. Lett. 2006, 8, 4153–4155. doi:10.1021/ol0618306
Return to citation in text: [1] -
Pineschi, M.; Del Moro, F.; Di Bussolo, V.; Macchia, F. Adv. Synth. Catal. 2006, 348, 301–304. doi:10.1002/adsc.200505309
Return to citation in text: [1] [2] -
Rodríguez-Fernández, M.; Yan, X.; Collados, J. F.; White, P. B.; Harutyunyan, S. R. J. Am. Chem. Soc. 2017, 139, 14224–14231. doi:10.1021/jacs.7b07344
Return to citation in text: [1] -
Guo, Y.; Kootstra, J.; Harutyunyan, S. R. Angew. Chem., Int. Ed. 2018, 57, 13547–13550. doi:10.1002/anie.201808392
Return to citation in text: [1] -
Endo, K.; Hamada, D.; Yakeishi, S.; Shibata, T. Angew. Chem., Int. Ed. 2013, 52, 606–610. doi:10.1002/anie.201206297
Return to citation in text: [1]
| 36. | Yoshida, M.; Ohmiya, H.; Sawamura, M. J. Am. Chem. Soc. 2012, 134, 11896–11899. doi:10.1021/ja304481a |
| 37. | Ohmiya, H.; Yoshida, M.; Sawamura, M. Org. Lett. 2011, 13, 482–485. doi:10.1021/ol102819k |
| 38. | Drissi-Amraoui, S.; Morin, M. S. T.; Crévisy, C.; Baslé, O.; Marcia de Figueiredo, R.; Mauduit, M.; Campagne, J.-M. Angew. Chem., Int. Ed. 2015, 54, 11830–11834. doi:10.1002/anie.201506189 |
| 1. | Mauduit, M.; Baslé, O.; Clavier, H.; Crévisy, C.; Denicourt-Nowicki, A. Metal-Catalyzed Asymmetric Nucleophilic Addition to Electron-Deficient Alkenes. In Comprehensive Organic Synthesis, 2nd ed.; Molander, G. A.; Knochel, P., Eds.; Elsevier: Amsterdam, Netherlands, 2014; Vol. 4, pp 189–341. doi:10.1016/b978-0-08-097742-3.00406-7 |
| 2. | Alexakis, A.; Bäckvall, J. E.; Krause, N.; Pàmies, O.; Diéguez, M. Chem. Rev. 2008, 108, 2796–2823. doi:10.1021/cr0683515 |
| 3. | Harutyunyan, S. R.; den Hartog, T.; Geurts, K.; Minnaard, A. J.; Feringa, B. L. Chem. Rev. 2008, 108, 2824–2852. doi:10.1021/cr068424k |
| 4. | Jerphagnon, T.; Pizzuti, M. G.; Minnaard, A. J.; Feringa, B. L. Chem. Soc. Rev. 2009, 38, 1039–1075. doi:10.1039/b816853a |
| 10. | Calvo, B. C.; Buter, J.; Minnaard, A. J. Applications to the synthesis of natural products. In Copper-Catalyzed Asymmetric Synthesis; Alexakis, A.; Krause, N.; Woodward, S., Eds.; Wiley-VCH: Weinheim, Germany, 2014; pp 373–448. doi:10.1002/9783527664573.ch14 |
| 25. | Des Mazery, R.; Pullez, M.; López, F.; Harutyunyan, S. R.; Minnaard, A. J.; Feringa, B. L. J. Am. Chem. Soc. 2005, 127, 9966–9967. doi:10.1021/ja053020f |
| 43. | Nishikori, H.; Ito, K.; Katsuki, T. Tetrahedron: Asymmetry 1998, 9, 1165–1170. doi:10.1016/s0957-4166(98)00080-9 |
| 7. | Tissot, M.; Li, H.; Alexakis, A. Copper-catalyzed asymmetric conjugate addition and allylic substitution of organometallic reagents to extended multiple bond systems. In Copper catalyzed asymmetric synthesis; Alexakis, A.; Krause, N.; Woodward, S., Eds.; Wiley-VCH: Weinheim, Germany, 2014; pp 69–84. doi:10.1002/9783527664573.ch3 |
| 8. | Schmid, T. E.; Drissi-Amraoui, S.; Crévisy, C.; Baslé, O.; Mauduit, M. Beilstein J. Org. Chem. 2015, 11, 2418–2434. doi:10.3762/bjoc.11.263 |
| 9. |
den Hartog, T.; Huang, Y.; Fañanás-Mastral, M.; Meuwese, A.; Rudolph, A.; Pérez, M.; Minnaard, A. J.; Feringa, B. L. ACS Catal. 2015, 5, 560–574. doi:10.1021/cs501297s
See for mechanistic considerations. |
| 26. | Maciá Ruiz, B.; Geurts, K.; Fernández-Ibáñez, M. Á.; ter Horst, B.; Minnaard, A. J.; Feringa, B. L. Org. Lett. 2007, 9, 5123–5126. doi:10.1021/ol702425a |
| 47. | Hird, A. W.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 42, 1276–1279. doi:10.1002/anie.200390328 |
| 6. | Martin, D.; Kehrli, S.; d'Augustin, M.; Clavier, H.; Mauduit, M.; Alexakis, A. J. Am. Chem. Soc. 2006, 128, 8416–8417. doi:10.1021/ja0629920 |
| 23. |
Fañanás-Mastral, M.; Feringa, B. L. J. Am. Chem. Soc. 2010, 132, 13152–13153. doi:10.1021/ja105585y
See for a relevant alternative copper catalytic process based on the in situ enantioselective allylic alkylation of α,β-unstaurated aldehydes leading to β-substituted aldehydes with high ee values (up to 94%). |
| 41. | Lauberteaux, J.; Crévisy, C.; Baslé, O.; de Figueiredo, R. M.; Mauduit, M.; Campagne, J.-M. Org. Lett. 2019, 21, 1872–1876. doi:10.1021/acs.orglett.9b00479 |
| 5. | Alexakis, A.; Frutos, J.; Mangeney, P. Tetrahedron: Asymmetry 1993, 4, 2427–2430. doi:10.1016/s0957-4166(00)82216-8 |
| 24. | Goncalves-Contal, S.; Gremaud, L.; Palais, L.; Babel, L.; Alexakis, A. Synthesis 2016, 48, 3301–3308. doi:10.1055/s-0035-1562487 |
| 42. | Kitajima, H.; Katsuki, T. Synlett 1997, 568–570. doi:10.1055/s-1997-3235 |
| 43. | Nishikori, H.; Ito, K.; Katsuki, T. Tetrahedron: Asymmetry 1998, 9, 1165–1170. doi:10.1016/s0957-4166(98)00080-9 |
| 44. | Evans, D. A.; Willis, M. C.; Johnston, J. N. Org. Lett. 1999, 1, 865–868. doi:10.1021/ol9901570 |
| 45. | Evans, D. A.; Rovis, T.; Johnson, J. S. Pure Appl. Chem. 1999, 71, 1407–1415. doi:10.1351/pac199971081407 |
| 46. | Evans, D. A.; Scheidt, K. A.; Johnston, J. N.; Willis, M. C. J. Am. Chem. Soc. 2001, 123, 4480–4491. doi:10.1021/ja010302g |
| 15. | Corey, E. J.; Boaz, N. W. Tetrahedron Lett. 1985, 26, 6015–6018. doi:10.1016/s0040-4039(00)95113-x |
| 16. | Corey, E. J.; Boaz, N. W. Tetrahedron Lett. 1985, 26, 6019–6022. doi:10.1016/s0040-4039(00)95114-1 |
| 17. | Alexakis, A.; Berlan, J.; Besace, Y. Tetrahedron Lett. 1986, 27, 1047–1050. doi:10.1016/s0040-4039(86)80044-2 |
| 18. | Nakamura, E.; Matsuzawa, S.; Horiguchi, Y.; Kuwajima, I. Tetrahedron Lett. 1986, 27, 4029–4032. doi:10.1016/s0040-4039(00)84902-3 |
| 19. | Chuit, C.; Foulon, J. P.; Normant, J. F. Tetrahedron 1980, 36, 2305–2310. doi:10.1016/0040-4020(80)80126-8 |
| 21. | Quintard, A.; Alexakis, A. Adv. Synth. Catal. 2010, 352, 1856–1860. doi:10.1002/adsc.201000309 |
| 38. | Drissi-Amraoui, S.; Morin, M. S. T.; Crévisy, C.; Baslé, O.; Marcia de Figueiredo, R.; Mauduit, M.; Campagne, J.-M. Angew. Chem., Int. Ed. 2015, 54, 11830–11834. doi:10.1002/anie.201506189 |
| 39. | Drissi-Amraoui, S.; Schmid, T. E.; Lauberteaux, J.; Crévisy, C.; Baslé, O.; de Figueiredo, R. M.; Halbert, S.; Gérard, H.; Mauduit, M.; Campagne, J.-M. Adv. Synth. Catal. 2016, 358, 2519–2540. doi:10.1002/adsc.201600458 |
| 14. | Palais, L.; Babel, L.; Quintard, A.; Belot, S.; Alexakis, A. Org. Lett. 2010, 12, 1988–1991. doi:10.1021/ol100441r |
| 22. | Afewerki, S.; Breistein, P.; Pirttilä, K.; Deiana, L.; Dziedzic, P.; Ibrahem, I.; Córdova, A. Chem. – Eur. J. 2011, 17, 8784–8788. doi:10.1002/chem.201100756 |
| 40. |
ter Horst, B.; Feringa, B. L.; Minnaard, A. J. Chem. Commun. 2010, 46, 2535–2547. doi:10.1039/b926265b
See for a review on iterative ECA processes. |
| 11. |
Jones, P.; Reddy, C. K.; Knochel, P. Tetrahedron 1998, 54, 1471–1490. doi:10.1016/s0040-4020(97)10382-9
See for previous results in copper-free conjugate addition of organometallic reagents to α,β-unsaturated aldehydes. |
| 12. |
Bräse, S.; Höfener, S. Angew. Chem., Int. Ed. 2005, 44, 7879–7881. doi:10.1002/anie.200501732
See for the first enantioselective copper-free conjugate addition of organometallic reagents to α,β-unsaturated aldehydes. |
| 13. |
Marshall, J. A.; Herold, M.; Eidam, H. S.; Eidam, P. Org. Lett. 2006, 8, 5505–5508. doi:10.1021/ol062154a
See for the first copper-catalyzed diastereoselective conjugate addition of organometallic reagents to α,β-unsaturated aldehydes. |
| 39. | Drissi-Amraoui, S.; Schmid, T. E.; Lauberteaux, J.; Crévisy, C.; Baslé, O.; de Figueiredo, R. M.; Halbert, S.; Gérard, H.; Mauduit, M.; Campagne, J.-M. Adv. Synth. Catal. 2016, 358, 2519–2540. doi:10.1002/adsc.201600458 |
| 10. | Calvo, B. C.; Buter, J.; Minnaard, A. J. Applications to the synthesis of natural products. In Copper-Catalyzed Asymmetric Synthesis; Alexakis, A.; Krause, N.; Woodward, S., Eds.; Wiley-VCH: Weinheim, Germany, 2014; pp 373–448. doi:10.1002/9783527664573.ch14 |
| 20. | Gremaud, L.; Palais, L.; Alexakis, A. Chimia 2012, 66, 196–200. doi:10.2533/chimia.2012.196 |
| 39. | Drissi-Amraoui, S.; Schmid, T. E.; Lauberteaux, J.; Crévisy, C.; Baslé, O.; de Figueiredo, R. M.; Halbert, S.; Gérard, H.; Mauduit, M.; Campagne, J.-M. Adv. Synth. Catal. 2016, 358, 2519–2540. doi:10.1002/adsc.201600458 |
| 28. | van Zijl, A. W.; Szymanski, W.; López, F.; Minnaard, A. J.; Feringa, B. L. J. Org. Chem. 2008, 73, 6994–7002. doi:10.1021/jo8010649 |
| 26. | Maciá Ruiz, B.; Geurts, K.; Fernández-Ibáñez, M. Á.; ter Horst, B.; Minnaard, A. J.; Feringa, B. L. Org. Lett. 2007, 9, 5123–5126. doi:10.1021/ol702425a |
| 48. | Hajra, A.; Yoshikai, N.; Nakamura, E. Org. Lett. 2006, 8, 4153–4155. doi:10.1021/ol0618306 |
| 27. | Bates, R. W.; Sridhar, S. J. Org. Chem. 2008, 73, 8104–8105. doi:10.1021/jo801433f |
| 49. | Pineschi, M.; Del Moro, F.; Di Bussolo, V.; Macchia, F. Adv. Synth. Catal. 2006, 348, 301–304. doi:10.1002/adsc.200505309 |
| 49. | Pineschi, M.; Del Moro, F.; Di Bussolo, V.; Macchia, F. Adv. Synth. Catal. 2006, 348, 301–304. doi:10.1002/adsc.200505309 |
| 34. | Evans, D. A.; Fandrick, K. R.; Song, H.-J. J. Am. Chem. Soc. 2005, 127, 8942–8943. doi:10.1021/ja052433d |
| 35. | Bauke Albada, H.; Rosati, F.; Coquière, D.; Roelfes, G.; Liskamp, R. M. J. Eur. J. Org. Chem. 2011, 1714–1720. doi:10.1002/ejoc.201001522 |
| 32. | Lee, J. C. H.; Hall, D. G. J. Am. Chem. Soc. 2010, 132, 5544–5545. doi:10.1021/ja9104057 |
| 33. | Gao, Z.; Fletcher, S. P. Chem. Commun. 2017, 53, 10216–10219. doi:10.1039/c7cc05433e |
| 9. |
den Hartog, T.; Huang, Y.; Fañanás-Mastral, M.; Meuwese, A.; Rudolph, A.; Pérez, M.; Minnaard, A. J.; Feringa, B. L. ACS Catal. 2015, 5, 560–574. doi:10.1021/cs501297s
See for mechanistic considerations. |
| 52. | Endo, K.; Hamada, D.; Yakeishi, S.; Shibata, T. Angew. Chem., Int. Ed. 2013, 52, 606–610. doi:10.1002/anie.201206297 |
| 31. | den Hartog, T.; Rudolph, A.; Maciá, B.; Minnaard, A. J.; Feringa, B. L. J. Am. Chem. Soc. 2010, 132, 14349–14351. doi:10.1021/ja105704m |
| 29. | den Hartog, T.; Harutyunyan, S. R.; Font, D.; Minnaard, A. J.; Feringa, B. L. Angew. Chem., Int. Ed. 2008, 47, 398–401. doi:10.1002/anie.200703702 |
| 30. | den Hartog, T.; Jan van Dijken, D.; Minnaard, A. J.; Feringa, B. L. Tetrahedron: Asymmetry 2010, 21, 1574–1584. doi:10.1016/j.tetasy.2010.04.057 |
| 50. | Rodríguez-Fernández, M.; Yan, X.; Collados, J. F.; White, P. B.; Harutyunyan, S. R. J. Am. Chem. Soc. 2017, 139, 14224–14231. doi:10.1021/jacs.7b07344 |
| 29. | den Hartog, T.; Harutyunyan, S. R.; Font, D.; Minnaard, A. J.; Feringa, B. L. Angew. Chem., Int. Ed. 2008, 47, 398–401. doi:10.1002/anie.200703702 |
| 51. | Guo, Y.; Kootstra, J.; Harutyunyan, S. R. Angew. Chem., Int. Ed. 2018, 57, 13547–13550. doi:10.1002/anie.201808392 |
© 2020 Pichon et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)