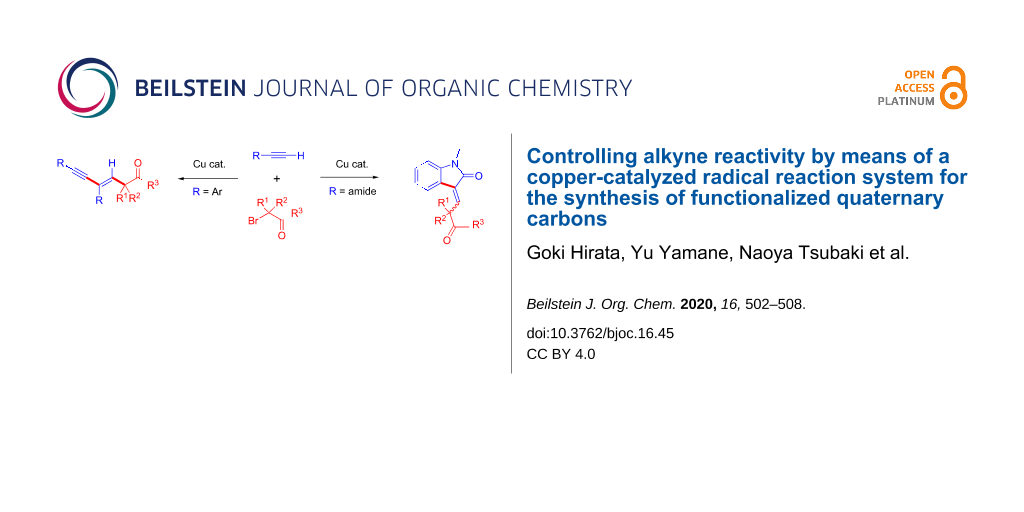
Abstract
A terminal alkyne is one of the most useful reactants for the synthesis of alkyne and alkene derivatives. Because an alkyne undergoes addition reaction at a C–C triple bond or cross-coupling at a terminal C–H bond. Combining those reaction patterns could realize a new reaction methodology to synthesize complex molecules including C–C multiple bonds. In this report, we found that the reaction of 3 equivalents of terminal alkyne 1 (aryl substituted alkyne) and an α-bromocarbonyl compound 2 (tertiary alkyl radical precursor) undergoes tandem alkyl radical addition/Sonogashira coupling to produce 1,3-enyne compound 3 possessing a quaternary carbon in the presence of a copper catalyst. Moreover, the reaction of α-bromocarbonyl compound 2 and an alkyne 4 possessing a carboxamide moiety undergoes tandem alkyl radical addition/C–H coupling to produce indolinone derivative 5.

Graphical Abstract
Introduction
Terminal alkynes are undoubtedly useful functional groups for organic synthesis, and they can undergo a variety of reactions [1]. The C–C triple bond of an alkyne is suitable for addition reactions, whereas the terminal hydrogen atom is a good target for cross-coupling by using Sonogashira and related coupling reactions [2-4]. Although there are many reports on alkyne transformations, one recent development in this area has been the reaction of alkynes with tertiary alkyl electrophiles to produce functionalized quaternary carbon atoms via addition [5-10] or coupling [11-16].
Recently, we have prepared quaternary carbon centers via radical reactions by using α-bromocarbonyl compounds (a tertiary alkyl source) and olefins or heteroatoms in the presence of a copper catalyst [17-19]. During our studies, we found that combinations of alkynes and tertiary alkyl radicals generated from the reaction of a copper catalyst and an α-bromocarbonyl compound can undergo i) Sonogashira type couplings via an alkynyl-Cu intermediate [20], ii) cis-hydro tertiary alkylations via 1-alkenyl-Cu [21], and iii) trans-hydro tertiary alkylations via atom-transfer radical addition (ATRA) [21] (Scheme 1, i–iii). Therefore, we postulated that if we could control the reactivities of the alkynyl–Cu and ATRA adducts, a tandem tertiary alkylation followed by an alkynylation could occur to produce a 1,3-enyne possessing a quaternary carbon center with good regio- and stereoselectivity (Scheme 1, this work). Similarly, Zhu’s group has reported that the reaction of an alkyne and an α-bromocarbonyl compound furnishes a highly functionalized 1,3-enyne compound via ATRA followed by an alkynylation reaction [22], but both Pd and Cu are required as catalysts in that case. Our methodology can realize a Pd-free catalyst system to prepare complex quaternary carbon atoms. Herein, we report the Cu-catalyzed control of the reactivity of an alkyne (addition and coupling) undergoing tandem tertiary alkylation and alkynylation to produce a 1,3-enyne containing a quaternary carbon center with good regio- and stereoselectivity.
![[1860-5397-16-45-i1]](/bjoc/content/inline/1860-5397-16-45-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
In our preliminary research, we tried various Cu salts, including CuI, CuBr, CuCl, CuOAc, and CuOTf, and ligands, including PPh3, 1,10-phenanthroline (1,10-Phen), N,N,N',N'',N''-pentamethyldiethylenetriamine, and tris(2-pyridylmethyl)amine, in the reaction of phenylacetylene (1a) and α-bromoester 2a. From these experiments, CuBr and 1,10-Phen acted as a good catalyst system for obtaining 1,3-enyne 3a in 62% yield with good regio- and stereoselectivity (Table 1, entry 1). On the basis of this preliminary result, we also tried other conditions. Toluene was very effective in our previous reaction system [17] but was not effective at all in this case (Table 1, entry 2). The reaction without NaI resulted in the formation of 3a-Br in 30% yield, instead of 3a (Table 1, entry 3). If KI was used instead of NaI, the yield of 3a decreased (Table 1, entry 4). We will discuss the proposed reaction mechanism later in the text, but the formation of 3a-I via ATRA could be important for the alkynylation reaction. Generally, the Sonogashira coupling requires both a Pd catalyst and a Cu co-catalyst [2-4]. However, couplings with terminal alkynes can be carried out in the absence of the Pd catalyst [23-32]; this is the so-called Castro–Stephens reaction [33]. The effect of the base was very important for producing the main product 3a (Table 1, entries 5–8). If the reaction was performed in the presence of a base other than Cs2CO3, a decreased yield of 3a was observed. Finally, an increased amount of catalyst was effective for obtaining the highest yield (Table 1, entry 9). The total yield was moderate, but the yields for each step of this two-step tandem reaction system (ATRA followed by Castro–Stephens coupling) should be over 80%.
Table 1: Optimization.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-45-i5.svg?max-width=637&scale=1.0)
|
||||
| entry | changes from standard conditions | 3a (%) | 3a-I (%) | 3a-Br (%) |
|---|---|---|---|---|
| 1 | none | 62 | 10 | trace |
| 2 | toluene instead of MeCN | trace | – | – |
| 3 | without NaI | <5 | 0 | 30 |
| 4 | KI instead of NaI | 30 | 7 | 7 |
| 5 | Hunig’s base instead of Cs2CO3 | trace | 60 | trace |
| 6 | iPr2NH instead of Cs2CO3 | trace | 65 | trace |
| 7 | K2CO3 instead of Cs2CO3 | 6 | 58 | trace |
| 8 | K3PO4 instead of Cs2CO3 | 26 | 20 | trace |
| 9 | 15 mol % CuBr and 30 mol % 1,10-Phen | 66(52)b | 7 | trace |
aConducted at 80 °C for 24 h in MeCN with CuBr (10 mol %), 1,10-Phen (20 mol %), Cs2CO3 (4.0 equiv), NaI (2.0 equiv), 1a (3.0 equiv) and 2a (1.0 equiv). Yields were determined by 1H NMR analysis. bGPC yield.
Under the optimized conditions, the reactivities of alkynes 1 and α-bromocarbonyl compounds 2 were examined (Figure 1). The two-step tandem alkyne transformation produced various 1,3-enynes 3 with concomitant formation of ATRA adducts as side-products. It was very difficult to separate the products by silica-gel column chromatography; therefore, we are reporting the 1H NMR and GPC yields of 3. (The pure products were obtained by gel permeation chromatography (GPC).) For example, α-bromocarbonyl compounds 2 possessing various degrees of steric bulkiness (ethyl groups) at the carbonyl α-position or a tert-butyl ester group reacted with 1a to produce 3b and 3c in moderate yields. Bromomalonate, bromolactone, and bromoketone derivatives resulted in the formation of 3d, 3e, and 3f in 40%, 53%, and 52% yield, respectively. Ortho and meta substituted arylalkynes 1 reacted with 2a to produce 3g and 3h, respectively. An arylalkyne 1 possessing an electron-withdrawing group (ester) yielded 3i without affecting the reactivity of 2. Sulfur functional groups tend to decrease the catalytic activity of copper salts, but thienyl-substituted alkyne 1 reacted with 2a to produce 3j in 48% yield.
![[1860-5397-16-45-1]](/bjoc/content/figures/1860-5397-16-45-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Substrate scope of 1 and 2. aConducted at 80 °C for 24 h in MeCN with CuBr (10 mol %), 1,10-Phen (20 mol %), Cs2CO3 (4.0 equiv), NaI (2.0 equiv), 1 (3.0 equiv) and 2 (1.0 equiv). Yields were determined by 1H NMR analysis.
Figure 1: Substrate scope of 1 and 2. aConducted at 80 °C for 24 h in MeCN with CuBr (10 mol %), 1,10-Phen (2...
Although the exact reaction mechanism is currently unclear, one possibility involves a radical pathway including cross-coupling with an alkynyl copper species (Scheme 2). After the generation of A, addition of A to 1 takes place to give the radical intermediate B. This then reacts with the Cu(II) species to produce intermediate C, with concomitant formation of a Cu(I) species. The brominated intermediate C undergoes a cross-coupling reaction with the alkynyl copper species to give the desired product 3. We have detected brominated intermediate C during the reaction. We have also examined the reaction between 3a-Br (intermediate C) and the alkynyl copper species (1a-Cu) (Scheme 3). The result showed that 3a-Br reacted with the alkynyl copper species to produce the desired product 3a in reasonable yield. This reaction should be a Sonogashira coupling without a Pd catalyst [23,24,30].
![[1860-5397-16-45-i2]](/bjoc/content/inline/1860-5397-16-45-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-45-i3]](/bjoc/content/inline/1860-5397-16-45-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Interestingly, if the reaction of 2a and electron-deficient alkyne 4a was performed under the conditions shown in Table 1, entry 9, the C–H cyclized product 5a was obtained instead of 3k. In this case, an alkyl radical addition followed by C–H cyclization via an alkenyl radical intermediate could be occur (Scheme 4). A Pd-catalyzed cascade reaction (C–C bond formation/C–H cyclization process) of N-arylpropynamide 4 for the preparation of indolinone derivatives 5 was previously reported by Li’s group [34,35]. In another report on C–H cyclization by Lei’s group, Ni-catalyzed aromatic C–H alkylation occurs via a radical reaction [36]. Both cases were helpful in our development of the current Cu-catalyzed cascade C–H cyclization system.
![[1860-5397-16-45-i4]](/bjoc/content/inline/1860-5397-16-45-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
After careful optimization, we found that CuI, 1,10-Phen, Cy2NMe as a base, and 1,4-dioxane were effective for obtaining the best yields of products 5 (Figure 2). In this examination, product isolation was difficult because of the formation of stereoisomers (E and Z stereoisomers of 5). The yields shown in Figure 2 were including isomers (We also put yield of pure E-5 as an NMR yield.). Although the chemical yields of 5 were moderate, the stereoselectivities in this reaction were good (the major stereoisomers of 5 produced were E). We tested compounds 2 possessing acyclic and cyclic structures and compounds 4 possessing MeO, F, and N-Et moieties as substrates for the reaction; however, no big differences were observed. The C–H cyclized products were not obtained from the reaction of compound 4 if it did not possess an alkyl substituent on the nitrogen atom of the amide bond.
![[1860-5397-16-45-2]](/bjoc/content/figures/1860-5397-16-45-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Substrate scope of 2 and 4. aConducted at 100 °C for 20 h in 1,4-dioxane with CuI (10 mol %), 1,10-Phen (10 mol %), Cy2NMe (1.0 equiv), 2 (1.0 equiv) and 4 (3.0 equiv). Yields were determined by 1H NMR analysis. bIncluding E and Z isomers.
Figure 2: Substrate scope of 2 and 4. aConducted at 100 °C for 20 h in 1,4-dioxane with CuI (10 mol %), 1,10-...
Conclusion
In summary, we have developed two types of tandem reactions catalyzed by a copper salt. The reaction of 2-bromocarbonyl compounds and aryl-substituted alkynes underwent alkyl radical addition at a C–C triple bond followed by Sonogashira coupling to produce 1,3-enyne compounds. On the other hand, the reaction with alkyne possessing a carboxamide moiety underwent tandem alkyl radical addition at the C–C triple bond followed by C–H coupling to produce indolinone derivatives. These results could suggest new aspects of alkyne transformations in a copper catalyzed alkyl radical reaction system.
Supporting Information
| Supporting Information File 1: Experimental procedures, compound characterization data, and NMR spectra. | ||
| Format: PDF | Size: 2.8 MB | Download |
References
-
Trost, B. M.; Li, C.-J. Modern Alkyne Chemistry: Catalytic and Atom- Economic Transformations; Wiley-VCH: Weinheim, Germany, 2015.
Return to citation in text: [1] -
Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874–922. doi:10.1021/cr050992x
Return to citation in text: [1] [2] -
Sonogashira, K.; Tohda, Y.; Hagihara, N. Tetrahedron Lett. 1975, 16, 4467–4470. doi:10.1016/s0040-4039(00)91094-3
Return to citation in text: [1] [2] -
Liu, W.; Li, L.; Li, C.-J. Nat. Commun. 2015, 6, 6526. doi:10.1038/ncomms7526
Return to citation in text: [1] [2] -
Liu, W.; Chen, Z.; Li, L.; Wang, H.; Li, C.-J. Chem. – Eur. J. 2016, 22, 5888–5893. doi:10.1002/chem.201600219
Return to citation in text: [1] -
Beringer, F. M.; Galton, S. A. J. Org. Chem. 1965, 30, 1930–1934. doi:10.1021/jo01017a053
Return to citation in text: [1] -
Ochiai, M.; Kunishima, M.; Nagao, Y.; Fuji, K.; Shiro, M.; Fujita, E. J. Am. Chem. Soc. 1986, 108, 8281–8283. doi:10.1021/ja00286a037
Return to citation in text: [1] -
Bachi, M. D.; Bar-Ner, N.; Crittell, C. M.; Stang, P. J.; Williamson, B. L. J. Org. Chem. 1991, 56, 3912–3915. doi:10.1021/jo00012a025
Return to citation in text: [1] -
Fernández González, D.; Brand, J. P.; Waser, J. Chem. – Eur. J. 2010, 16, 9457–9461. doi:10.1002/chem.201001539
Return to citation in text: [1] -
Brand, J. P.; Waser, J. Chem. Soc. Rev. 2012, 41, 4165–4179. doi:10.1039/c2cs35034c
Return to citation in text: [1] -
Dong, W.; Yuan, Y.; Gao, X.; Keranmu, M.; Li, W.; Xie, X.; Zhang, Z. Org. Lett. 2018, 20, 5762–5765. doi:10.1021/acs.orglett.8b02463
Return to citation in text: [1] -
Gao, Y.; Zhang, P.; Li, G.; Zhao, Y. J. Org. Chem. 2018, 83, 13726–13733. doi:10.1021/acs.joc.8b02001
Return to citation in text: [1] -
Liu, B.; Yu, J.-X.; Li, Y.; Li, J.-H.; He, D.-L. Org. Lett. 2018, 20, 2129–2132. doi:10.1021/acs.orglett.8b00236
Return to citation in text: [1] -
Gao, Y.; Zhang, P.; Ji, Z.; Tang, G.; Zhao, Y. ACS Catal. 2017, 7, 186–190. doi:10.1021/acscatal.6b03033
Return to citation in text: [1] -
Che, C.; Huang, Q.; Zheng, H.; Zhu, G. Chem. Sci. 2016, 7, 4134–4139. doi:10.1039/c5sc04980f
Return to citation in text: [1] -
Cheung, C. W.; Zhurkin, F. E.; Hu, X. J. Am. Chem. Soc. 2015, 137, 4932–4935. doi:10.1021/jacs.5b01784
Return to citation in text: [1] -
Nishikata, T.; Noda, Y.; Fujimoto, R.; Sakashita, T. J. Am. Chem. Soc. 2013, 135, 16372–16375. doi:10.1021/ja409661n
Return to citation in text: [1] [2] -
Nishikata, T.; Ishida, S.; Fujimoto, R. Angew. Chem., Int. Ed. 2016, 55, 10008–10012. doi:10.1002/anie.201603426
Return to citation in text: [1] -
Ishida, S.; Takeuchi, K.; Taniyama, N.; Sunada, Y.; Nishikata, T. Angew. Chem., Int. Ed. 2017, 56, 11610–11614. doi:10.1002/anie.201706293
Return to citation in text: [1] -
Yamane, Y.; Miwa, N.; Nishikata, T. ACS Catal. 2017, 7, 6872–6876. doi:10.1021/acscatal.7b02615
Return to citation in text: [1] -
Nakamura, K.; Nishikata, T. ACS Catal. 2017, 7, 1049–1052. doi:10.1021/acscatal.6b03343
Return to citation in text: [1] [2] -
Che, C.; Zheng, H.; Zhu, G. Org. Lett. 2015, 17, 1617–1620. doi:10.1021/acs.orglett.5b00546
Return to citation in text: [1] -
Okuro, K.; Furuune, M.; Enna, M.; Miura, M.; Nomura, M. J. Org. Chem. 1993, 58, 4716–4721. doi:10.1021/jo00069a040
Return to citation in text: [1] [2] -
Gujadhur, R. K.; Bates, C. G.; Venkataraman, D. Org. Lett. 2001, 3, 4315–4317. doi:10.1021/ol0170105
Return to citation in text: [1] [2] -
Thathagar, M. B.; Beckers, J.; Rothenberg, G. Green Chem. 2004, 6, 215–218. doi:10.1039/b401586j
Return to citation in text: [1] -
Ma, D.; Liu, F. Chem. Commun. 2004, 1934–1935. doi:10.1039/b407090a
Return to citation in text: [1] -
Wang, Y. F.; Deng, W.; Liu, L.; Guo, Q. X. Chin. Chem. Lett. 2005, 16, 1197–1200.
Return to citation in text: [1] -
Saejueng, P.; Bates, C. G.; Venkataraman, D. Synthesis 2005, 1706–1712. doi:10.1055/s-2005-869893
Return to citation in text: [1] -
Xie, Y.-X.; Deng, C.-L.; Pi, S.-F.; Li, J.-H.; Yin, D.-L. Chin. J. Chem. 2006, 24, 1290–1294. doi:10.1002/cjoc.200690240
Return to citation in text: [1] -
Li, J.-H.; Li, J.-L.; Wang, D.-P.; Pi, S.-F.; Xie, Y.-X.; Zhang, M.-B.; Hu, X.-C. J. Org. Chem. 2007, 72, 2053–2057. doi:10.1021/jo0623742
Return to citation in text: [1] [2] -
Tang, B.-X.; Wang, F.; Li, J.-H.; Xie, Y.-X.; Zhang, M.-B. J. Org. Chem. 2007, 72, 6294–6297. doi:10.1021/jo070538o
Return to citation in text: [1] -
Guan, J. T.; Yu, G.-A.; Chen, L.; Weng, T. Q.; Yuan, J. J.; Liu, S. H. Appl. Organomet. Chem. 2009, 23, 75–77. doi:10.1002/aoc.1474
Return to citation in text: [1] -
Stephens, R. D.; Castro, C. E. J. Org. Chem. 1963, 28, 3313–3315. doi:10.1021/jo01047a008
Return to citation in text: [1] -
Tang, S.; Peng, P.; Zhong, P.; Li, J.-H. J. Org. Chem. 2008, 73, 5476–5480. doi:10.1021/jo8008808
Return to citation in text: [1] -
Jiang, T.-S.; Tang, R.-Y.; Zhang, X.-G.; Li, X.-H.; Li, J.-H. J. Org. Chem. 2009, 74, 8834–8837. doi:10.1021/jo901963g
Return to citation in text: [1] -
Liu, C.; Liu, D.; Zhang, W.; Zhou, L.; Lei, A. Org. Lett. 2013, 15, 6166–6169. doi:10.1021/ol403021p
Return to citation in text: [1]
| 1. | Trost, B. M.; Li, C.-J. Modern Alkyne Chemistry: Catalytic and Atom- Economic Transformations; Wiley-VCH: Weinheim, Germany, 2015. |
| 17. | Nishikata, T.; Noda, Y.; Fujimoto, R.; Sakashita, T. J. Am. Chem. Soc. 2013, 135, 16372–16375. doi:10.1021/ja409661n |
| 18. | Nishikata, T.; Ishida, S.; Fujimoto, R. Angew. Chem., Int. Ed. 2016, 55, 10008–10012. doi:10.1002/anie.201603426 |
| 19. | Ishida, S.; Takeuchi, K.; Taniyama, N.; Sunada, Y.; Nishikata, T. Angew. Chem., Int. Ed. 2017, 56, 11610–11614. doi:10.1002/anie.201706293 |
| 34. | Tang, S.; Peng, P.; Zhong, P.; Li, J.-H. J. Org. Chem. 2008, 73, 5476–5480. doi:10.1021/jo8008808 |
| 35. | Jiang, T.-S.; Tang, R.-Y.; Zhang, X.-G.; Li, X.-H.; Li, J.-H. J. Org. Chem. 2009, 74, 8834–8837. doi:10.1021/jo901963g |
| 11. | Dong, W.; Yuan, Y.; Gao, X.; Keranmu, M.; Li, W.; Xie, X.; Zhang, Z. Org. Lett. 2018, 20, 5762–5765. doi:10.1021/acs.orglett.8b02463 |
| 12. | Gao, Y.; Zhang, P.; Li, G.; Zhao, Y. J. Org. Chem. 2018, 83, 13726–13733. doi:10.1021/acs.joc.8b02001 |
| 13. | Liu, B.; Yu, J.-X.; Li, Y.; Li, J.-H.; He, D.-L. Org. Lett. 2018, 20, 2129–2132. doi:10.1021/acs.orglett.8b00236 |
| 14. | Gao, Y.; Zhang, P.; Ji, Z.; Tang, G.; Zhao, Y. ACS Catal. 2017, 7, 186–190. doi:10.1021/acscatal.6b03033 |
| 15. | Che, C.; Huang, Q.; Zheng, H.; Zhu, G. Chem. Sci. 2016, 7, 4134–4139. doi:10.1039/c5sc04980f |
| 16. | Cheung, C. W.; Zhurkin, F. E.; Hu, X. J. Am. Chem. Soc. 2015, 137, 4932–4935. doi:10.1021/jacs.5b01784 |
| 36. | Liu, C.; Liu, D.; Zhang, W.; Zhou, L.; Lei, A. Org. Lett. 2013, 15, 6166–6169. doi:10.1021/ol403021p |
| 5. | Liu, W.; Chen, Z.; Li, L.; Wang, H.; Li, C.-J. Chem. – Eur. J. 2016, 22, 5888–5893. doi:10.1002/chem.201600219 |
| 6. | Beringer, F. M.; Galton, S. A. J. Org. Chem. 1965, 30, 1930–1934. doi:10.1021/jo01017a053 |
| 7. | Ochiai, M.; Kunishima, M.; Nagao, Y.; Fuji, K.; Shiro, M.; Fujita, E. J. Am. Chem. Soc. 1986, 108, 8281–8283. doi:10.1021/ja00286a037 |
| 8. | Bachi, M. D.; Bar-Ner, N.; Crittell, C. M.; Stang, P. J.; Williamson, B. L. J. Org. Chem. 1991, 56, 3912–3915. doi:10.1021/jo00012a025 |
| 9. | Fernández González, D.; Brand, J. P.; Waser, J. Chem. – Eur. J. 2010, 16, 9457–9461. doi:10.1002/chem.201001539 |
| 10. | Brand, J. P.; Waser, J. Chem. Soc. Rev. 2012, 41, 4165–4179. doi:10.1039/c2cs35034c |
| 33. | Stephens, R. D.; Castro, C. E. J. Org. Chem. 1963, 28, 3313–3315. doi:10.1021/jo01047a008 |
| 2. | Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874–922. doi:10.1021/cr050992x |
| 3. | Sonogashira, K.; Tohda, Y.; Hagihara, N. Tetrahedron Lett. 1975, 16, 4467–4470. doi:10.1016/s0040-4039(00)91094-3 |
| 4. | Liu, W.; Li, L.; Li, C.-J. Nat. Commun. 2015, 6, 6526. doi:10.1038/ncomms7526 |
| 23. | Okuro, K.; Furuune, M.; Enna, M.; Miura, M.; Nomura, M. J. Org. Chem. 1993, 58, 4716–4721. doi:10.1021/jo00069a040 |
| 24. | Gujadhur, R. K.; Bates, C. G.; Venkataraman, D. Org. Lett. 2001, 3, 4315–4317. doi:10.1021/ol0170105 |
| 30. | Li, J.-H.; Li, J.-L.; Wang, D.-P.; Pi, S.-F.; Xie, Y.-X.; Zhang, M.-B.; Hu, X.-C. J. Org. Chem. 2007, 72, 2053–2057. doi:10.1021/jo0623742 |
| 22. | Che, C.; Zheng, H.; Zhu, G. Org. Lett. 2015, 17, 1617–1620. doi:10.1021/acs.orglett.5b00546 |
| 2. | Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874–922. doi:10.1021/cr050992x |
| 3. | Sonogashira, K.; Tohda, Y.; Hagihara, N. Tetrahedron Lett. 1975, 16, 4467–4470. doi:10.1016/s0040-4039(00)91094-3 |
| 4. | Liu, W.; Li, L.; Li, C.-J. Nat. Commun. 2015, 6, 6526. doi:10.1038/ncomms7526 |
| 21. | Nakamura, K.; Nishikata, T. ACS Catal. 2017, 7, 1049–1052. doi:10.1021/acscatal.6b03343 |
| 23. | Okuro, K.; Furuune, M.; Enna, M.; Miura, M.; Nomura, M. J. Org. Chem. 1993, 58, 4716–4721. doi:10.1021/jo00069a040 |
| 24. | Gujadhur, R. K.; Bates, C. G.; Venkataraman, D. Org. Lett. 2001, 3, 4315–4317. doi:10.1021/ol0170105 |
| 25. | Thathagar, M. B.; Beckers, J.; Rothenberg, G. Green Chem. 2004, 6, 215–218. doi:10.1039/b401586j |
| 26. | Ma, D.; Liu, F. Chem. Commun. 2004, 1934–1935. doi:10.1039/b407090a |
| 27. | Wang, Y. F.; Deng, W.; Liu, L.; Guo, Q. X. Chin. Chem. Lett. 2005, 16, 1197–1200. |
| 28. | Saejueng, P.; Bates, C. G.; Venkataraman, D. Synthesis 2005, 1706–1712. doi:10.1055/s-2005-869893 |
| 29. | Xie, Y.-X.; Deng, C.-L.; Pi, S.-F.; Li, J.-H.; Yin, D.-L. Chin. J. Chem. 2006, 24, 1290–1294. doi:10.1002/cjoc.200690240 |
| 30. | Li, J.-H.; Li, J.-L.; Wang, D.-P.; Pi, S.-F.; Xie, Y.-X.; Zhang, M.-B.; Hu, X.-C. J. Org. Chem. 2007, 72, 2053–2057. doi:10.1021/jo0623742 |
| 31. | Tang, B.-X.; Wang, F.; Li, J.-H.; Xie, Y.-X.; Zhang, M.-B. J. Org. Chem. 2007, 72, 6294–6297. doi:10.1021/jo070538o |
| 32. | Guan, J. T.; Yu, G.-A.; Chen, L.; Weng, T. Q.; Yuan, J. J.; Liu, S. H. Appl. Organomet. Chem. 2009, 23, 75–77. doi:10.1002/aoc.1474 |
| 21. | Nakamura, K.; Nishikata, T. ACS Catal. 2017, 7, 1049–1052. doi:10.1021/acscatal.6b03343 |
| 20. | Yamane, Y.; Miwa, N.; Nishikata, T. ACS Catal. 2017, 7, 6872–6876. doi:10.1021/acscatal.7b02615 |
| 17. | Nishikata, T.; Noda, Y.; Fujimoto, R.; Sakashita, T. J. Am. Chem. Soc. 2013, 135, 16372–16375. doi:10.1021/ja409661n |
© 2020 Hirata et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)