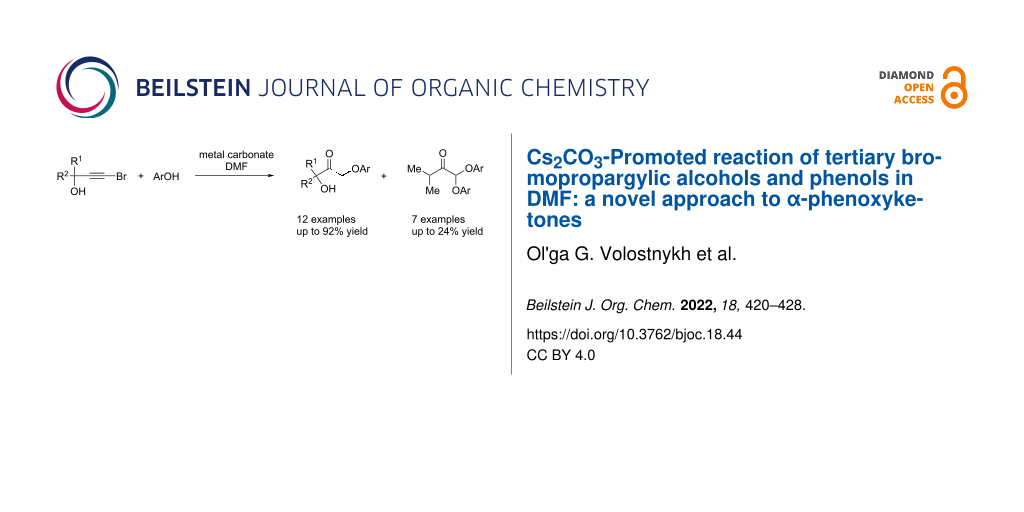
Abstract
The reaction of bromopropargylic alcohols with phenols in the presence of Cs2CO3/DMF affords α-phenoxy-α’-hydroxyketones (1:1 adducts) and α,α-diphenoxyketones (1:2 adducts) in up to 92% and 24% yields, respectively. Both products are formed via ring opening of the same intermediates, 1,3-dioxolan-2-ones, generated in situ from bromopropargylic alcohols and Cs2CO3.

Graphical Abstract
Introduction
Due to the relative stability, ease of handling and the presence of reactive sites, bromoacetylenes are widely applied in synthetic organic chemistry. They are known to be involved in various transformations including homo- and cross-coupling [1-7], addition [1,8,9], cycloaddition [1,10,11] and other reactions. Of particular synthetic value is the addition to the triple bond of bromoacetylenes to provide vinyl adducts, which can undergo numerous transformations. For example, bromoacetylenes were demonstrated to add imidazoles, imidazolines [12], and benzimidazoles [13,14] to give vinyl bromides. Sulfonamides reacted with bromoacetylenes to deliver N-bromovinyl-p-toluenesulfonamides that under Heck reaction conditions afforded N-(p-toluenesulfonyl)pyrroles [15]. The CsF-promoted nucleophilic addition of isocyanides to bromoacetylenes furnished the functionalized bromovinyl amides followed by Pd-catalyzed formation of 5-iminopyrrolone [16]. Sequential nucleophilic addition/intramolecular cyclization of amidine with bromoacetylenes led to imidazoles [17]. Also, M2CO3-catalyzed (M = K or Cs) addition of phenols to bromoacetylenes produced bromovinyl phenyl ethers, which were converted into 4H-chromen-4-ones, benzo[b]furans, etc. [18-21]. The latter reaction attracted our attention and prompted us to explore the interaction of phenols and bromopropargylic alcohols under the reported conditions. The bromopropargylic alcohols are readily available from acetylenic alcohols and hypobromite [22] or N-bromosuccinimide [23]. The presence of the hydroxy group expands the synthetic potential of these bromoacetylenes. Thus, we have recently demonstrated a highly selective hydration/acylation of tertiary bromopropargylic alcohols with carboxylic acids promoted by alkali metal carbonates [24]. The reaction proceeds via the ring-opening of 1,3-dioxolan-2-one intermediates formed with hydroxy and alkynyl groups of bromopropargylic alcohol and alkali metal carbonate. In the light of the above, it was unclear, in which direction would proceed the reaction of bromopropargylic alcohols and phenols. In the present paper, we report on the results of these studies.
Results and Discussion
Initially, bromopropargylic alcohol 1a and phenol (2a) were chosen as the model substrates for our investigation (Table 1). Completion of the reaction was monitored by IR and 1H NMR spectroscopy by the disappearance of the bands at 2196–2212 cm–1 (–C≡C–Br) and signals of the bromopropargylic alcohol 1a, respectively. Under the conditions previously used [18-21] for the addition of phenols to bromoacetylenes (K2CO3 or Cs2CO3, DMF, 110 °C), the reaction turned out to be non-selective: along with the expected bromovinyl phenyl ether 3a (3–9%) and phenoxyhydroxyketone 4a (25–39%), diphenoxyketone 5a was isolated in 9–24% yield (Table 1, entries 1–3). At 50–55 °C, the reaction slowed down and became more selective (Table 1, entries 4 and 5). With Cs2CO3 (1 equiv) at 50–55 °C, the reaction proceeded for 3 h, the yield of the phenoxyhydroxyketone 4a increased up to 55% and 5-phenoxymethylene-1,3-dioxolan-2-one 7, one of the probable intermediates, was isolated in 5% preparative yield (Table 1, entry 4), whereas the use of 2 equiv of Cs2CO3 led to slightly more selective reaction (Table 1, entry 5). Further lowering the temperature reduces the selectivity toward phenoxyketone 4a. At room temperature, the full conversion of bromopropargylic alcohol 1a took 15 h and yields of phenoxyketones 4a and 5a decreased (Table 1, entry 7). In the presence of K2CO3 (1 equiv) at 50–55 °C, the same reaction was completed for 8 h, the yields and selectivity being not improved (Table 1, entry 10). In these cases, 5-phenoxymethylene-1,3-dioxolan-2-one 7 was also isolated in 6–9% preparative yield. Hydrocarbonates CsHCO3 and KHCO3 were also tested in the reaction, which gave 5-bromomethylene-1,3-dioxolan-2-one 6a as a major product in 29–36% yield (Table 1, entries 11 and 12). Considering that hydration occurs during the formation of phenoxyketone 4a, we added water to the reaction system. It was shown that the reaction of 1a with 2a in aqueous DMF (1 equiv of Cs2CO3, DMF/H2O, 10:1, 50–55 °C) was highly selective to deliver phenoxyhydroxyketone 4a in 78% yield and dihydroxyketone 8a as a side product (Table 1, entry 6). Hence, the addition of phenol to the triple bond is a minor direction for the reaction of bromopropargylic alcohols and phenol in the presence of Cs2CO3/DMF, which was completely suppressed by addition of water. When DMF was replaced by DMSO (Table 1, entry 13), the preparative yield of reaction products decreased possibly due to product losses during extraction. No reaction was observed in CHCl3 (Table 1, entry 14) or utilizing organic bases (Et3N, DBU) (Table 1, entries 16–18). The efforts to increase the yield of diphenoxyketone 5a using 2 equivalents of phenol (2a) in the reaction with bromopropargylic alcohol 1a (Table 1, entries 8 and 9) failed.
Table 1: Screening of the conditions for reaction of bromopropargylic alcohol 1a and phenol (2a)a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-44-i11.svg?max-width=637&scale=1.0)
|
|||||||||
| Entry | Alkali metal carbonate (equiv) | Т (°С) | Time (h) | 3ab | 4ab | 5ab | 6ab | 7b | 8ab |
| 1 | K2CO3 (1) | 110 | 1 | 9 | 31 | 21 | – | – | – |
| 2 | Cs2CO3 (1) | 110 | 1 | 3 | 39 | 24 | – | – | – |
| 3 | Cs2CO3 (2) | 110 | 1 | 9 | 25 | 9 | – | – | – |
| 4 | Cs2CO3 (1) | 50–55 | 3 | 4 | 55 | 22 | – | 5 | – |
| 5 | Cs2CO3 (2) | 50–55 | 3 | – | 44 | 24 | – | – | – |
| 6c | Cs2CO3 (1) | 50–55 | 3 | – | 78 | – | – | – | 5 |
| 7 | Cs2CO3 (1) | rt | 15 | 4 | 29 | 16 | – | 6 | – |
| 8d | Cs2CO3 (1) | 110 | 1 | 4 | 58 | 19 | – | – | – |
| 9d | Cs2CO3 (2) | 110 | 1 | 17 | 9 | – | – | – | |
| 10 | K2CO3 (1) | 50–55 | 8 | – | 30 | 10 | – | 9 | – |
| 11 | KHCO3 (1) | 110 | 1 | – | 8 | 5 | 29 | – | – |
| 12 | CsHCO3 (1) | 110 | 1 | – | 6 | 8 | 36 | – | – |
| 13e | Cs2CO3 (1) | 50–55 | 3 | – | 25 | 18 | – | 9 | – |
| 14f | Cs2CO3 (1) | 50–55 | 3 | – | – | – | – | – | – |
| 15 | Na2CO3 (1) | 110 | 1 | – | – | – | – | – | – |
| 16 | Et3N | 50–55 | 3 | – | – | – | – | – | – |
| 17 | DBU | 50–55 | 3 | – | – | – | – | – | – |
| 18 | DBU | 110 | 3 | – | – | – | – | – | – |
aReaction conditions: 1a (1.2 mmol), 2a (1 mmol), alkali metal carbonate (1–2 equiv) in DMF (5 mL); the products were separated by column chromatography; bYields (%) are for the isolated products; cIn DMF/H2O (10:1); dWith 2 equiv of phenol; eIn DMSO; fIn CHCl3.
Employing the reaction conditions similar to those given in entries 4 and 6 (Table 1), we examined the substrate scope of the process relative to other phenols (Scheme 1). It was found that the electronic character of the substituents and the steric hindrance affected the reaction outcome. α-Naphthol (2b) and β-naphthol (2c) reacted with bromopropargylic alcohol 1a in DMF or DMF/H2O to furnish naphthoxyhydroxyketones 4b,c in preparative yields (up to 81%) comparable to those of 4a. The introduction of an electron-withdrawing substituent (p-NO2) at the benzene ring gave a better result: p-nitrophenoxyhydroxyketone 4d was formed in 65% (DMF) and 92% (DMF/H2O) isolated yields. However, the reaction of bromopropargylic alcohol 1a with o-nitrophenol (2e) afforded o-nitrophenoxyhydroxyketone 4e in only 48% (DMF) and 33% yields (DMF/H2O). The presence of an electron-donating group in p-cresol (2f), p-metoxyphenol (2g) and eugenol (2i) decreased the yields of phenoxyhydroxyketones 4f,g, and i in comparison with phenol. Bromovinyl phenyl ethers 3 were not isolated. In DMF, diphenoxyketones 5b–g were obtained in almost all the cases, the reaction with nitrophenols 2d,e being the only exception. When the reactions of bromopropargylic alcohol 1a with phenols 2b–i were carried out in DMF/H2O, dihydroxyketone 8a was isolated as a side product.
![[1860-5397-18-44-i1]](/bjoc/content/inline/1860-5397-18-44-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Scope of the reaction of bromopropargylic alcohol 1a and phenols 2b–i.
Scheme 1: Scope of the reaction of bromopropargylic alcohol 1a and phenols 2b–i.
Next, several experiments were carried out to evaluate the role of the steric effects of the alkyl substituents in bromopropargylic alcohols. The reaction of bromopropargylic alcohol 1b bearing a cyclohexyl substituent with phenol (2a) in DMF/H2O (1 equiv of Cs2CO3, 50–55 °C, 3 h) gave phenoxyhydroxyketone 4j in 60% yield (Scheme 2). Dihydroxyketone 8b was isolated as side product in 5% yield. The reaction of bromopropargylic alcohol 1b with p-nitrophenol (2d, DMF/H2O, 50–55 °C, 3 h) furnished product 8b (14% yield) along with phenoxyhydroxyketone 4k (78% isolated yield).
![[1860-5397-18-44-i2]](/bjoc/content/inline/1860-5397-18-44-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction of bromopropargylic alcohol 1b and phenols 2a and 2d.
Scheme 2: Reaction of bromopropargylic alcohol 1b and phenols 2a and 2d.
Bromopropargylic alcohol 1c having a tert-butyl group reacted with phenol (2a) in DMF for 3 h to give phenoxyhydroxyketone 4l in only 34% yield, 5-bromomethylene-1,3-dioxolan-2-one 6b (5%) being isolated (Scheme 3). In DMF/H2O (3 h), the conversion of 1c was incomplete (50%) and phenoxyhydroxyketone 4l was obtained in 39% yield. So, the steric hindrances of the bulky groups noticeably affect the reaction.
![[1860-5397-18-44-i3]](/bjoc/content/inline/1860-5397-18-44-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of bromopropargylic alcohol 1c and phenol (2a).
Scheme 3: Reaction of bromopropargylic alcohol 1c and phenol (2a).
The reaction of secondary and primary bromopropargylic alcohols (4-bromobut-3-yn-2-ol and 3-bromoprop-2-yn-1-ol) and phenol (2a) with 1 equiv of Cs2CO3, DMF, 50–55 °C, for 3 h did not gave any products, the competitive polymerization of bromopropargylic alcohols 1 being predominant.
Finally, chloroacetylenic alcohol was involved in the reaction with phenol (2a, 1 equiv Cs2CO3, DMF, 50–55 °C, 3 h) to afford the corresponding product 4a in 29% isolated yield (Scheme 4).
![[1860-5397-18-44-i4]](/bjoc/content/inline/1860-5397-18-44-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of chloropropargylic alcohol and phenol (2a).
Scheme 4: Reaction of chloropropargylic alcohol and phenol (2a).
We tested aniline and 2-naphthylamine as nucleophiles (DMF, 50–55 °C) in the reaction of bromopropargylic alcohol 1a (Scheme 5). But such a protocol turned out to be ineffective providing no desired products.
![[1860-5397-18-44-i5]](/bjoc/content/inline/1860-5397-18-44-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reaction of bromopropargylic alcohol 1a and anilines.
Scheme 5: Reaction of bromopropargylic alcohol 1a and anilines.
Several control experiments were performed to gain insight into the reaction mechanism (Scheme 6). When the reaction of 5-bromomethylene-1,3-dioxolan-2-one 6a and phenol (2a) was carried out with KOH, the conversion of the starting 6a was 55% and crude product contained phenoxyketone 4a, diphenoxyketone 5a and 5-phenoxymethylene-1,3-dioxolan-2-one 7. Using 2 equivalents of phenol (2a) in the reaction of 5-bromomethylene-1,3-dioxolan-2-one 6a (Cs2CO3, DMF, 110 °C, 20 min) gave phenoxyketone 4a and diphenoxyketone 5a in 40 and 16% yields, correspondingly. These results confirm that compound 6a is the main intermediate to form phenoxyketones. Next, we carried out the experiment using CO2 gas with DBU as a base. In comparison with reactions without CO2 (Table 1, entries 17 and 18), bromopropargylic alcohol 1a with free CO2 gas in the presence of 100 mol % of DBU and phenol (2a) (DMF, 50–55 °C, 3 h) afforded phenoxyketone 4a, 5-bromomethylene-1,3-dioxolan-2-one 6a and 5-phenoxymethylene-1,3-dioxolan-2-one 7 in 27, 4 and 19% yields, respectively. This result suggest that Cs2CO3 acts as a source of CO2 for the formation of 5-bromomethylene-1,3-dioxolan-2-one 6a.
![[1860-5397-18-44-i6]](/bjoc/content/inline/1860-5397-18-44-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Obviously, the formation of phenoxyhydroxyketone 4 proceeds via 1,3-dioxolan-2-one 6 generated from bromopropargylic alcohol 1 and Cs2CO3. Then, Br-substitution/hydration of 6 and the release of CO2 give product 4 (Scheme 7).
![[1860-5397-18-44-i7]](/bjoc/content/inline/1860-5397-18-44-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: A plausible mechanism for the formation of phenoxyhydroxyketone 4.
Scheme 7: A plausible mechanism for the formation of phenoxyhydroxyketone 4.
Apparently, diphenoxyketone 5 results from decarboxylative conversion of 1,3-dioxolan-2-one 7 leading to intermediate A, nucleophilic attack of phenolate at the less sterically hindered carbon of the above zwitterion A and subsequent protonation of anion B (Scheme 8).
![[1860-5397-18-44-i8]](/bjoc/content/inline/1860-5397-18-44-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: A plausible mechanism for the formation of diphenoxyketone 5.
Scheme 8: A plausible mechanism for the formation of diphenoxyketone 5.
Based on these plausible mechanisms for the formation of phenoxyketones, it can be assumed that a decline of the Cs+ concentration after Cs2CO3 convertion to CsBr (because of the very poor solubility of CsBr in DMF) has an influence on the rate of diphenoxyketone formation. In addition, the suppression of the di(nitrophenoxy)ketone formation can be due to the lower basicity of a reaction mixture since nitrophenols 2d,e are more acidic than phenols 2a–c,f–i (pKa values: 9.99 [25,26] phenol (2a), 9.40 [27] α-naphthol (2b), 9.57 [27] β-naphthol (2c), 7.18 [25,26] p-nitrophenol (2d), 7.23 [25,26] o-nitrophenol (2e), 10.28 [25,26] p-cresol (2f), 10.27 [25,26] p-methoxyphenol (2g), 9.36 [25,26] p-bromophenol (2h), 10.19 eugenol (2i)). Addition of water to the reaction mixture also reduces the pH of the medium and simultaneously increases the concentration of hydroxide ions, therefore, diphenoxyketones 5 were not produced and dihydroxyketones 8 were formed as side products in these cases.
Among the approaches to produce α-phenoxyketones, the most common methodologies are base-catalyzed alkylation of the corresponding phenols with halo- [28-30] and mesyl [31-33] ketones (Scheme 9), the preparation of which are not always selective and high-yielded. The ring opening of ArOCH2-epoxides [34,35], the SmI2-catalyzed reductive coupling of acid halides with ketones [36,37] and acetolyses of α-phenoxy-α-diazoketones [38] were also employed.
![[1860-5397-18-44-i9]](/bjoc/content/inline/1860-5397-18-44-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Examples of representative preparation of phenoxyketones 4.
Scheme 9: Examples of representative preparation of phenoxyketones 4.
Recently, F. P. Cossío et al. [39] have described a method for the preparation of benzo[b]furans by thermal heating of a dispersion of α-phenoxyketones in Al2O3. We involved the synthesized α-phenoxyketones 4 in this reaction. The results showed that instead of benzo[b]furan formation, α-ketol rearrangement of phenoxyketones 4a,f occurred to afford β-phenoxyketones 9a,b in 55–60% yields (Scheme 10).
![[1860-5397-18-44-i10]](/bjoc/content/inline/1860-5397-18-44-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: α-Ketol rearrangement of phenoxyketones 4a and 4f.
Scheme 10: α-Ketol rearrangement of phenoxyketones 4a and 4f.
Conclusion
We have shown that the main direction of the reaction of bromopropargylic alcohols and phenols in Cs2CO3/DMF is the hydration/phenoxylation of bromopropargylic alcohols to afford phenoxyketones. This step-economical process takes place under mild reaction conditions using simple readily available starting materials. The synthesized phenoxyketones are of interest as valuable building blocks for the production of other important molecules (e.g., amino alcohols, diols, etc.) [40-46] and potential pharmaceuticals. α-Hydroxyketones are structural subunits of natural products [47-49] and compounds possessing immunosuppressant [50], antidepressant [51], amyloid-β protein production inhibitory [52], urease inhibitory [53], farnesyl transferase inhibitory (kurasoin A and B) [54,55], antitumor and antibacterial (doxorubicin, olivomycin A, chromomycin A3, carminomycin I, epothilones) [56-58] activities.
Experimental
General information
1Н and 13С NMR spectra were recorded on a Bruker DPX-400 spectrometer (400.1 and 100.6 MHz, respectively) in CDCl3 or (CD3)2CO using hexamethyldisiloxane as internal reference at 20–25 °C. IR spectra were measured on a Varian 3100 FT-IR Excalibur series instrument as thin films or KВr pellets. Microanalyses were performed on a Flash 2000 elemental analyzer. Melting points were determined using a Kofler micro hot stage apparatus. Mass spectra were recorded on a GCMS-QP5050A spectrometer made by Shimadzu Company. Chromatographic column parameters were as follows: SPBТМ-5, length 60 m, internal diameter 0.25 mm, thickness of stationary phase film 0.25 μm; injector temperature 250 °C, gas carrier – helium, flow rate 0.7 mL/min; detector temperature 250 °C; mass analyzer: quadrupole, electron ionization, electron energy: 70 eV, ion source temperature 200 °C; mass range 34–650 Da. The solvent was distilled DMF. Column chromatography was performed on silica gel 60 (230–400 mesh, particle size 0.040–0.063 mm, Merck). Bromopropargylic alcohols 1a–c and chloropropargylic alcohol were prepared according to published methods [22,23,59]. Phenol (2a), naphthalen-1-ol (2b), naphthalen-2-ol (2c), 4-nitrophenol (2d), 2-nitrophenol (2e), p-cresol (2f), 4-methoxyphenol (2g), 4-bromophenol (2h), 4-allyl-2-methoxyphenol (2i) are commercial reagents. Commercially available starting materials were used without further purification. The structures of synthesized products have been proven by 1H, 13C and 2D (NOESY, 1Н,13С HSQC, 1Н,13С HMBC) NMR techniques, as well as IR spectra.
Typical procedure for preparation of phenoxyhydroxyketones 4 in DMF, 50–55 °C. To a stirred solution of Cs2CO3 (326 mg, 1 mmol) and phenol (2a; 94 mg, 1 mmol) in DMF (5 mL) 4-bromo-2-methylbut-3-yn-2-ol (1a; 196 mg, 1.2 mmol) was added dropwise. The reaction mixture was stirred at 50–55 °C for 3 h, filtered and concentrated. The residue was purified by flash column chromatography on silica gel (5.0 × 4.0 cm, gradient elution, C6H14/Et2O, 2:1 followed by Et2O, Me2CO) to give products 3a (10 mg, 4%), 4a (214 mg, 55%), 5a (30 mg, 22%) and 7 (11 mg, 5%).
Supporting Information
| Supporting Information File 1: General information, synthetic procedures and additional optimization results, NMR spectra and characterization of synthesized compounds. | ||
| Format: PDF | Size: 3.1 MB | Download |
References
-
Wu, W.; Jiang, H. Acc. Chem. Res. 2014, 47, 2483–2504. doi:10.1021/ar5001499
Return to citation in text: [1] [2] [3] -
Wang, S.; Yu, L.; Li, P.; Meng, L.; Wang, L. Synthesis 2011, 1541–1546. doi:10.1055/s-0030-1259998
Return to citation in text: [1] -
de Orbe, M. E.; Zanini, M.; Quinonero, O.; Echavarren, A. M. ACS Catal. 2019, 9, 7817–7822. doi:10.1021/acscatal.9b02314
Return to citation in text: [1] -
Cornelissen, L.; Lefrancq, M.; Riant, O. Org. Lett. 2014, 16, 3024–3027. doi:10.1021/ol501140p
Return to citation in text: [1] -
Krishnan, K. K.; Ujwaldev, S. M.; Thankachan, A. P.; Harry, N. A.; Gopinathan, A. Mol. Catal. 2017, 440, 140–147. doi:10.1016/j.mcat.2017.07.021
Return to citation in text: [1] -
Sindhu, K. S.; Thankachan, A. P.; Sajitha, P. S.; Anilkumar, G. Org. Biomol. Chem. 2015, 13, 6891–6905. doi:10.1039/c5ob00697j
Return to citation in text: [1] -
Porey, S.; Zhang, X.; Bhowmick, S.; Singh, V. K.; Guin, S.; Paton, R. S.; Maiti, D. J. Am. Chem. Soc. 2020, 142, 3762–3774. doi:10.1021/jacs.9b10646
Return to citation in text: [1] -
Cadierno, V. Eur. J. Inorg. Chem. 2020, 886–898. doi:10.1002/ejic.201901016
Return to citation in text: [1] -
Volkov, A. N.; Volkova, K. A. Russ. J. Org. Chem. 2007, 43, 161–169. doi:10.1134/s1070428007020017
Return to citation in text: [1] -
Luo, Q.; Jia, G.; Sun, J.; Lin, Z. J. Org. Chem. 2014, 79, 11970–11980. doi:10.1021/jo5018348
Return to citation in text: [1] -
Okuda, Y.; Imafuku, K.; Tsuchida, Y.; Seo, T.; Akashi, H.; Orita, A. Org. Lett. 2020, 22, 5099–5103. doi:10.1021/acs.orglett.0c01681
Return to citation in text: [1] -
Yamagishi, M.; Okazaki, J.; Nishigai, K.; Hata, T.; Urabe, H. Org. Lett. 2012, 14, 34–37. doi:10.1021/ol2027448
Return to citation in text: [1] -
Peng, J.; Shang, G.; Chen, C.; Miao, Z.; Li, B. J. Org. Chem. 2013, 78, 1242–1248. doi:10.1021/jo302471z
Return to citation in text: [1] -
Chen, X.; Sun, P.; Mo, B.; Chen, C.; Peng, J. J. Org. Chem. 2021, 86, 352–366. doi:10.1021/acs.joc.0c02126
Return to citation in text: [1] -
Yamagishi, M.; Nishigai, K.; Hata, T.; Urabe, H. Org. Lett. 2011, 13, 4873–4875. doi:10.1021/ol201952b
Return to citation in text: [1] -
Li, Y.; Zhao, J.; Chen, H.; Liu, B.; Jiang, H. Chem. Commun. 2012, 48, 3545–3547. doi:10.1039/c2cc17717j
Return to citation in text: [1] -
Chen, X. Y.; Englert, U.; Bolm, C. Chem. – Eur. J. 2015, 21, 13221–13224. doi:10.1002/chem.201502707
Return to citation in text: [1] -
Speck, K.; Magauer, T. Chem. – Eur. J. 2017, 23, 1157–1165. doi:10.1002/chem.201605029
Return to citation in text: [1] [2] -
Speck, K.; Wildermuth, R.; Magauer, T. Angew. Chem., Int. Ed. 2016, 55, 14131–14135. doi:10.1002/anie.201608040
Return to citation in text: [1] [2] -
Luo, Y.; Yuan, C.; Xu, J.; Li, Y.; Liu, H.; Semin, S.; Rasing, T.; Yang, W.; Li, Y. ACS Appl. Mater. Interfaces 2017, 9, 30862–30871. doi:10.1021/acsami.7b10109
Return to citation in text: [1] [2] -
Wang, S.; Li, P.; Yu, L.; Wang, L. Org. Lett. 2011, 13, 5968–5971. doi:10.1021/ol202383z
Return to citation in text: [1] [2] -
Nazarov, I. V.; Shvekhgeimer, G. A. Zh. Obshch. Khim. 1959, 29, 457.
Return to citation in text: [1] [2] -
Hofmeister, H.; Annen, K.; Laurent, H.; Wiechert, R. Angew. Chem. 1984, 96, 720–722. doi:10.1002/ange.19840960932
Return to citation in text: [1] [2] -
Shemyakina, O. A.; Volostnykh, O. G.; Stepanov, A. V.; Ushakov, I. A. Eur. J. Org. Chem. 2019, 7117–7121. doi:10.1002/ejoc.201901226
Return to citation in text: [1] -
Jover, J.; Bosque, R.; Sales, J. QSAR Comb. Sci. 2007, 26, 385–397. doi:10.1002/qsar.200610088
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Roy, K.; Popelier, P. L. A. J. Phys. Org. Chem. 2009, 22, 186–196. doi:10.1002/poc.1447
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Bryson, A.; Matthews, R. W. Aust. J. Chem. 1963, 16, 401–410. doi:10.1071/ch9630401
Return to citation in text: [1] [2] -
Li, J.-Q.; Wang, Z.-P.; Gao, Y.; Zhao, W.-G. RSC Adv. 2016, 6, 82131–82137. doi:10.1039/c6ra17908h
Return to citation in text: [1] -
Boehm, M. F.; Fitzgerald, P.; Zou, A.; Elgort, M. G.; Bischoff, E. D.; Mere, L.; Mais, D. E.; Bissonnette, R. P.; Heyman, R. A.; Nadzan, A. M.; Reichman, M.; Allegretto, E. A. Chem. Biol. 1999, 6, 265–275. doi:10.1016/s1074-5521(99)80072-6
Return to citation in text: [1] -
Piantadosi, C.; Hall, I. H.; Wyrick, S. D.; Ishaq, K. S. J. Med. Chem. 1976, 19, 222–229. doi:10.1021/jm00224a006
Return to citation in text: [1] -
Han, A.; Olsen, O.; D’Souza, C.; Shan, J.; Zhao, F.; Yanolatos, J.; Hovhannisyan, Z.; Haxhinasto, S.; Delfino, F.; Olson, W. J. Med. Chem. 2021, 64, 11958–11971. doi:10.1021/acs.jmedchem.1c00541
Return to citation in text: [1] -
Purakkattle, B. J.; Berlin, M. Y.; Lim, Y.-H.; Bitar, R. D.; McCormick, K. D.; Aslanian, R. G.; Lee, Y. J.; Zheng, J.; Huang, Y.; Won, W. Novel [3,2-c] heteroaryl steroids as glucocorticoid receptor agonists compositions and uses thereof. U.S. Pat. Appl. US20120171126A1, July 5, 2012.
Return to citation in text: [1] -
Simons, S. S., Jr.; Pons, M.; Johnson, D. F. J. Org. Chem. 1980, 45, 3084–3088. doi:10.1021/jo01303a030
Return to citation in text: [1] -
Connolly, S.; Bennion, C.; Botterell, S.; Croshaw, P. J.; Hallam, C.; Hardy, K.; Hartopp, P.; Jackson, C. G.; King, S. J.; Lawrence, L.; Mete, A.; Murray, D.; Robinson, D. H.; Smith, G. M.; Stein, L.; Walters, I.; Wells, E.; Withnall, W. J. J. Med. Chem. 2002, 45, 1348–1362. doi:10.1021/jm011050x
Return to citation in text: [1] -
Ismail, N.; Rao, R. N. Chem. Lett. 2000, 29, 844–845. doi:10.1246/cl.2000.844
Return to citation in text: [1] -
Uneyama, K.; Isimura, A.; Fujii, K.; Torii, S. Tetrahedron Lett. 1983, 24, 2857–2860. doi:10.1016/s0040-4039(00)88043-0
Return to citation in text: [1] -
Ruder, S. M. Tetrahedron Lett. 1992, 33, 2621–2624. doi:10.1016/s0040-4039(00)79041-1
Return to citation in text: [1] -
Pusino, A.; Rosnati, V.; Solinas, C.; Vettori, U. Tetrahedron 1983, 39, 2259–2263. doi:10.1016/s0040-4020(01)91950-7
Return to citation in text: [1] -
Arias, L.; Vara, Y.; Cossío, F. P. J. Org. Chem. 2012, 77, 266–275. doi:10.1021/jo201841y
Return to citation in text: [1] -
Song, W.; Liu, P.; Lei, M.; You, H.; Chen, X.; Chen, H.; Ma, L.; Hu, L. Synth. Commun. 2012, 42, 236–245. doi:10.1080/00397911.2010.523489
Return to citation in text: [1] -
Fang, Q. K.; Han, Z.; Grover, P.; Kessler, D.; Senanayake, C. H.; Wald, S. A. Tetrahedron: Asymmetry 2000, 11, 3659–3663. doi:10.1016/s0957-4166(00)00349-9
Return to citation in text: [1] -
Pusino, A.; Saba, A.; Rosnati, V. Tetrahedron 1986, 42, 4319–4324. doi:10.1016/s0040-4020(01)87658-4
Return to citation in text: [1] -
Ma, Z.; Zhou, M.; Ma, L.; Zhang, M. J. Chem. Res. 2020, 44, 426–436. doi:10.1177/1747519820907244
Return to citation in text: [1] -
Jia, S.; Lei, Y.; Song, L.; Krishna Reddy, A. G.; Xing, D.; Hu, W. Adv. Synth. Catal. 2017, 359, 58–63. doi:10.1002/adsc.201600998
Return to citation in text: [1] -
Trost, B. M.; Hisaindee, S. Org. Lett. 2006, 8, 6003–6005. doi:10.1021/ol062485n
Return to citation in text: [1] -
Harada, S.; Kumagai, N.; Kinoshita, T.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2003, 125, 2582–2590. doi:10.1021/ja028928+
Return to citation in text: [1] -
Tiaden, A.; Hilbi, H. Sensors 2012, 12, 2899–2919. doi:10.3390/s120302899
Return to citation in text: [1] -
Tiaden, A.; Spirig, T.; Hilbi, H. Trends Microbiol. 2010, 18, 288–297. doi:10.1016/j.tim.2010.03.004
Return to citation in text: [1] -
Huang, S.-X.; Powell, E.; Rajski, S. R.; Zhao, L.-X.; Jiang, C.-L.; Duan, Y.; Xu, W.; Shen, B. Org. Lett. 2010, 12, 3525–3527. doi:10.1021/ol1013526
Return to citation in text: [1] -
Mann, J. Nat. Prod. Rep. 2001, 18, 417–430. doi:10.1039/b001720p
Return to citation in text: [1] -
Hoyos, P.; Sinisterra, J.-V.; Molinari, F.; Alcántara, A. R.; Domínguez de María, P. Acc. Chem. Res. 2010, 43, 288–299. doi:10.1021/ar900196n
Return to citation in text: [1] -
Wallace, O. B.; Smith, D. W.; Deshpande, M. S.; Polson, C.; Felsenstein, K. M. Bioorg. Med. Chem. Lett. 2003, 13, 1203–1206. doi:10.1016/s0960-894x(02)01058-2
Return to citation in text: [1] -
Tanaka, T.; Kawase, M.; Tani, S. Bioorg. Med. Chem. 2004, 12, 501–505. doi:10.1016/j.bmc.2003.10.017
Return to citation in text: [1] -
Uchida, R.; Shiomi, K.; Sunazuka, T.; Inokoshi, J.; Nishizawa, A.; Hirose, T.; Tanaka, H.; Iwai, Y.; Omura, S. J. Antibiot. 1996, 49, 886–889. doi:10.7164/antibiotics.49.886
Return to citation in text: [1] -
Koblan, K. S.; Kohl, N. E.; Omer, C. A.; Anthony, N. J.; Conner, M. W.; de Solms, S. J.; Williams, T. M.; Graham, S. L.; Hartman, G. D.; Oliff, A.; Gibbs, J. B. Biochem. Soc. Trans. 1996, 24, 688–692. doi:10.1042/bst0240688
Return to citation in text: [1] -
Slavik, M.; Carter, S. K. Adv. Pharmacol. Chemother. 1975, 12, 1–30. doi:10.1016/s1054-3589(08)60218-5
Return to citation in text: [1] -
Pettit, G. R.; Einck, J. J.; Herald, C. L.; Ode, R. H.; Von Dreele, R. B.; Brown, P.; Brazhnikova, M. G.; Gauze, G. F. J. Am. Chem. Soc. 1975, 97, 7387–7388. doi:10.1021/ja00858a036
Return to citation in text: [1] -
Hortobágyi, G. N. Drugs 1997, 54, 1–7. doi:10.2165/00003495-199700544-00003
Return to citation in text: [1] -
Shi, D.; Liu, Z.; Zhang, Z.; Shi, W.; Chen, H. ChemCatChem 2015, 7, 1424–1426. doi:10.1002/cctc.201500243
Return to citation in text: [1]
| 40. | Song, W.; Liu, P.; Lei, M.; You, H.; Chen, X.; Chen, H.; Ma, L.; Hu, L. Synth. Commun. 2012, 42, 236–245. doi:10.1080/00397911.2010.523489 |
| 41. | Fang, Q. K.; Han, Z.; Grover, P.; Kessler, D.; Senanayake, C. H.; Wald, S. A. Tetrahedron: Asymmetry 2000, 11, 3659–3663. doi:10.1016/s0957-4166(00)00349-9 |
| 42. | Pusino, A.; Saba, A.; Rosnati, V. Tetrahedron 1986, 42, 4319–4324. doi:10.1016/s0040-4020(01)87658-4 |
| 43. | Ma, Z.; Zhou, M.; Ma, L.; Zhang, M. J. Chem. Res. 2020, 44, 426–436. doi:10.1177/1747519820907244 |
| 44. | Jia, S.; Lei, Y.; Song, L.; Krishna Reddy, A. G.; Xing, D.; Hu, W. Adv. Synth. Catal. 2017, 359, 58–63. doi:10.1002/adsc.201600998 |
| 45. | Trost, B. M.; Hisaindee, S. Org. Lett. 2006, 8, 6003–6005. doi:10.1021/ol062485n |
| 46. | Harada, S.; Kumagai, N.; Kinoshita, T.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2003, 125, 2582–2590. doi:10.1021/ja028928+ |
| 47. | Tiaden, A.; Hilbi, H. Sensors 2012, 12, 2899–2919. doi:10.3390/s120302899 |
| 48. | Tiaden, A.; Spirig, T.; Hilbi, H. Trends Microbiol. 2010, 18, 288–297. doi:10.1016/j.tim.2010.03.004 |
| 49. | Huang, S.-X.; Powell, E.; Rajski, S. R.; Zhao, L.-X.; Jiang, C.-L.; Duan, Y.; Xu, W.; Shen, B. Org. Lett. 2010, 12, 3525–3527. doi:10.1021/ol1013526 |
| 1. | Wu, W.; Jiang, H. Acc. Chem. Res. 2014, 47, 2483–2504. doi:10.1021/ar5001499 |
| 2. | Wang, S.; Yu, L.; Li, P.; Meng, L.; Wang, L. Synthesis 2011, 1541–1546. doi:10.1055/s-0030-1259998 |
| 3. | de Orbe, M. E.; Zanini, M.; Quinonero, O.; Echavarren, A. M. ACS Catal. 2019, 9, 7817–7822. doi:10.1021/acscatal.9b02314 |
| 4. | Cornelissen, L.; Lefrancq, M.; Riant, O. Org. Lett. 2014, 16, 3024–3027. doi:10.1021/ol501140p |
| 5. | Krishnan, K. K.; Ujwaldev, S. M.; Thankachan, A. P.; Harry, N. A.; Gopinathan, A. Mol. Catal. 2017, 440, 140–147. doi:10.1016/j.mcat.2017.07.021 |
| 6. | Sindhu, K. S.; Thankachan, A. P.; Sajitha, P. S.; Anilkumar, G. Org. Biomol. Chem. 2015, 13, 6891–6905. doi:10.1039/c5ob00697j |
| 7. | Porey, S.; Zhang, X.; Bhowmick, S.; Singh, V. K.; Guin, S.; Paton, R. S.; Maiti, D. J. Am. Chem. Soc. 2020, 142, 3762–3774. doi:10.1021/jacs.9b10646 |
| 13. | Peng, J.; Shang, G.; Chen, C.; Miao, Z.; Li, B. J. Org. Chem. 2013, 78, 1242–1248. doi:10.1021/jo302471z |
| 14. | Chen, X.; Sun, P.; Mo, B.; Chen, C.; Peng, J. J. Org. Chem. 2021, 86, 352–366. doi:10.1021/acs.joc.0c02126 |
| 27. | Bryson, A.; Matthews, R. W. Aust. J. Chem. 1963, 16, 401–410. doi:10.1071/ch9630401 |
| 12. | Yamagishi, M.; Okazaki, J.; Nishigai, K.; Hata, T.; Urabe, H. Org. Lett. 2012, 14, 34–37. doi:10.1021/ol2027448 |
| 27. | Bryson, A.; Matthews, R. W. Aust. J. Chem. 1963, 16, 401–410. doi:10.1071/ch9630401 |
| 1. | Wu, W.; Jiang, H. Acc. Chem. Res. 2014, 47, 2483–2504. doi:10.1021/ar5001499 |
| 10. | Luo, Q.; Jia, G.; Sun, J.; Lin, Z. J. Org. Chem. 2014, 79, 11970–11980. doi:10.1021/jo5018348 |
| 11. | Okuda, Y.; Imafuku, K.; Tsuchida, Y.; Seo, T.; Akashi, H.; Orita, A. Org. Lett. 2020, 22, 5099–5103. doi:10.1021/acs.orglett.0c01681 |
| 18. | Speck, K.; Magauer, T. Chem. – Eur. J. 2017, 23, 1157–1165. doi:10.1002/chem.201605029 |
| 19. | Speck, K.; Wildermuth, R.; Magauer, T. Angew. Chem., Int. Ed. 2016, 55, 14131–14135. doi:10.1002/anie.201608040 |
| 20. | Luo, Y.; Yuan, C.; Xu, J.; Li, Y.; Liu, H.; Semin, S.; Rasing, T.; Yang, W.; Li, Y. ACS Appl. Mater. Interfaces 2017, 9, 30862–30871. doi:10.1021/acsami.7b10109 |
| 21. | Wang, S.; Li, P.; Yu, L.; Wang, L. Org. Lett. 2011, 13, 5968–5971. doi:10.1021/ol202383z |
| 56. | Slavik, M.; Carter, S. K. Adv. Pharmacol. Chemother. 1975, 12, 1–30. doi:10.1016/s1054-3589(08)60218-5 |
| 57. | Pettit, G. R.; Einck, J. J.; Herald, C. L.; Ode, R. H.; Von Dreele, R. B.; Brown, P.; Brazhnikova, M. G.; Gauze, G. F. J. Am. Chem. Soc. 1975, 97, 7387–7388. doi:10.1021/ja00858a036 |
| 58. | Hortobágyi, G. N. Drugs 1997, 54, 1–7. doi:10.2165/00003495-199700544-00003 |
| 1. | Wu, W.; Jiang, H. Acc. Chem. Res. 2014, 47, 2483–2504. doi:10.1021/ar5001499 |
| 8. | Cadierno, V. Eur. J. Inorg. Chem. 2020, 886–898. doi:10.1002/ejic.201901016 |
| 9. | Volkov, A. N.; Volkova, K. A. Russ. J. Org. Chem. 2007, 43, 161–169. doi:10.1134/s1070428007020017 |
| 25. | Jover, J.; Bosque, R.; Sales, J. QSAR Comb. Sci. 2007, 26, 385–397. doi:10.1002/qsar.200610088 |
| 26. | Roy, K.; Popelier, P. L. A. J. Phys. Org. Chem. 2009, 22, 186–196. doi:10.1002/poc.1447 |
| 22. | Nazarov, I. V.; Shvekhgeimer, G. A. Zh. Obshch. Khim. 1959, 29, 457. |
| 23. | Hofmeister, H.; Annen, K.; Laurent, H.; Wiechert, R. Angew. Chem. 1984, 96, 720–722. doi:10.1002/ange.19840960932 |
| 59. | Shi, D.; Liu, Z.; Zhang, Z.; Shi, W.; Chen, H. ChemCatChem 2015, 7, 1424–1426. doi:10.1002/cctc.201500243 |
| 18. | Speck, K.; Magauer, T. Chem. – Eur. J. 2017, 23, 1157–1165. doi:10.1002/chem.201605029 |
| 19. | Speck, K.; Wildermuth, R.; Magauer, T. Angew. Chem., Int. Ed. 2016, 55, 14131–14135. doi:10.1002/anie.201608040 |
| 20. | Luo, Y.; Yuan, C.; Xu, J.; Li, Y.; Liu, H.; Semin, S.; Rasing, T.; Yang, W.; Li, Y. ACS Appl. Mater. Interfaces 2017, 9, 30862–30871. doi:10.1021/acsami.7b10109 |
| 21. | Wang, S.; Li, P.; Yu, L.; Wang, L. Org. Lett. 2011, 13, 5968–5971. doi:10.1021/ol202383z |
| 23. | Hofmeister, H.; Annen, K.; Laurent, H.; Wiechert, R. Angew. Chem. 1984, 96, 720–722. doi:10.1002/ange.19840960932 |
| 53. | Tanaka, T.; Kawase, M.; Tani, S. Bioorg. Med. Chem. 2004, 12, 501–505. doi:10.1016/j.bmc.2003.10.017 |
| 17. | Chen, X. Y.; Englert, U.; Bolm, C. Chem. – Eur. J. 2015, 21, 13221–13224. doi:10.1002/chem.201502707 |
| 24. | Shemyakina, O. A.; Volostnykh, O. G.; Stepanov, A. V.; Ushakov, I. A. Eur. J. Org. Chem. 2019, 7117–7121. doi:10.1002/ejoc.201901226 |
| 54. | Uchida, R.; Shiomi, K.; Sunazuka, T.; Inokoshi, J.; Nishizawa, A.; Hirose, T.; Tanaka, H.; Iwai, Y.; Omura, S. J. Antibiot. 1996, 49, 886–889. doi:10.7164/antibiotics.49.886 |
| 55. | Koblan, K. S.; Kohl, N. E.; Omer, C. A.; Anthony, N. J.; Conner, M. W.; de Solms, S. J.; Williams, T. M.; Graham, S. L.; Hartman, G. D.; Oliff, A.; Gibbs, J. B. Biochem. Soc. Trans. 1996, 24, 688–692. doi:10.1042/bst0240688 |
| 16. | Li, Y.; Zhao, J.; Chen, H.; Liu, B.; Jiang, H. Chem. Commun. 2012, 48, 3545–3547. doi:10.1039/c2cc17717j |
| 51. | Hoyos, P.; Sinisterra, J.-V.; Molinari, F.; Alcántara, A. R.; Domínguez de María, P. Acc. Chem. Res. 2010, 43, 288–299. doi:10.1021/ar900196n |
| 15. | Yamagishi, M.; Nishigai, K.; Hata, T.; Urabe, H. Org. Lett. 2011, 13, 4873–4875. doi:10.1021/ol201952b |
| 52. | Wallace, O. B.; Smith, D. W.; Deshpande, M. S.; Polson, C.; Felsenstein, K. M. Bioorg. Med. Chem. Lett. 2003, 13, 1203–1206. doi:10.1016/s0960-894x(02)01058-2 |
| 25. | Jover, J.; Bosque, R.; Sales, J. QSAR Comb. Sci. 2007, 26, 385–397. doi:10.1002/qsar.200610088 |
| 26. | Roy, K.; Popelier, P. L. A. J. Phys. Org. Chem. 2009, 22, 186–196. doi:10.1002/poc.1447 |
| 25. | Jover, J.; Bosque, R.; Sales, J. QSAR Comb. Sci. 2007, 26, 385–397. doi:10.1002/qsar.200610088 |
| 26. | Roy, K.; Popelier, P. L. A. J. Phys. Org. Chem. 2009, 22, 186–196. doi:10.1002/poc.1447 |
| 25. | Jover, J.; Bosque, R.; Sales, J. QSAR Comb. Sci. 2007, 26, 385–397. doi:10.1002/qsar.200610088 |
| 26. | Roy, K.; Popelier, P. L. A. J. Phys. Org. Chem. 2009, 22, 186–196. doi:10.1002/poc.1447 |
| 38. | Pusino, A.; Rosnati, V.; Solinas, C.; Vettori, U. Tetrahedron 1983, 39, 2259–2263. doi:10.1016/s0040-4020(01)91950-7 |
| 39. | Arias, L.; Vara, Y.; Cossío, F. P. J. Org. Chem. 2012, 77, 266–275. doi:10.1021/jo201841y |
| 34. | Connolly, S.; Bennion, C.; Botterell, S.; Croshaw, P. J.; Hallam, C.; Hardy, K.; Hartopp, P.; Jackson, C. G.; King, S. J.; Lawrence, L.; Mete, A.; Murray, D.; Robinson, D. H.; Smith, G. M.; Stein, L.; Walters, I.; Wells, E.; Withnall, W. J. J. Med. Chem. 2002, 45, 1348–1362. doi:10.1021/jm011050x |
| 35. | Ismail, N.; Rao, R. N. Chem. Lett. 2000, 29, 844–845. doi:10.1246/cl.2000.844 |
| 36. | Uneyama, K.; Isimura, A.; Fujii, K.; Torii, S. Tetrahedron Lett. 1983, 24, 2857–2860. doi:10.1016/s0040-4039(00)88043-0 |
| 37. | Ruder, S. M. Tetrahedron Lett. 1992, 33, 2621–2624. doi:10.1016/s0040-4039(00)79041-1 |
| 28. | Li, J.-Q.; Wang, Z.-P.; Gao, Y.; Zhao, W.-G. RSC Adv. 2016, 6, 82131–82137. doi:10.1039/c6ra17908h |
| 29. | Boehm, M. F.; Fitzgerald, P.; Zou, A.; Elgort, M. G.; Bischoff, E. D.; Mere, L.; Mais, D. E.; Bissonnette, R. P.; Heyman, R. A.; Nadzan, A. M.; Reichman, M.; Allegretto, E. A. Chem. Biol. 1999, 6, 265–275. doi:10.1016/s1074-5521(99)80072-6 |
| 30. | Piantadosi, C.; Hall, I. H.; Wyrick, S. D.; Ishaq, K. S. J. Med. Chem. 1976, 19, 222–229. doi:10.1021/jm00224a006 |
| 31. | Han, A.; Olsen, O.; D’Souza, C.; Shan, J.; Zhao, F.; Yanolatos, J.; Hovhannisyan, Z.; Haxhinasto, S.; Delfino, F.; Olson, W. J. Med. Chem. 2021, 64, 11958–11971. doi:10.1021/acs.jmedchem.1c00541 |
| 32. | Purakkattle, B. J.; Berlin, M. Y.; Lim, Y.-H.; Bitar, R. D.; McCormick, K. D.; Aslanian, R. G.; Lee, Y. J.; Zheng, J.; Huang, Y.; Won, W. Novel [3,2-c] heteroaryl steroids as glucocorticoid receptor agonists compositions and uses thereof. U.S. Pat. Appl. US20120171126A1, July 5, 2012. |
| 33. | Simons, S. S., Jr.; Pons, M.; Johnson, D. F. J. Org. Chem. 1980, 45, 3084–3088. doi:10.1021/jo01303a030 |
| 25. | Jover, J.; Bosque, R.; Sales, J. QSAR Comb. Sci. 2007, 26, 385–397. doi:10.1002/qsar.200610088 |
| 26. | Roy, K.; Popelier, P. L. A. J. Phys. Org. Chem. 2009, 22, 186–196. doi:10.1002/poc.1447 |
| 25. | Jover, J.; Bosque, R.; Sales, J. QSAR Comb. Sci. 2007, 26, 385–397. doi:10.1002/qsar.200610088 |
| 26. | Roy, K.; Popelier, P. L. A. J. Phys. Org. Chem. 2009, 22, 186–196. doi:10.1002/poc.1447 |
© 2022 Volostnykh et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.