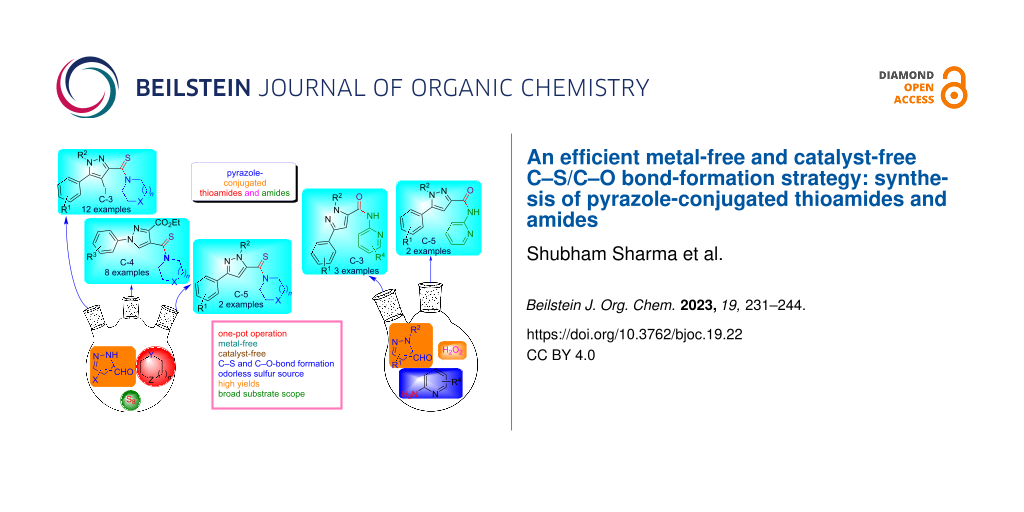
Abstract
An operationally simple and metal-free approach is described for the synthesis of pyrazole-tethered thioamide and amide conjugates. The thioamides were generated by employing a three-component reaction of diverse pyrazole C-3/4/5 carbaldehydes, secondary amines, and elemental sulfur in a single synthetic operation. The advantages of this developed protocol refer to the broad substrate scope, metal-free and easy to perform reaction conditions. Moreover, the pyrazole C-3/5-linked amide conjugates were also synthesized via an oxidative amination of pyrazole carbaldehydes and 2-aminopyridines using hydrogen peroxide as an oxidant.

Graphical Abstract
Introduction
During the past years, the significance of pyrazole chemistry has been notably escalated which is attributed to the discovery of their amazing biological properties. Among the heterocyclic molecules, pyrazoles are considered as privileged scaffolds for the design and construction of pharmacologically relevant compounds [1-3]. Their effectiveness has been witnessed in agrochemicals [4-6], chemicals, and pharmaceutical industries. Moreover, recent findings have affirmed the potential of the pyrazole nucleus as CB1 receptor antagonists [7], estrogen receptor ligands [8], A2A receptor antagonists [9], and DNA intercalating agents [10]. Importantly, pyrazole derivatives can be traced in a spectrum of well-established drug candidates of various categories with diverse therapeutic properties such as antipyretic [11], antibacterial [12], anticancer [13-15], antiviral [16], analgesic [17], antioxidants, antimicrobial [18], antidiabetic, anticonvulsant [19], antihelminthic [20], and antiarrhythmic activities. The pyrazole nucleus is a core unit in several FDA-approved marketed drugs such as sildenafil [21-23], celebrex [24,25], difenamizole [26], epirizole [27], rimonabant [28] etc. (Figure 1). Additionally, pyrazole derivatives hold a prominent position in the field of materials science as a result of their numerous applications in products like brightening agents [29], semiconductors [30], and organic light-emitting diodes [31]. Substituted pyrazoles are also of considerable interest because of their synthetic utility as chiral auxiliaries [32], synthetic reagents in multicomponent reactions [33,34], and guanylating agents [35].
![[1860-5397-19-22-1]](/bjoc/content/figures/1860-5397-19-22-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Representative drug molecules based on pyrazole, thioamide, and amide derivatives.
Figure 1: Representative drug molecules based on pyrazole, thioamide, and amide derivatives.
The installation of a thioamide functionality has attracted an immense attention in medicinal chemistry, due to various biological activities [36-39]. Accordingly, a broad spectrum of effective and useful methods has been acknowledged in the literature for their preparation [40-42]. In this regard, a review article by Jagodzinski et al. based on the examination of a massive virtual library synthesized with frequently occurring pharmacophores originating from drug components comes to the conclusion that the thioamide linkage establishes an intriguing class of biologically significant compounds amenable to combinatorial chemistry [43]. This organic functional group is found in vital biological and pharmaceutical molecules such as N-cyclohexylethyl-ETAsV [44], carbimazole, methimazole, propylthiouracil [45], and closthioamide [46] (Figure 1). Moreover, they also find widespread applications as intermediates for the construction of five- and six-membered heterocyclic compounds [47] and active pharmaceutical ingredients [48] such as fenclosic acid, fentiazac, and febuxostate.
Similarly, in contemporary chemistry, the amide functionality is one of the most studied functional groups. Specifically, this moiety is vital for the formation of the backbone of structural proteins and enzymes [49]. The amide linkage is present in several naturally occurring compounds and it is also one of the most productive functional groups in current pharmaceutical drugs [50,51]. As prime examples; atorvastatin [52], valsartan [53] and N-cyclohexylethyl-ETAsV are successfully utilized to treat various life challenging diseases (Figure 1). Accordingly, as a part of our ongoing research project, it was planned to incorporate thioamide and amide functional groups into a pyrazole framework to develop new scaffolds.
An extensive literature survey revealed that several approaches are well-documented for the construction of the thioamide functionality including base-catalyzed Willgerodt–Kindler reaction [54], Kindler reaction in the presence of sulfated tungstate [55], thionation of amides using thionating reagents [56] and thionation of amides using TsCl (4-toluenesulfonyl chloride) or PSCl3-mediated Beckmann rearrangement [57]. Although, these protocols are useful and have exhibited wide applications in organic synthesis (Figure 2), the scope of these reported methods may suffer from drawbacks such as harsh reaction conditions, use of expensive reagents, prolonged reaction times, low product yields, and cumbersome product isolation procedures [58-62]. In the recent past, our group also reported two methods towards the exploration of elemental sulfur for the formation of a sulfur-containing framework; however, these methods suffer from some drawbacks such as lack of diversity in starting substrate, need of base/catalyst and limitation of starting reagents [63,64]. Our current work was completed with the exploration of the position of the pyrazole ring like C-3, C-4 and C-5 and we also employed the pyrazole-based AXB3s (4-iodo-C-3 and 4-iodo-C-5). Moreover, we also disclosed the synthesis of pyrazole C-3/C-5 amide conjugates.
![[1860-5397-19-22-2]](/bjoc/content/figures/1860-5397-19-22-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Previous and present findings for the synthesis of thioamide derivatives.
Figure 2: Previous and present findings for the synthesis of thioamide derivatives.
Fascinated by the immense pharmacological profiles of pyrazole, thioamide and amide derivatives, it was envisaged to develop a practical approach towards the synthesis of pyrazole-thioamide and pyrazole-amide conjugates. Elemental sulfur was explored as a sulfurating reagent for the generation of thioamides owing to its nontoxic, odorless nature and versatile reactivity profile [65-76]. To the best of our knowledge, the syntheses of pyrazole C-3/4/5-linked thioamide and amide conjugates have not been reported. Herein, we report an operationally simple one-pot procedure for the preparation of highly diversified thioamide and amide-linked pyrazole analogues.
Results and Discussion
Initially, the synthesis of pyrazole C-3/4/5 carbaldehydes and 4-iodopyrazole-3-carbaldehydes was achieved by employing the recently reported procedures [77-80]. Thereafter, the pyrazole-3-carbaldehyde 1, morpholine (C) and elemental sulfur were selected as the model substrates towards the preparation of pyrazole-linked thioamide derivatives. To begin with, an experiment was executed with model reactants in the presence of catalytic amounts of β-cyclodextrin (β-CD) [81] under aqueous conditions at room temperature as well as under heating at 100 °C (entries 1 and 2, Table 1). Unfortunately, the model reactants remained unreacted and similar observations were recorded using a mixture of H2O/MeOH 1:4, and methanol as a reaction medium (entries 3 and 4, Table 1). Moreover, it was also investigated that various organic solvents in combination with β-CD at room temperature were inactive towards accomplishment of this transformation (entries 5–8, Table 1). Fortunately, when the reaction was performed in CH3CN at 60 °C; a polar product was obtained, which was isolated after a short silica gel column chromatography (entry 9, Table 1). To our delight, the spectroscopic analysis revealed the structure of the purified product as (5-(4-fluorophenyl)-1-phenyl-1H-pyrazol-3-yl)(morpholino)methanethione (1C), which was obtained in 64% isolated yield. Encouraged by these preliminary results, we next assembled the model reactants in DMF as a solvent in the presence of β-CD at 60 °C. It was learned that the outcome of the reaction was slightly better (reaction time was reduced and the yield of the product 1C was increased to 70%, entry 10, Table 1), which indicated the superiority of DMF over other solvents. Subsequently, we examined the effects of La(OTf)3 as a catalyst in combination with DMF as a solvent. However, the targeted prototype 1C was obtained only in 20% yield at 60 °C after 24 hours of reaction time (entries 11 and 12, Table 1). Next, ZnO nanoparticles were screened for the thioamidation of pyrazole-3-carbaldehyde. The desired thioamide-conjugated pyrazole 1C was afforded in 30% yield, as the starting substrates were not completely consumed even after 24 hours of reaction time (entry 13, Table 1). On the basis of the experimental results above, we concluded that CH3CN and DMF were the ideal solvents for this transformation towards the effective formation of the product. As per literature reports, K2CO3 shows remarkable efficacy in various organic transformations [82]. Hence, this reaction was also examined under the influences of K2CO3 (2 equiv) in CH3CN, but the reaction conditions were inactive towards the formation of pyrazole-tethered thioamide 1C (entry 14, Table 1). Surprisingly, when the reaction was carried out in DMF at ambient temperature, the desired product 1C was obtained in 80% yield (entry 15, Table 1). However, the same reaction under heating conditions at 70 °C, afforded the desired product 1C in 82% yield with a drastic reduction in the reaction time to 1 hour (entry 16, Table 1). Moreover, an increase in the amount of base had a negligible effect on the yield of the thioamide conjugate 1C (entries 17 and 18, Table 1). To check the role of K2CO3, we executed a model reaction in DMF without base (K2CO3) and it was noted that pyrazole-linked thioamide 1C was obtained in excellent yield (90%) after 2 hours of reaction time (entry 19, Table 1). This experiment indicated that the K2CO3 was not mandatory for the desired thioamidation reaction. After that, DMSO and NMP were also screened as solvents in the absence of base, but a longer reaction time was required for similar transformation (7 h) (entries 20 and 21, Table 1). A reaction of model substrates under neat conditions delivered product 1C in poor yield (entry 22, Table 1). Based on these screening experiments, it was concluded that the reaction proceeded smoothly in DMF as the reaction medium at 70 °C for 2 hours, and these were considered as the optimal conditions for further investigation of the scope of the developed strategy (entry 19, Table 1).
Table 1: Screening of reaction conditions towards the formation of pyrazole-conjugated thioamide.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-22-i10.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Catalyst/reagent (equiv) | solventb | temp. (oC) | time (h) | isolated yieldc,d |
|---|---|---|---|---|---|
| 1 | β-CD (0.2) | H2O | rt | 7 | NRe |
| 2 | β-CD (0.2) | H2O | 100 | 7 | NR |
| 3 | β-CD (0.2) | MeOH | rt | 7 | NR |
| 4 | β-CD (0.2) | H2O/MeOH 1:4 | rt | 7 | NR |
| 5 | β-CD (0.2) | DCE | rt | 7 | NR |
| 6 | β-CD (0.2) | AcOH | rt | 3 | NR |
| 7 | β-CD (0.2) | CH3CN | rt | 3 | NR |
| 8 | β-CD (0.2) | toluene | rt | 3 | NR |
| 9 | β-CD (0.2) | CH3CN | 60 | 7 | 64% |
| 10 | β-CD (0.2) | DMF | 60 | 3 | 70% |
| 11 | La(OTf)3 (0.1) | DMF | rt | 24 | NR |
| 12 | La(OTf)3 (0.1) | DMF | 60 | 24 | 20% |
| 13 | ZnO NPs (0.1) | DMF | rt | 24 | 30% + 1 |
| 14 | K2CO3 (2.0) | CH3CN | rt | 18 | NR |
| 15 | K2CO3 (1.0) | DMF | rt | 24 | 80% |
| 16 | K2CO3 (1.0) | DMF | 70 | 1 | 82% |
| 17 | K2CO3 (2.0) | DMF | 70 | 1 | 80% |
| 18 | K2CO3 (3.0) | DMF | 70 | 1 | 79% |
| 19 | – | DMF | 70 | 2 | 90% |
| 20 | – | DMSO | 70 | 7 | 88% |
| 21 | – | NMP | 70 | 7 | 85% |
| 22 | – | neat | 70 | 29 | 13% + 1 |
aAll reactions were optimized with 0.07 mmol (1 equiv) of 1, 0.08 mmol (1.1 equiv) of C, 0.28 mmol (4 equiv) of sulfur in 2 mL of solvent; ball reactions were performed in anhydrous solvents (except entries 1, 2, 4, and 22); cisolated yields of the purified product 1C; dNR = no reaction; ethe model substrates remained intact.
Having established the optimal reaction conditions, we explored the generality and the scope of this metal- and catalyst-free approach by employing pyrazole C-3 carbaldehydes 1–4, secondary amines A–E and elemental sulfur as substrates. It was observed that the reaction conditions were compatible with different pyrazole-3-carbaldehydes and various secondary amines for the synthesis of pyrazole C-3-tethered thioamides 1A–E and 2–4C with the yield ranging from 53–90% (Scheme 1). Notably, 1-methylpiperazine (E) afforded the product in low yield (53%). The electronic nature of the substituents located at the N-1 and C-5 positions of the pyrazole ring exerted unnoticeable impacts on the yields of the desired products.
![[1860-5397-19-22-i1]](/bjoc/content/inline/1860-5397-19-22-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of pyrazole C-3-tethered thioamides.
Scheme 1: Synthesis of pyrazole C-3-tethered thioamides.
Encouraged by these successful results, we further investigated the thioamidation reaction of various pyrazole-4-carbaldehydes 5–8 using the optimal conditions as illustrated in Scheme 2. The pyrazole-4-carbaldehydes 5–8 were found to be suitable substrates for this operation. It is pertinent to mention that the substrate 5 reacted with cyclic secondary amines A–C to yield the designed prototypes in moderate to good yields (49–76%), whereas thiomorpholine (D) delivered the thioamide conjugate 5D in low yield (34%). During the preparation of pyrazole C-4-conjugated thioamides 5A–E and 6–8C, it was also noticed that when the reaction was exercised with morpholine (C), the reaction was accomplished in lesser time (36 min to 1 h) as compared to other secondary amines.
![[1860-5397-19-22-i2]](/bjoc/content/inline/1860-5397-19-22-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of pyrazole C-4-tethered thioamides.
Scheme 2: Synthesis of pyrazole C-4-tethered thioamides.
To further validate the synthetic flexibility of this methodology, we employed pyrazole C-5 carbaldehydes 9 and 10 for the synthesis of thioamide conjugates. It was noticed that the pyrazole-5-carbaldehydes 9 and 10 were more reactive as compared to pyrazole C-3 and C-4 carbaldehydes, leading to the formation of products 9C and 10A in high yields (67–71%) within 1 hour of reaction time as depicted in Scheme 3.
![[1860-5397-19-22-i3]](/bjoc/content/inline/1860-5397-19-22-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Metal- and catalyst-free preparation of pyrazole C-5-linked thioamide conjugates.
Scheme 3: Metal- and catalyst-free preparation of pyrazole C-5-linked thioamide conjugates.
Thereafter, the substrates 4-iodopyrazole-3-carbaldehydes were further investigated for this metal- and catalyst-free sulfur insertion reaction as shown in Scheme 4. It was found that 4-iodopyrazole C-3 carbaldehydes 11 and 12 were also tolerated well for this thioamidation process and furnished the anticipated products 11A,B,E, and 12C in good to excellent yields (58–92%) within 40 min to 4 hours.
![[1860-5397-19-22-i4]](/bjoc/content/inline/1860-5397-19-22-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 4-iodopyrazole C-3-tethered thioamides.
Scheme 4: Synthesis of 4-iodopyrazole C-3-tethered thioamides.
To check the industrial scope of the current protocol, we conducted a gram-scale reaction between pyrazole-3-carbaldehyde 1, morpholine (C) and elemental sulfur under the standard reaction conditions as depicted in Scheme 5. It was noticed that this one-pot operation was completed within 2.5 hours and smoothly furnished the desired product, (5-(4-fluorophenyl)-1-phenyl-1H-pyrazol-3-yl)(morpholino)methanethione (1C) in 86% yield.
![[1860-5397-19-22-i5]](/bjoc/content/inline/1860-5397-19-22-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Gram-scale scope of the current protocol.
Scheme 5: Gram-scale scope of the current protocol.
To find out more information about the mechanistic route of the reaction, we performed a control experiment in the presence of TEMPO as a radical scavenger as depicted in Scheme 6. The reaction of pyrazole-3-carbaldehyde 1, pyrrolidine (A) and elemental sulfur in the presence of 1.1 equiv of TEMPO delivered the targeted product in 76% yield. On the basis of this experiment, it was concluded that TEMPO did not affect the progress of the reaction and the formation of product 1A. Hence, a radical mechanism of the reaction may be ruled out.
![[1860-5397-19-22-i6]](/bjoc/content/inline/1860-5397-19-22-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
The successful synthesis of pyrazole C-3/4/5-tethered thioamides inspired us to generate analogous pyrazole-pyridine conjugates having an amide linkage. For this purpose, 5-(4-fluorophenyl)-1-phenyl-1H-pyrazole-3-carbaldehyde (1) and 2-aminopyridine (F) were selected as the model reactants to explore this transformation. Initially, we conducted an oxidative amidation reaction of pyrazole-3-carbaldehyde 1 and 2-aminopyridine (F) in the presence of TBHP in DMSO as a solvent at 130 °C (entry 1, Table 2). However, the reaction required longer time (20 h) for the completion, and afforded a product in 29% yield only. It was realized that the isolated product was the desired product, 5-(4-fluorophenyl)-1-phenyl-N-(pyridin-2-yl)-1H-pyrazole-3-carboxamide (1F), as analyzed by spectroscopic data.
Table 2: Optimization of the reaction conditions towards the formation of pyrazole-pyridine conjugates having an amide linkage.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-19-22-i11.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Oxidant (equiv) | Solventb | Temp. (°C) | Time (h) | Isolated yieldc |
|---|---|---|---|---|---|
| 1 | TBHP (3.0) | DMSO | 130 | 20 | 29% |
| 2 | TBHP (3.0) | DMF (10.0 equiv) | 130 | 10 | 35% + 1 |
| 3 | TBHP (3.0) | CH3CN (10.0 equiv) | 100 | 10 | 37% + 1 |
| 4 | TBHP (3.0) | THF (10.0 equiv) | 100 | 9 | 42% + 1 |
| 5 | TBHP (3.0) | MeOH (10.0 equiv) | 80 | 8 | 40% + 1 |
| 6 | TBHP (10.0) | DMSO (2.0 equiv) | 70 | 18 | 36% + 1 |
| 7 | H2O2 (25.0) | neat | rt | 19 | 30% |
| 8 | H2O2 (5.0) | DMSO | 70 | 19 | 10% + 1 |
| 9 | H2O2 (25.0) | DMSO (2.0 equiv) | 70 | 5 | 40% |
| 10 | H2O2 (10.0) | DMSO (2.0 equiv) | 70 | 4 | 50% |
| 11 | H2O2 (10.0) | DMF (2.0 equiv) | 70 | 8 | 33% + 1 |
| 12 | H2O2 (10.0) | CH3CN (2.0 equiv) | 70 | 5 | 56% |
| 13 | H2O2 (10.0) | THF (2.0 equiv) | 70 | 7 | 58% |
| 14 | H2O2 (10.0) | MeOH (2.0 equiv) | 70 | 6 | 45% |
| 15 | H2O2 (10.0) | CH3CN | 70 | 4 | 54% |
| 16 | H2O2 (10.0) | THF | 70 | 4 | 61% |
aAll the optimization reactions were conducted with 0.07 mmol (1.0 equiv) of 1, 0.08 mmol (1.1 equiv) of F; ball the reactions were examined in dry solvents (except entry 7); cisolated yields of 1F.
Next, we screened other organic solvents including DMF, CH3CN, THF, and MeOH to improve the yield of the desired product 1F, but only a slight improvement in the yield was observed (entries 2–5, Table 2). The oxidant TBHP (10 equiv) failed to deliver the anticipated product in good yield (36%, entry 6, Table 2). Similar results were obtained with H2O2 (25.0 equiv) under neat reaction conditions (entry 7, Table 2). Next, we performed the oxidative amidation reaction with 5.0 equiv of H2O2 in DMSO as the reaction medium under heating, whereas, a poor yield of the product was obtained (entry 8, Table 2). Moreover, different combinations of H2O2 and DMSO were examined for the oxidative amidation of pyrazole-3-carbaldehyde 1 (entries 9 and 10, Table 2). Interestingly, a significant reduction in the reaction time was detected with 25 equiv as well as 10 equiv of H2O2. Next, we screened DMF, CH3CN, THF, and MeOH (2.0 equiv) with 10.0 equiv of H2O2 to increase the yield of the designed prototype 1F. An acceptable enhancement was observed in the yield of the desired compound (58%) 1F (entries 11–14, Table 2). After that, we subjected all the starting substrates to an excess amount of CH3CN and THF as reaction solvents (entries 15 and 16, Table 2). It was noticed from these two experiments that THF was the outstanding solvent for our current transformation (entry 16, Table 2). From the above screening experiments, it was concluded that 10.0 equiv of hydrogen peroxide in THF at 70 °C proved to be the optimal conditions for the construction of the pyrazole-pyridine conjugate with an amide linkage (entry 16, Table 2).
Having the optimized conditions in hand, we employed pyrazole-3-carbaldehydes 1 and 4 for the reaction with different 2-aminopyridines F and G towards the preparation of amide tethers as displayed in Scheme 7. The pyrazole-3-carbaldehydes 1 and 4 reacted efficiently with 2-aminopyridine (F) to deliver the pyrazole conjugated amides 1F and 4F in good yields (61 and 70%), whereas, in the case of 5-nitro-2-aminopyridine (G), the anticipated product 1G was obtained in low yield (34%).
![[1860-5397-19-22-i7]](/bjoc/content/inline/1860-5397-19-22-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: H2O2-mediated synthesis of pyrazole-pyridine conjugates with amide tethers.
Scheme 7: H2O2-mediated synthesis of pyrazole-pyridine conjugates with amide tethers.
To check the synthetic versatility of this oxidative amidation approach, we tested the scope of the methodology with pyrazole-5-carbaldehydes 9 and 10. Using this method, 3-(4-chlorophenyl)-1-phenyl-N-(pyridin-2-yl)-1H-pyrazole-5-carboxamide (10F) was produced in good yield (62%), while 9F was generated in low yield (36%) as depicted in Scheme 8.
![[1860-5397-19-22-i8]](/bjoc/content/inline/1860-5397-19-22-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of pyrazole-pyridine conjugates 9F and 10F having amide tethers.
Scheme 8: Synthesis of pyrazole-pyridine conjugates 9F and 10F having amide tethers.
Based on the current experimental observations and literature reports [62,83] a plausible mechanistic pathway is outlined in Scheme 9 for the formation of the thioamide and amide-linked pyrazole derivatives 1C and 1F. It is proposed that initially pyrazole-3-carbaldehyde 1 reacted with morpholine (C) to furnish the iminium intermediate 13. Meanwhile, the intermediate polysulfide 14 formed by the nucleophilic attack of morpholine (C) on elemental sulfur may react with the intermediate 13 to afford another intermediate 15, which undergoes oxidation to release the thioamide-tethered pyrazole 1C. On the other hand, the pyrazole carbaldehyde 1 forms imine intermediate 16 by condensation with 2-aminopyridine. Thereafter, a nucleophilic attack of H2O2 on the imine carbon may afford the intermediate 17. Finally, the loss of a water molecule from the intermediate 17 may generate the pyrazole-pyridine conjugate with amide linkage 1F.
![[1860-5397-19-22-i9]](/bjoc/content/inline/1860-5397-19-22-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: A tentative mechanism for the formation of pyrazole conjugates with thioamide and amide linkage.
Scheme 9: A tentative mechanism for the formation of pyrazole conjugates with thioamide and amide linkage.
Conclusion
In summary, a simple, straightforward and efficient approach for the construction of biologically interesting highly diversified pyrazole-linked thioamide and amide conjugates has been developed. The pyrazole C-3/4/5-tethered thioamide conjugates were prepared via a one-pot reaction between highly diversified pyrazole carbaldehydes, cyclic secondary amines, and elemental sulfur under metal and catalyst-free conditions. The salient features of the current protocol may be attributed to the broad substrate scope, commercially available secondary amines, operational simplicity, multicomponent character of the reaction, easy isolation of products, short reaction time, and good to excellent yields of the desired molecules. Moreover, a practical synthetic utility of pyrazole-3/5-carbaldehydes has been explored through the formation of amide bond-tethered pyrazole-pyridine conjugates. This developed methodology was successfully carried out by employing commercially available substituted 2-aminopyridines and hydrogen peroxide as an oxidant. The biological evaluation of the thioamide and amide conjugates is underway in our laboratory.
Experimental
General information
All chemicals and reagents were purchased from Sigma-Aldrich, Acros, Avera Synthesis, Spectrochem Pvt. Ltd., and used without further purification. Commercially available anhydrous solvents (THF, DMF, benzene, toluene, MeOH, EtOH, and CH2Cl2 Spectrochem) were used in the reactions. Thin-layer chromatography (TLC) was performed using precoated aluminum plates purchased from E. Merck (silica gel 60 PF254, 0.25 mm). Column chromatography was performed using Spectrochem silica gel (60–120 mesh). Melting points were determined in open capillary tubes on the Precision Digital melting point apparatus (LABCO make) containing silicone oil, and the results are uncorrected. IR spectra (neat) were recorded on an Agilent FTIR spectrophotometer. 1H and 13C NMR spectra were recorded either on an Avance III Bruker or a JEOL JNM-ECS spectrometer at operating frequencies of 200/400/500 MHz (1H) and or 100/125/150 MHz (13C) as indicated in the individual spectra using TMS as an internal standard. Elemental analyses were performed on a Carlo-Erba 108 or an Elementar Vario EL III microanalyzer. The room temperature varied between 25 °C and 30 °C. The multiplicities in the 1HNMR spectra are presented as s for singlet, d for doublet, dd for doublet of the doublet, td for a triplet of doublet, t for triplet and m for multiplet. The multiplicity in the 13C NMR spectra is presented as d for doublet.
Experimental procedures
General procedure for the synthesis of compounds 1A–E, 2–4C, 5A–E, 6–8C, 9C, 10A, 11A,B, 11E, and 12C as exemplified for (5-(4-fluorophenyl)-1-phenyl-1H-pyrazol-3-yl)(morpholino)methanethione (1C): In a dry round-bottomed flask, pyrazole-3-carbaldehyde 1 (0.20 g, 0.75 mmol), morpholine (C, 0.072 g, 0.83 mmol), and sulfur powder (0.096 g, 3 mmol) were added to dry DMF (2 mL) at room temperature. The reaction flask was heated at 70 °C in an oil bath for 1 h. After completion of the reaction, as determined by TLC, cold water was added to the reaction mixture at room temperature which resulted in precipitation of the product. The product was collected by filtration under reduced pressure using a Büchner funnel and further purified by silica gel column chromatography (60–120 mesh silica gel) using hexane and ethyl acetate as an eluent (80:20, v/v) to give the final product 1C (0.247 g, 90%; Rf 0.19 (hexane/EtOAc 90:10, v/v)).
Gram-scale synthesis of (5-(4-fluorophenyl)-1-phenyl-1H-pyrazol-3-yl)(morpholino)methanethione (1C): A 50 mL round-bottomed flask was charged with pyrazole-3-carbaldehyde 1 (1 g, 3.74 mmol), morpholine (C, 0.36 g, 4.14 mmol), and elemental sulfur (0.48 g, 15 mmol) in dry DMF (10 mL) followed by heating of the reaction mixture at 70 °C for 2.5 h. On completion of the reaction, as determined by TLC, the reaction content was cooled to room temperature and poured into ice-cold water under stirring, which resulted in the formation of a precipitate. The solid was collected under vacuum using a Büchner funnel and further purified by silica gel column chromatography (60–120 mesh silica gel) using hexane and ethyl acetate (80:20, v/v) as an eluent to give the analytically pure product 1C (1.18 g from 1 g, 86%; Rf 0.19 (hexane/EtOAc 90:10, v/v)).
Procedure for the synthesis of (5-(4-fluorophenyl)-1-phenyl-1H-pyrazol-3-yl)(pyrrolidin-1-yl)methanethione (1A) through control experiment: In a dry round-bottomed flask, pyrazole-3-carbaldehyde 1 (0.05 g, 0.19 mmol), pyrrolidine (A, 0.015 g, 0.21 mmol), and sulfur powder (0.024 g, 0.75 mmol) were added to dry DMF (2 mL) at room temperature. The reaction flask was heated at 70 °C in an oil bath for 3.5 h. After completion of the reaction, as monitored by the TLC, cold water was added to the reaction mixture at room temperature which resulted in the formation of a precipitate. The product was collected by filtration under reduced pressure using a Büchner funnel and further purified by silica gel column chromatography (60–120 mesh silica gel) using hexane and ethyl acetate as an eluent (80:20, v/v) to give final product 1A (0.049 g, 76%; Rf 0.68, (hexane/EtOAc 70:30, v/v)).
Typical procedure for the synthesis of compounds 1F, 1G, 4F, 9F, and 10F as exemplified for 5-(4-fluorophenyl)-1-phenyl-N-(pyridin-2-yl)-1H-pyrazole-3-carboxamide (1F): To a stirred solution of compound 1 (0.10 g, 0.37 mmol) and 2-aminopyridine (F, 0.04 g, 0.42 mmol) in dry THF, H2O2 (0.087 mL, 3.73 mmol) was added dropwise at room temperature and the reaction mixture was heated at 70 °C for 20 h. Upon completion of the reaction, as monitored by TLC, the reaction mixture was cooled to room temperature, water was added, and the product was extracted with ethyl acetate (3 × 25 mL). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure to afford crude product 1F. This material was purified by silica gel column chromatography (60–120 mesh) using hexane and ethyl acetate as an eluent (95:05, v/v) to get the analytically pure product 1F (0.082 g, 61%; Rf 0.63, (hexane/EtOAc 90:10, v/v)).
Supporting Information
| Supporting Information File 1: Analytical data and copies of spectra. | ||
| Format: PDF | Size: 3.3 MB | Download |
Acknowledgements
S. S. and V. S. gratefully acknowledges the CIL, Central University of Punjab, Bathinda and Advanced Material and Research Centre (AMRC) at the Indian Institute of Technology Mandi, HP, India for recording the spectroscopic data.
Funding
S.S. acknowledges the Ministry of Human Resource Development (MHRD), New Delhi, India, and CSIR, New Delhi for Junior Research Fellowships. S. K. and V. S. gratefully acknowledges the financial support in the form of research grants from CSIR (02 (0202) /14/EMR-II), DST (CS-361/ 2011), and DST-FIST (CSI-228/2011) New Delhi (India).
References
-
Da Costa, L.; Scheers, E.; Coluccia, A.; Casulli, A.; Roche, M.; Di Giorgio, C.; Neyts, J.; Terme, T.; Cirilli, R.; La Regina, G.; Silvestri, R.; Mirabelli, C.; Vanelle, P. J. Med. Chem. 2018, 61, 8402–8416. doi:10.1021/acs.jmedchem.8b00931
Return to citation in text: [1] -
Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y. N.; Al-aizari, F. A.; Ansar, M. Molecules 2018, 23, 134–220. doi:10.3390/molecules23010134
Return to citation in text: [1] -
Nandi, G. C.; Singh, M. S.; Ila, H.; Junjappa, H. Eur. J. Org. Chem. 2012, 967–974. doi:10.1002/ejoc.201101397
Return to citation in text: [1] -
Yoshikawa, Y.; Katsuta, H.; Kishi, J.; Yanase, Y. J. Pestic. Sci. (Tokyo, Jpn.) 2011, 36, 347–356. doi:10.1584/jpestics.g10-70
Return to citation in text: [1] -
Numata, A.; Tanima, D.; Ando, M.; Iwawaki, Y. Pyrazole derivatives and pesticides. US2013/0338367A1, Dec 19, 2013.
Return to citation in text: [1] -
Wang, M.-M.; Huang, H.; Shu, L.; Liu, J.-M.; Zhang, J.-Q.; Yan, Y.-L.; Zhang, D.-Y. Beilstein J. Org. Chem. 2020, 16, 233–247. doi:10.3762/bjoc.16.25
Return to citation in text: [1] -
Wang, H.; Duffy, R. A.; Boykow, G. C.; Chackalamannil, S.; Madison, V. S. J. Med. Chem. 2008, 51, 2439–2446. doi:10.1021/jm701519h
Return to citation in text: [1] -
Naoum, F.; Kasiotis, K. M.; Magiatis, P.; Haroutounian, S. A. Molecules 2007, 12, 1259–1273. doi:10.3390/12071259
Return to citation in text: [1] -
Slee, D. H.; Moorjani, M.; Zhang, X.; Lin, E.; Lanier, M. C.; Chen, Y.; Rueter, J. K.; Lechner, S. M.; Markison, S.; Malany, S.; Joswig, T.; Santos, M.; Gross, R. S.; Williams, J. P.; Castro-Palomino, J. C.; Crespo, M. I.; Prat, M.; Gual, S.; Díaz, J.-L.; Jalali, K.; Sai, Y.; Zuo, Z.; Yang, C.; Wen, J.; O’Brien, Z.; Petroski, R.; Saunders, J. J. Med. Chem. 2008, 51, 1730–1739. doi:10.1021/jm701187w
Return to citation in text: [1] -
Lauria, A.; Abbate, I.; Patella, C.; Gambino, N.; Silvestri, A.; Barone, G.; Almerico, A. M. Tetrahedron Lett. 2008, 49, 5125–5128. doi:10.1016/j.tetlet.2008.06.104
Return to citation in text: [1] -
Chandra, T.; Garg, N.; Lata, S.; Saxena, K. K.; Kumar, A. Eur. J. Med. Chem. 2010, 45, 1772–1776. doi:10.1016/j.ejmech.2010.01.009
Return to citation in text: [1] -
Abdel-Hafez, E.-S. M. N.; Abuo-Rahma, G. E.-D. A. A.; Abdel-Aziz, M.; Radwan, M. F.; Farag, H. H. Bioorg. Med. Chem. 2009, 17, 3829–3837. doi:10.1016/j.bmc.2009.04.037
Return to citation in text: [1] -
Lv, P.-C.; Li, H.-Q.; Sun, J.; Zhou, Y.; Zhu, H.-L. Bioorg. Med. Chem. 2010, 18, 4606–4614. doi:10.1016/j.bmc.2010.05.034
Return to citation in text: [1] -
Balbi, A.; Anzaldi, M.; Macciò, C.; Aiello, C.; Mazzei, M.; Gangemi, R.; Castagnola, P.; Miele, M.; Rosano, C.; Viale, M. Eur. J. Med. Chem. 2011, 46, 5293–5309. doi:10.1016/j.ejmech.2011.08.014
Return to citation in text: [1] -
Hura, N.; Naaz, A.; Prassanawar, S. S.; Guchhait S. K; Panda, D. ACS Omega 2018, 3, 1955–1969. doi:10.1021/acsomega.7b01784
Return to citation in text: [1] -
El-Sabbagh, O. I.; Baraka, M. M.; Ibrahim, S. M.; Pannecouque, C.; Andrei, G.; Snoeck, R.; Balzarini, J.; Rashad, A. A. Eur. J. Med. Chem. 2009, 44, 3746–3753. doi:10.1016/j.ejmech.2009.03.038
Return to citation in text: [1] -
Hall, A.; Billinton, A.; Brown, S. H.; Clayton, N. M.; Chowdhury, A.; Giblin, G. M. P.; Goldsmith, P.; Hayhow, T. G.; Hurst, D. N.; Kilford, I. R.; Naylor, A.; Passingham, B.; Winyard, L. Bioorg. Med. Chem. Lett. 2008, 18, 3392–3399. doi:10.1016/j.bmcl.2008.04.018
Return to citation in text: [1] -
Sharshira, E. M.; Hamada, N. M. M. Molecules 2012, 17, 4962–4971. doi:10.3390/molecules17054962
Return to citation in text: [1] -
Chimenti, F.; Bolasco, A.; Manna, F.; Secci, D.; Chimenti, P.; Befani, O.; Turini, P.; Giovannini, V.; Mondovì, B.; Cirilli, R.; La Torre, F. J. Med. Chem. 2004, 47, 2071–2074. doi:10.1021/jm031042b
Return to citation in text: [1] -
Partridge, F. A.; Forman, R.; Bataille, C. J. R.; Wynne, G. M.; Nick, M.; Russell, A. J.; Else, K. J.; Sattelle, D. B. Beilstein J. Org. Chem. 2020, 16, 1203–1224. doi:10.3762/bjoc.16.105
Return to citation in text: [1] -
Dale, D. J.; Dunn, P. J.; Golightly, C.; Hughes, M. L.; Levett, P. C.; Pearce, A. K.; Searle, P. M.; Ward, G.; Wood, A. S. Org. Process Res. Dev. 2000, 4, 17–22. doi:10.1021/op9900683
Return to citation in text: [1] -
Dunn, P. J.; Galvin, S.; Hettenbach, K. Green Chem. 2004, 6, 43–48. doi:10.1039/b312329d
Return to citation in text: [1] -
Ghozlan, S. A. S.; Badahdah, K. O.; Abdelhamid, I. A. Beilstein J. Org. Chem. 2007, 3, No. 15. doi:10.1186/1860-5397-3-15
Return to citation in text: [1] -
Penning, T. D.; Talley, J. J.; Bertenshaw, S. R.; Carter, J. S.; Collins, P. W.; Docter, S.; Graneto, M. J.; Lee, L. F.; Malecha, J. W.; Miyashiro, J. M.; Rogers, R. S.; Rogier, D. J.; Yu, S. S.; Anderson, G. D.; Burton, E. G.; Cogburn, J. N.; Gregory, S. A.; Koboldt, C. M.; Perkins, W. E.; Seibert, K.; Veenhuizen, A. W.; Zhang, Y. Y.; Isakson, P. C. J. Med. Chem. 1997, 40, 1347–1365. doi:10.1021/jm960803q
Return to citation in text: [1] -
Baumann, M.; Baxendale, I. R.; Ley, S. V.; Nikbin, N. Beilstein J. Org. Chem. 2011, 7, 442–495. doi:10.3762/bjoc.7.57
Return to citation in text: [1] -
Kameyama, T.; Nabeshima, T. Neuropharmacology 1978, 17, 249–256. doi:10.1016/0028-3908(78)90108-9
Return to citation in text: [1] -
Chen, C.-M.; Kositprapa, U. Pharmaceutical formulations containing a non-steroidal antiinflammatory drug and a proton pump inhibitor. US Patent US6869615B2, March 22, 2005.
Return to citation in text: [1] -
Kotagiri, V. K.; Suthrapu, S.; Reddy, J. M.; Rao, C. P.; Bollugoddu, V.; Bhattacharya, A.; Bandichhor, R. Org. Process Res. Dev. 2007, 11, 910–912. doi:10.1021/op700110b
Return to citation in text: [1] -
Wang, M.; Zhang, J.; Liu, J.; Xu, C.; Ju, H. J. Lumin. 2002, 99, 79–83. doi:10.1016/s0022-2313(01)00204-6
Return to citation in text: [1] -
Burschka, J.; Kessler, F.; Nazeeruddin, M. K.; Grätzel, M. Chem. Mater. 2013, 25, 2986–2990. doi:10.1021/cm400796u
Return to citation in text: [1] -
Chou, P.-T.; Chi, Y. Chem. – Eur. J. 2007, 13, 380–395. doi:10.1002/chem.200601272
Return to citation in text: [1] -
Molteni, G.; Buttero, P. D. Tetrahedron: Asymmetry 2005, 16, 1983–1987. doi:10.1016/j.tetasy.2005.04.014
Return to citation in text: [1] -
Tu, X.-J.; Hao, W.-J.; Ye, Q.; Wang, S.-S.; Jiang, B.; Li, G.; Tu, S.-J. J. Org. Chem. 2014, 79, 11110–11118. doi:10.1021/jo502096t
Return to citation in text: [1] -
Yadav, P.; Awasthi, A.; Gokulnath, S.; Tiwari, D. K. J. Org. Chem. 2021, 86, 2658–2666. doi:10.1021/acs.joc.0c02696
Return to citation in text: [1] -
Castagnolo, D.; Schenone, S.; Botta, M. Chem. Rev. 2011, 111, 5247–5300. doi:10.1021/cr100423x
Return to citation in text: [1] -
Bartlett, P. A.; Spear, K. L.; Jacobsen, N. E. Biochemistry 1982, 21, 1608–1611. doi:10.1021/bi00536a022
Return to citation in text: [1] -
Yu, K.-L.; Torri, A. F.; Luo, G.; Cianci, C.; Grant-Young, K.; Danetz, S.; Tiley, L.; Krystal, M.; Meanwell, N. A. Bioorg. Med. Chem. Lett. 2002, 12, 3379–3382. doi:10.1016/s0960-894x(02)00761-8
Return to citation in text: [1] -
Gannon, M. K.; Holt, J. J.; Bennett, S. M.; Wetzel, B. R.; Loo, T. W.; Bartlett, M. C.; Clarke, D. M.; Sawada, G. A.; Higgins, J. W.; Tombline, G.; Raub, T. J.; Detty, M. R. J. Med. Chem. 2009, 52, 3328–3341. doi:10.1021/jm900253g
Return to citation in text: [1] -
Banala, S.; Süssmuth, R. D. ChemBioChem 2010, 11, 1335–1337. doi:10.1002/cbic.201000266
Return to citation in text: [1] -
Mahanta, N.; Szantai-Kis, D. M.; Petersson, E. J.; Mitchell, D. A. ACS Chem. Biol. 2019, 14, 142–163. doi:10.1021/acschembio.8b01022
Return to citation in text: [1] -
Kumar, K.; Konar, D.; Goyal, S.; Gangar, M.; Chouhan, M.; Rawal, R. K.; Nair, V. A. ChemistrySelect 2016, 1, 3228–3231. doi:10.1002/slct.201600601
Return to citation in text: [1] -
Chaubey, T. N.; Borpatra, P. J.; Sharma, A.; Pandey, S. K. Org. Lett. 2022, 24, 8062–8066. doi:10.1021/acs.orglett.2c03371
Return to citation in text: [1] -
Jagodziński, T. S. Chem. Rev. 2003, 103, 197–228. doi:10.1021/cr0200015
Return to citation in text: [1] -
Bach, A.; Eildal, J. N. N.; Stuhr-Hansen, N.; Deeskamp, R.; Gottschalk, M.; Pedersen, S. W.; Kristensen, A. S.; Strømgaard, K. J. Med. Chem. 2011, 54, 1333–1346. doi:10.1021/jm1013924
Return to citation in text: [1] -
Nakamura, H.; Noh, J. Y.; Itoh, K.; Fukata, S.; Miyauchi, A.; Hamada, N. J. Clin. Endocrinol. Metab. 2007, 92, 2157–2162. doi:10.1210/jc.2006-2135
Return to citation in text: [1] -
Lincke, T.; Behnken, S.; Ishida, K.; Roth, M.; Hertweck, C. Angew. Chem. 2010, 122, 2055–2057. doi:10.1002/ange.200906114
Return to citation in text: [1] -
Hisano, T.; Yabuta, Y. Chem. Pharm. Bull. 1973, 21, 511–517. doi:10.1248/cpb.21.511
Return to citation in text: [1] -
Lednicer, D. Strategies for Organic Drug Synthesis and Design; John Wiley: Hoboken, 2008. doi:10.1002/9780470399613
Return to citation in text: [1] -
Montalbetti, C. A. G. N.; Falque, V. Tetrahedron 2005, 61, 10827–10852. doi:10.1016/j.tet.2005.08.031
Return to citation in text: [1] -
Dunetz, J. R.; Magano, J.; Weisenburger, G. A. Org. Process Res. Dev. 2016, 20, 140–177. doi:10.1021/op500305s
Return to citation in text: [1] -
Nandi, G. C.; Samai, S.; Singh, M. S. J. Org. Chem. 2010, 75, 7785–7795. doi:10.1021/jo101572c
Return to citation in text: [1] -
Lau, W. C.; Waskell, L. A.; Watkins, P. B.; Neer, C. J.; Horowitz, K.; Hopp, A. S.; Tait, A. R.; Carville, D. G. M.; Guyer, K. E.; Bates, E. R. Circulation 2003, 107, 32–37. doi:10.1161/01.cir.0000047060.60595.cc
Return to citation in text: [1] -
Abraham, I.; MacDonald, K.; Hermans, C.; Aerts, A.; Lee, C.; Brie, H.; Vancayzeele, S. Vasc. Health Risk Manage. 2011, 7, 209–235. doi:10.2147/vhrm.s9434
Return to citation in text: [1] -
Okamoto, K.; Yamamoto, T.; Kanbara, T. Synlett 2007, 2687–2690. doi:10.1055/s-2007-991073
Return to citation in text: [1] -
Pathare, S. P.; Chaudhari, P. S.; Akamanchi, K. G. Appl. Catal., A 2012, 425-426, 125–129. doi:10.1016/j.apcata.2012.03.012
Return to citation in text: [1] -
Curphey, T. J. J. Org. Chem. 2002, 67, 6461–6473. doi:10.1021/jo0256742
Return to citation in text: [1] -
Pathak, U.; Pandey, L. K.; Mathur, S.; Suryanarayana, M. V. S. Chem. Commun. 2009, 5409–5411. doi:10.1039/b911844f
Return to citation in text: [1] -
Manaka, A.; Sato, M. Synth. Commun. 2005, 35, 761–764. doi:10.1081/scc-200050393
Return to citation in text: [1] -
Xu, H.; Deng, H.; Li, Z.; Xiang, H.; Zhou, X. Eur. J. Org. Chem. 2013, 7054–7057. doi:10.1002/ejoc.201301148
Return to citation in text: [1] -
Yin, Z.; Zheng, B. J. Sulfur Chem. 2013, 34, 527–531. doi:10.1080/17415993.2013.765429
Return to citation in text: [1] -
Wei, J.; Li, Y.; Jiang, X. Org. Lett. 2016, 18, 340–343. doi:10.1021/acs.orglett.5b03541
Return to citation in text: [1] -
Tayade, Y. A.; Jangale, A. D.; Dalal, D. S. ChemistrySelect 2018, 3, 8895–8900. doi:10.1002/slct.201801553
Return to citation in text: [1] [2] -
Sharma, S.; Malakar, C. C.; Singh, V. Asian J. Org. Chem. 2020, 9, 1857–1868. doi:10.1002/ajoc.202000390
Return to citation in text: [1] -
Singh, M.; Vaishali; Kumar, R.; Singh, V. ChemistrySelect 2020, 5, 5172–5179. doi:10.1002/slct.202001149
Return to citation in text: [1] -
Nguyen, T. B. Adv. Synth. Catal. 2017, 359, 1066–1130. doi:10.1002/adsc.201601329
Return to citation in text: [1] -
Nguyen, T. B. Adv. Synth. Catal. 2020, 362, 3448–3484. doi:10.1002/adsc.202000535
Return to citation in text: [1] -
Singh, M.; Awasthi, P.; Singh, V. Eur. J. Org. Chem. 2020, 1023–1041. doi:10.1002/ejoc.201901908
Return to citation in text: [1] -
Wu, K.; Ling, Y.; Ding, A.; Jin, L.; Sun, N.; Hu, B.; Shen, Z.; Hu, X. Beilstein J. Org. Chem. 2021, 17, 805–812. doi:10.3762/bjoc.17.69
Return to citation in text: [1] -
Ilkin, V.; Berseneva, V.; Beryozkina, T.; Glukhareva, T.; Dianova, L.; Dehaen, W.; Seliverstova, E.; Bakulev, V. Beilstein J. Org. Chem. 2020, 16, 2937–2947. doi:10.3762/bjoc.16.243
Return to citation in text: [1] -
Tang, Q.; Yin, X.; Kuchukulla, R. R.; Zeng, Q. Chem. Rec. 2021, 21, 893–905. doi:10.1002/tcr.202100026
Return to citation in text: [1] -
Liu, S.; Deng, G.-J.; Huang, H. Synlett 2021, 32, 142–158. doi:10.1055/s-0040-1707217
Return to citation in text: [1] -
Beletskaya, I. P.; Ananikov, V. P. Chem. Rev. 2022, 122, 16110–16293. doi:10.1021/acs.chemrev.1c00836
Return to citation in text: [1] -
Yu, Z.; Su, J.; Huang, C.; Wei, J.; Han, D. L.; Ye, D. Q.; Li, Y. Asian J. Org. Chem. 2022, 11, e202200288. doi:10.1002/ajoc.202200288
Return to citation in text: [1] -
Koyanagi, A.; Murata, Y.; Hayakawa, S.; Matsumura, M.; Yasuike, S. Beilstein J. Org. Chem. 2022, 18, 1479–1487. doi:10.3762/bjoc.18.155
Return to citation in text: [1] -
Pošta, M.; Zima, V.; Poštová Slavětínská, L.; Matoušová, M.; Beier, P. Beilstein J. Org. Chem. 2022, 18, 549–554. doi:10.3762/bjoc.18.57
Return to citation in text: [1] -
Pathania, S.; Narang, R. K.; Rawal, R. K. Eur. J. Med. Chem. 2019, 180, 486–508. doi:10.1016/j.ejmech.2019.07.043
Return to citation in text: [1] -
Sharma, S.; Paul, A. K.; Singh, V. New J. Chem. 2020, 44, 684–694. doi:10.1039/c9nj05426j
Return to citation in text: [1] -
Devi, N.; Singh, D.; Sunkaria, R. K.; Malakar, C. C.; Mehra, S.; Rawal, R. K.; Singh, V. ChemistrySelect 2016, 1, 4696–4703. doi:10.1002/slct.201601133
Return to citation in text: [1] -
Devi, N.; Shankar, R.; Singh, V. J. Heterocycl. Chem. 2018, 55, 373–390. doi:10.1002/jhet.3045
Return to citation in text: [1] -
Nag, S.; Singh, V.; Batra, S. ARKIVOC 2007, No. 14, 185–203. doi:10.3998/ark.5550190.0008.e18
Return to citation in text: [1] -
Mondal, R.; Mallik, A. K. Org. Prep. Proced. Int. 2014, 46, 391–434. doi:10.1080/00304948.2014.944402
Return to citation in text: [1] -
Dheer, D.; Rawal, R. K.; Singh, V.; Sangwan, P. L.; Das, P.; Shankar, R. Tetrahedron 2017, 73, 4295–4306. doi:10.1016/j.tet.2017.05.081
Return to citation in text: [1] -
Sankari Devi, E.; Alanthadka, A.; Tamilselvi, A.; Nagarajan, S.; Sridharan, V.; Maheswari, C. U. Org. Biomol. Chem. 2016, 14, 8228–8231. doi:10.1039/c6ob01454b
Return to citation in text: [1]
| 44. | Bach, A.; Eildal, J. N. N.; Stuhr-Hansen, N.; Deeskamp, R.; Gottschalk, M.; Pedersen, S. W.; Kristensen, A. S.; Strømgaard, K. J. Med. Chem. 2011, 54, 1333–1346. doi:10.1021/jm1013924 |
| 45. | Nakamura, H.; Noh, J. Y.; Itoh, K.; Fukata, S.; Miyauchi, A.; Hamada, N. J. Clin. Endocrinol. Metab. 2007, 92, 2157–2162. doi:10.1210/jc.2006-2135 |
| 1. | Da Costa, L.; Scheers, E.; Coluccia, A.; Casulli, A.; Roche, M.; Di Giorgio, C.; Neyts, J.; Terme, T.; Cirilli, R.; La Regina, G.; Silvestri, R.; Mirabelli, C.; Vanelle, P. J. Med. Chem. 2018, 61, 8402–8416. doi:10.1021/acs.jmedchem.8b00931 |
| 2. | Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y. N.; Al-aizari, F. A.; Ansar, M. Molecules 2018, 23, 134–220. doi:10.3390/molecules23010134 |
| 3. | Nandi, G. C.; Singh, M. S.; Ila, H.; Junjappa, H. Eur. J. Org. Chem. 2012, 967–974. doi:10.1002/ejoc.201101397 |
| 9. | Slee, D. H.; Moorjani, M.; Zhang, X.; Lin, E.; Lanier, M. C.; Chen, Y.; Rueter, J. K.; Lechner, S. M.; Markison, S.; Malany, S.; Joswig, T.; Santos, M.; Gross, R. S.; Williams, J. P.; Castro-Palomino, J. C.; Crespo, M. I.; Prat, M.; Gual, S.; Díaz, J.-L.; Jalali, K.; Sai, Y.; Zuo, Z.; Yang, C.; Wen, J.; O’Brien, Z.; Petroski, R.; Saunders, J. J. Med. Chem. 2008, 51, 1730–1739. doi:10.1021/jm701187w |
| 21. | Dale, D. J.; Dunn, P. J.; Golightly, C.; Hughes, M. L.; Levett, P. C.; Pearce, A. K.; Searle, P. M.; Ward, G.; Wood, A. S. Org. Process Res. Dev. 2000, 4, 17–22. doi:10.1021/op9900683 |
| 22. | Dunn, P. J.; Galvin, S.; Hettenbach, K. Green Chem. 2004, 6, 43–48. doi:10.1039/b312329d |
| 23. | Ghozlan, S. A. S.; Badahdah, K. O.; Abdelhamid, I. A. Beilstein J. Org. Chem. 2007, 3, No. 15. doi:10.1186/1860-5397-3-15 |
| 53. | Abraham, I.; MacDonald, K.; Hermans, C.; Aerts, A.; Lee, C.; Brie, H.; Vancayzeele, S. Vasc. Health Risk Manage. 2011, 7, 209–235. doi:10.2147/vhrm.s9434 |
| 8. | Naoum, F.; Kasiotis, K. M.; Magiatis, P.; Haroutounian, S. A. Molecules 2007, 12, 1259–1273. doi:10.3390/12071259 |
| 24. | Penning, T. D.; Talley, J. J.; Bertenshaw, S. R.; Carter, J. S.; Collins, P. W.; Docter, S.; Graneto, M. J.; Lee, L. F.; Malecha, J. W.; Miyashiro, J. M.; Rogers, R. S.; Rogier, D. J.; Yu, S. S.; Anderson, G. D.; Burton, E. G.; Cogburn, J. N.; Gregory, S. A.; Koboldt, C. M.; Perkins, W. E.; Seibert, K.; Veenhuizen, A. W.; Zhang, Y. Y.; Isakson, P. C. J. Med. Chem. 1997, 40, 1347–1365. doi:10.1021/jm960803q |
| 25. | Baumann, M.; Baxendale, I. R.; Ley, S. V.; Nikbin, N. Beilstein J. Org. Chem. 2011, 7, 442–495. doi:10.3762/bjoc.7.57 |
| 54. | Okamoto, K.; Yamamoto, T.; Kanbara, T. Synlett 2007, 2687–2690. doi:10.1055/s-2007-991073 |
| 7. | Wang, H.; Duffy, R. A.; Boykow, G. C.; Chackalamannil, S.; Madison, V. S. J. Med. Chem. 2008, 51, 2439–2446. doi:10.1021/jm701519h |
| 19. | Chimenti, F.; Bolasco, A.; Manna, F.; Secci, D.; Chimenti, P.; Befani, O.; Turini, P.; Giovannini, V.; Mondovì, B.; Cirilli, R.; La Torre, F. J. Med. Chem. 2004, 47, 2071–2074. doi:10.1021/jm031042b |
| 50. | Dunetz, J. R.; Magano, J.; Weisenburger, G. A. Org. Process Res. Dev. 2016, 20, 140–177. doi:10.1021/op500305s |
| 51. | Nandi, G. C.; Samai, S.; Singh, M. S. J. Org. Chem. 2010, 75, 7785–7795. doi:10.1021/jo101572c |
| 4. | Yoshikawa, Y.; Katsuta, H.; Kishi, J.; Yanase, Y. J. Pestic. Sci. (Tokyo, Jpn.) 2011, 36, 347–356. doi:10.1584/jpestics.g10-70 |
| 5. | Numata, A.; Tanima, D.; Ando, M.; Iwawaki, Y. Pyrazole derivatives and pesticides. US2013/0338367A1, Dec 19, 2013. |
| 6. | Wang, M.-M.; Huang, H.; Shu, L.; Liu, J.-M.; Zhang, J.-Q.; Yan, Y.-L.; Zhang, D.-Y. Beilstein J. Org. Chem. 2020, 16, 233–247. doi:10.3762/bjoc.16.25 |
| 20. | Partridge, F. A.; Forman, R.; Bataille, C. J. R.; Wynne, G. M.; Nick, M.; Russell, A. J.; Else, K. J.; Sattelle, D. B. Beilstein J. Org. Chem. 2020, 16, 1203–1224. doi:10.3762/bjoc.16.105 |
| 52. | Lau, W. C.; Waskell, L. A.; Watkins, P. B.; Neer, C. J.; Horowitz, K.; Hopp, A. S.; Tait, A. R.; Carville, D. G. M.; Guyer, K. E.; Bates, E. R. Circulation 2003, 107, 32–37. doi:10.1161/01.cir.0000047060.60595.cc |
| 13. | Lv, P.-C.; Li, H.-Q.; Sun, J.; Zhou, Y.; Zhu, H.-L. Bioorg. Med. Chem. 2010, 18, 4606–4614. doi:10.1016/j.bmc.2010.05.034 |
| 14. | Balbi, A.; Anzaldi, M.; Macciò, C.; Aiello, C.; Mazzei, M.; Gangemi, R.; Castagnola, P.; Miele, M.; Rosano, C.; Viale, M. Eur. J. Med. Chem. 2011, 46, 5293–5309. doi:10.1016/j.ejmech.2011.08.014 |
| 15. | Hura, N.; Naaz, A.; Prassanawar, S. S.; Guchhait S. K; Panda, D. ACS Omega 2018, 3, 1955–1969. doi:10.1021/acsomega.7b01784 |
| 17. | Hall, A.; Billinton, A.; Brown, S. H.; Clayton, N. M.; Chowdhury, A.; Giblin, G. M. P.; Goldsmith, P.; Hayhow, T. G.; Hurst, D. N.; Kilford, I. R.; Naylor, A.; Passingham, B.; Winyard, L. Bioorg. Med. Chem. Lett. 2008, 18, 3392–3399. doi:10.1016/j.bmcl.2008.04.018 |
| 48. | Lednicer, D. Strategies for Organic Drug Synthesis and Design; John Wiley: Hoboken, 2008. doi:10.1002/9780470399613 |
| 12. | Abdel-Hafez, E.-S. M. N.; Abuo-Rahma, G. E.-D. A. A.; Abdel-Aziz, M.; Radwan, M. F.; Farag, H. H. Bioorg. Med. Chem. 2009, 17, 3829–3837. doi:10.1016/j.bmc.2009.04.037 |
| 18. | Sharshira, E. M.; Hamada, N. M. M. Molecules 2012, 17, 4962–4971. doi:10.3390/molecules17054962 |
| 49. | Montalbetti, C. A. G. N.; Falque, V. Tetrahedron 2005, 61, 10827–10852. doi:10.1016/j.tet.2005.08.031 |
| 11. | Chandra, T.; Garg, N.; Lata, S.; Saxena, K. K.; Kumar, A. Eur. J. Med. Chem. 2010, 45, 1772–1776. doi:10.1016/j.ejmech.2010.01.009 |
| 46. | Lincke, T.; Behnken, S.; Ishida, K.; Roth, M.; Hertweck, C. Angew. Chem. 2010, 122, 2055–2057. doi:10.1002/ange.200906114 |
| 10. | Lauria, A.; Abbate, I.; Patella, C.; Gambino, N.; Silvestri, A.; Barone, G.; Almerico, A. M. Tetrahedron Lett. 2008, 49, 5125–5128. doi:10.1016/j.tetlet.2008.06.104 |
| 16. | El-Sabbagh, O. I.; Baraka, M. M.; Ibrahim, S. M.; Pannecouque, C.; Andrei, G.; Snoeck, R.; Balzarini, J.; Rashad, A. A. Eur. J. Med. Chem. 2009, 44, 3746–3753. doi:10.1016/j.ejmech.2009.03.038 |
| 47. | Hisano, T.; Yabuta, Y. Chem. Pharm. Bull. 1973, 21, 511–517. doi:10.1248/cpb.21.511 |
| 28. | Kotagiri, V. K.; Suthrapu, S.; Reddy, J. M.; Rao, C. P.; Bollugoddu, V.; Bhattacharya, A.; Bandichhor, R. Org. Process Res. Dev. 2007, 11, 910–912. doi:10.1021/op700110b |
| 26. | Kameyama, T.; Nabeshima, T. Neuropharmacology 1978, 17, 249–256. doi:10.1016/0028-3908(78)90108-9 |
| 55. | Pathare, S. P.; Chaudhari, P. S.; Akamanchi, K. G. Appl. Catal., A 2012, 425-426, 125–129. doi:10.1016/j.apcata.2012.03.012 |
| 27. | Chen, C.-M.; Kositprapa, U. Pharmaceutical formulations containing a non-steroidal antiinflammatory drug and a proton pump inhibitor. US Patent US6869615B2, March 22, 2005. |
| 57. | Pathak, U.; Pandey, L. K.; Mathur, S.; Suryanarayana, M. V. S. Chem. Commun. 2009, 5409–5411. doi:10.1039/b911844f |
| 36. | Bartlett, P. A.; Spear, K. L.; Jacobsen, N. E. Biochemistry 1982, 21, 1608–1611. doi:10.1021/bi00536a022 |
| 37. | Yu, K.-L.; Torri, A. F.; Luo, G.; Cianci, C.; Grant-Young, K.; Danetz, S.; Tiley, L.; Krystal, M.; Meanwell, N. A. Bioorg. Med. Chem. Lett. 2002, 12, 3379–3382. doi:10.1016/s0960-894x(02)00761-8 |
| 38. | Gannon, M. K.; Holt, J. J.; Bennett, S. M.; Wetzel, B. R.; Loo, T. W.; Bartlett, M. C.; Clarke, D. M.; Sawada, G. A.; Higgins, J. W.; Tombline, G.; Raub, T. J.; Detty, M. R. J. Med. Chem. 2009, 52, 3328–3341. doi:10.1021/jm900253g |
| 39. | Banala, S.; Süssmuth, R. D. ChemBioChem 2010, 11, 1335–1337. doi:10.1002/cbic.201000266 |
| 62. | Tayade, Y. A.; Jangale, A. D.; Dalal, D. S. ChemistrySelect 2018, 3, 8895–8900. doi:10.1002/slct.201801553 |
| 83. | Sankari Devi, E.; Alanthadka, A.; Tamilselvi, A.; Nagarajan, S.; Sridharan, V.; Maheswari, C. U. Org. Biomol. Chem. 2016, 14, 8228–8231. doi:10.1039/c6ob01454b |
| 40. | Mahanta, N.; Szantai-Kis, D. M.; Petersson, E. J.; Mitchell, D. A. ACS Chem. Biol. 2019, 14, 142–163. doi:10.1021/acschembio.8b01022 |
| 41. | Kumar, K.; Konar, D.; Goyal, S.; Gangar, M.; Chouhan, M.; Rawal, R. K.; Nair, V. A. ChemistrySelect 2016, 1, 3228–3231. doi:10.1002/slct.201600601 |
| 42. | Chaubey, T. N.; Borpatra, P. J.; Sharma, A.; Pandey, S. K. Org. Lett. 2022, 24, 8062–8066. doi:10.1021/acs.orglett.2c03371 |
| 33. | Tu, X.-J.; Hao, W.-J.; Ye, Q.; Wang, S.-S.; Jiang, B.; Li, G.; Tu, S.-J. J. Org. Chem. 2014, 79, 11110–11118. doi:10.1021/jo502096t |
| 34. | Yadav, P.; Awasthi, A.; Gokulnath, S.; Tiwari, D. K. J. Org. Chem. 2021, 86, 2658–2666. doi:10.1021/acs.joc.0c02696 |
| 81. | Mondal, R.; Mallik, A. K. Org. Prep. Proced. Int. 2014, 46, 391–434. doi:10.1080/00304948.2014.944402 |
| 35. | Castagnolo, D.; Schenone, S.; Botta, M. Chem. Rev. 2011, 111, 5247–5300. doi:10.1021/cr100423x |
| 82. | Dheer, D.; Rawal, R. K.; Singh, V.; Sangwan, P. L.; Das, P.; Shankar, R. Tetrahedron 2017, 73, 4295–4306. doi:10.1016/j.tet.2017.05.081 |
| 31. | Chou, P.-T.; Chi, Y. Chem. – Eur. J. 2007, 13, 380–395. doi:10.1002/chem.200601272 |
| 65. | Nguyen, T. B. Adv. Synth. Catal. 2017, 359, 1066–1130. doi:10.1002/adsc.201601329 |
| 66. | Nguyen, T. B. Adv. Synth. Catal. 2020, 362, 3448–3484. doi:10.1002/adsc.202000535 |
| 67. | Singh, M.; Awasthi, P.; Singh, V. Eur. J. Org. Chem. 2020, 1023–1041. doi:10.1002/ejoc.201901908 |
| 68. | Wu, K.; Ling, Y.; Ding, A.; Jin, L.; Sun, N.; Hu, B.; Shen, Z.; Hu, X. Beilstein J. Org. Chem. 2021, 17, 805–812. doi:10.3762/bjoc.17.69 |
| 69. | Ilkin, V.; Berseneva, V.; Beryozkina, T.; Glukhareva, T.; Dianova, L.; Dehaen, W.; Seliverstova, E.; Bakulev, V. Beilstein J. Org. Chem. 2020, 16, 2937–2947. doi:10.3762/bjoc.16.243 |
| 70. | Tang, Q.; Yin, X.; Kuchukulla, R. R.; Zeng, Q. Chem. Rec. 2021, 21, 893–905. doi:10.1002/tcr.202100026 |
| 71. | Liu, S.; Deng, G.-J.; Huang, H. Synlett 2021, 32, 142–158. doi:10.1055/s-0040-1707217 |
| 72. | Beletskaya, I. P.; Ananikov, V. P. Chem. Rev. 2022, 122, 16110–16293. doi:10.1021/acs.chemrev.1c00836 |
| 73. | Yu, Z.; Su, J.; Huang, C.; Wei, J.; Han, D. L.; Ye, D. Q.; Li, Y. Asian J. Org. Chem. 2022, 11, e202200288. doi:10.1002/ajoc.202200288 |
| 74. | Koyanagi, A.; Murata, Y.; Hayakawa, S.; Matsumura, M.; Yasuike, S. Beilstein J. Org. Chem. 2022, 18, 1479–1487. doi:10.3762/bjoc.18.155 |
| 75. | Pošta, M.; Zima, V.; Poštová Slavětínská, L.; Matoušová, M.; Beier, P. Beilstein J. Org. Chem. 2022, 18, 549–554. doi:10.3762/bjoc.18.57 |
| 76. | Pathania, S.; Narang, R. K.; Rawal, R. K. Eur. J. Med. Chem. 2019, 180, 486–508. doi:10.1016/j.ejmech.2019.07.043 |
| 32. | Molteni, G.; Buttero, P. D. Tetrahedron: Asymmetry 2005, 16, 1983–1987. doi:10.1016/j.tetasy.2005.04.014 |
| 77. | Sharma, S.; Paul, A. K.; Singh, V. New J. Chem. 2020, 44, 684–694. doi:10.1039/c9nj05426j |
| 78. | Devi, N.; Singh, D.; Sunkaria, R. K.; Malakar, C. C.; Mehra, S.; Rawal, R. K.; Singh, V. ChemistrySelect 2016, 1, 4696–4703. doi:10.1002/slct.201601133 |
| 79. | Devi, N.; Shankar, R.; Singh, V. J. Heterocycl. Chem. 2018, 55, 373–390. doi:10.1002/jhet.3045 |
| 80. | Nag, S.; Singh, V.; Batra, S. ARKIVOC 2007, No. 14, 185–203. doi:10.3998/ark.5550190.0008.e18 |
| 29. | Wang, M.; Zhang, J.; Liu, J.; Xu, C.; Ju, H. J. Lumin. 2002, 99, 79–83. doi:10.1016/s0022-2313(01)00204-6 |
| 58. | Manaka, A.; Sato, M. Synth. Commun. 2005, 35, 761–764. doi:10.1081/scc-200050393 |
| 59. | Xu, H.; Deng, H.; Li, Z.; Xiang, H.; Zhou, X. Eur. J. Org. Chem. 2013, 7054–7057. doi:10.1002/ejoc.201301148 |
| 60. | Yin, Z.; Zheng, B. J. Sulfur Chem. 2013, 34, 527–531. doi:10.1080/17415993.2013.765429 |
| 61. | Wei, J.; Li, Y.; Jiang, X. Org. Lett. 2016, 18, 340–343. doi:10.1021/acs.orglett.5b03541 |
| 62. | Tayade, Y. A.; Jangale, A. D.; Dalal, D. S. ChemistrySelect 2018, 3, 8895–8900. doi:10.1002/slct.201801553 |
| 30. | Burschka, J.; Kessler, F.; Nazeeruddin, M. K.; Grätzel, M. Chem. Mater. 2013, 25, 2986–2990. doi:10.1021/cm400796u |
| 63. | Sharma, S.; Malakar, C. C.; Singh, V. Asian J. Org. Chem. 2020, 9, 1857–1868. doi:10.1002/ajoc.202000390 |
| 64. | Singh, M.; Vaishali; Kumar, R.; Singh, V. ChemistrySelect 2020, 5, 5172–5179. doi:10.1002/slct.202001149 |
© 2023 Sharma et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.