Abstract
Diethyl 2-nitro-(pentafluorosulfanyl)benzylphosphonates, available by the vicarious nucleophilic substitution reaction of meta- and para-nitro-(pentafluorosulfanyl)benzenes and diethyl chloromethylphosphonate, undergo Horner–Wadsworth–Emmons reaction with aldehydes in the presence of potassium hydroxide in acetonitrile at ambient temperature to give (E)-2-nitro-1-alkenyl-(pentafluorosulfanyl)benzenes in good yields and high stereoselectivities. Follow-up transformations of the primary products provided (E)-1-alkenyl-(pentafluorosulfanyl)benzenes and 2-(2-arylethyl)-(pentafluorosulfanyl)anilines.

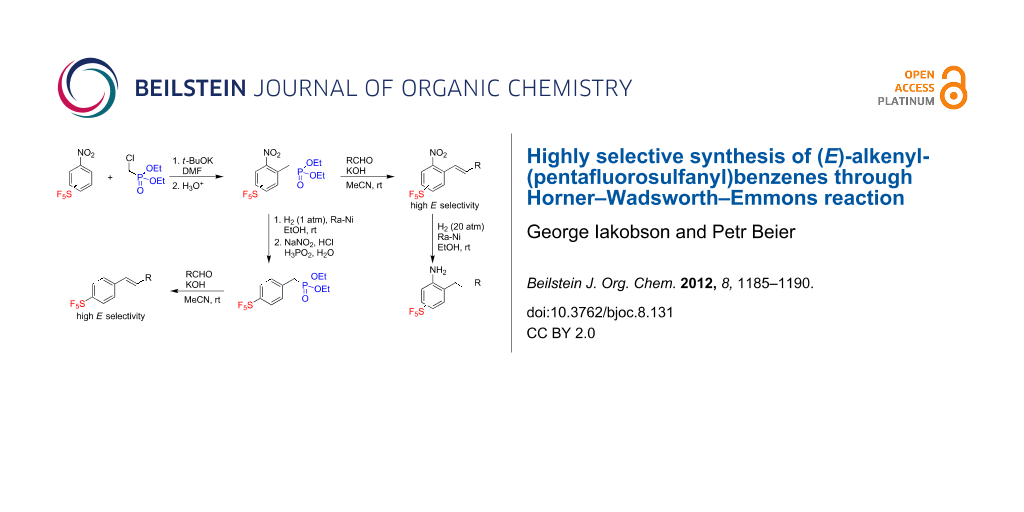
Graphical Abstract
Introduction
Although the pentafluorosulfanyl (SF5) containing compounds have been known for more than half a century [1-4], they remain a relatively underdeveloped class of compounds. This is so despite the unusual combination of properties that the SF5 group possesses, such as high thermal and chemical stability, high electronegativity and strong lipophilic character [2-7]. The availability of SF5-containing compounds is very limited. Aliphatic SF5-containing compounds are available through free-radical addition of toxic and expensive SF5Cl to unsaturated compounds [8,9], and aromatic meta- and para-nitro-(pentafluorosufanyl)benzenes (1 and 2) are made by direct fluorination of the corresponding bis(nitrophenyl)disulfides [10-12]. A recent report also described a two step synthesis of SF5-benzenes from diaryl disulfides avoiding the use of elemental fluorine [13]. While the only known SEAr of 1 or 2 is the nitration of 2 under harsh conditions and in low yield [14], we have recently described SNAr of the nitro group in compounds 1 and 2 with alkoxides and thiolates [15], vicarious nucleophilic substitution (VNS) of the hydrogen with carbon [16], oxygen [17] and nitrogen [18] nucleophiles, and the oxidative nucleophilic substitution with Grignard and alkyllithium reagents [19]. Reduction of the nitro group in 1 or 2 to (pentafluorosulfanyl)anilines followed by acylation, SEAr halogenation or diazotization (with follow-up reactions) has also been described [11,20-22].
Alkenyl substituted SF5-benzenes or SF5-containing stilbene derivatives are not known and would represent basic synthetic intermediates towards more elaborate structures. We envisioned a synthetic route towards these compounds through Horner–Wadsworth–Emmons (HWE) reaction of phosphonates 3 and 4, which are available by vicarious nucleophilic substitution (VNS) of commercial nitrobenzenes 1 and 2 with diethyl chloromethylphosphonate [16]. If required, the nitro group can be removed by a reduction/diazotization/reduction sequence before or after the HWE reaction (Scheme 1).
![[1860-5397-8-131-i1]](/bjoc/content/inline/1860-5397-8-131-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Proposed synthesis of alkenyl-(pentafluorosulfanyl)benzenes.
Scheme 1: Proposed synthesis of alkenyl-(pentafluorosulfanyl)benzenes.
The HWE reaction is a modification of the Wittig olefination in which a phosphoryl-stabilized carbanion reacts with an aldehyde or ketone to form an alkene and a water-soluble phosphate ester [23-25]. In general, this reaction preferentially gives more stable E-disubstituted alkenes, although several successful attempts have been made to favor Z-alkenes [26-28].
Results and Discussion
The HWE reaction of phosphonate 3 with benzaldehyde in the presence of a base giving stilbene derivative 5a was investigated. At first, attempts were made to form 5a directly from 1 and diethyl chloromethylphosphonate by a two-step one-pot process, involving VNS reaction in DMF with a three-fold excess of t-BuOK (−60 °C, 10 min), followed by the addition of benzaldehyde (1.5 equiv) and warming of the reaction mixture to 50 °C (Table 1, entry 1). These conditions provided the expected 5a in good GCMS yield and high E/Z selectivity, but called for rather long reaction time. By changing the solvent from DMF to THF, we observed a less efficient first step (the VNS reaction) with many unidentified side products being formed (Table 1, entry 2). Therefore, all other experiments were carried out starting from isolated phosphonate 3. We found that various bases mediate the HWE reaction. t-BuOK in THF gave good results; however, the reaction required heating to 50 °C for at least one hour. With n-BuLi the reaction is complete at ambient temperature in about half an hour. By using an extended reaction time, much less basic potassium or caesium carbonate in acetonitrile could also be used successfully. Finally, we have identified potassium hydroxide as a very inexpensive and convenient base. The best results were obtained by using 1.8 equiv of KOH in acetonitrile. The addition of small amounts of water increased the reaction rate, presumably by better solubilization of KOH and the formed potassium diethylphosphate, and gave the required product 5a in 84% isolated yield and high E/Z selectivity (Table 1, entry 13).
Table 1: Optimization of HWE reaction of phosphonate 3 with benzaldehyde.
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-131-i6.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Base (equiv) | Solvent | T (°C) | t (min) | 5a, Yield (%)a | E/Zb |
|---|---|---|---|---|---|---|
| 1c | t-BuOK (3.0) | DMF | 50 | 960 | 83 | 97:3 |
| 2c | t-BuOK (3.0) | THF | 50 | 120 | 58 | >98:2 |
| 3 | t-BuOK (3.0) | THF | 50 | 240 | 98 | 95:5 |
| 4 | t-BuOK (1.8) | THF | 50 | 60 | 98 | 94:6 |
| 5 | t-BuOK (1.3) | THF | 50 | 60 | 94 (68) | 95:5 |
| 6 | n-BuLi (2.0) | THF | rt | 30 | 93 | 92:8 |
| 7 | LiHMDS (2.0) | THF | rt | 60 | 59 | 92:8 |
| 8 | K2CO3 (3.0) | MeCN | 60 | 960 | 81 | 91:9 |
| 9 | Cs2CO3 (1.8) | MeCN | rt | 360 | 97 | 93:7 |
| 10 | KOH (1.3) | THF | rt | 7 | 89 | 95:5 |
| 11 | KOH (1.9) | THF | rt | 7 | 92 | 95:5 |
| 12 | KOH (1.8) | MeCN | rt | 60 | 90 (67) | 97:3 |
| 13 | KOH (1.8) | MeCNd | rt | 30 | 98 (84) | 98:2 |
aDetermined by GCMS analysis (in brackets isolated yield). bDetermined by GCMS analysis of the crude reaction mixture. cPhosphonate 3 was prepared in situ from 1 and diethyl chloromethylphosphonate (−60 °C, 10 min). dWater (8 equiv) was added.
Using optimized reaction conditions (Table 1, entry 13), the scope of the HWE reaction of various aldehydes with phosphonate 3 was explored (Table 2). Aromatic aldehydes with electron-donating groups required longer reaction times than those with electron-acceptor groups. All tested aromatic aldehydes provided compounds 5 in high isolated yields and selectivities. Application of (E)-cinnamaldehyde (3f) led to the formation of 5f in only 43% yield. Compound 5f was configurationally stable in solid form; however, we observed slow isomerization in solution (CDCl3) at ambient temperature and in daylight (from E/Z 93:7 to 66:33 after 10 days). Reactions with ketones, even electrophilic and non-enolizable ones such as 4,4'-dichlorobenzophenone or 2,2,2-trifluoroacetophenone, did not provide the expected alkene products.
Table 2: HWE reactions of phosphonate 3 with aldehydes.
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-131-i7.svg?max-width=637&scale=1.0)
|
||||
| Entry | R (equiv) | t (min) | 5, Yield (%)a | 5, E/Zb |
|---|---|---|---|---|
| 1 | Ph (1.5) | 30 | 5a, 84 | 98:2 |
| 2 | 4-NO2C6H4 (1.7) | 5 | 5b, 84 | >99:1 |
| 3 | 4-ClC6H4 (1.1) | 30 | 5c, 85 | 98:2 |
| 4 | 4-MeOC6H4 (1.5) | 90 | 5d, 86 | 99:1 |
| 5 | 1-Naphthyl (1.2) | 30 | 5e, 80 | 92:8 |
| 6 | (E)-PhCH=CH (1.2) | 40 | 5f, 43c | 93:7d |
| 7 | Et (1.5) | 90 | 5g, 67 | 94:6 |
aIsolated yield refers to the pure E-isomer unless noted otherwise. bDetermined by GCMS analysis of the crude reaction mixture. cIsolated yield referring to the 93:7 E/Z mixture. dThis ratio changed to 66:33 upon storage in CDCl3 solution at rt for 10 d.
Next, we investigated analogous HWE reactions of isomeric phosphonate 4 with various aldehydes. Good yields of products 6 were obtained with both aromatic and aliphatic aldehydes. In contrast to most of the reactions with 3, phosphonate 4 gave exclusively E-isomers of 6 (Table 3). This improved selectivity can be explained by relative differences in the stabilities and reactivities of carbanions derived from phosphonates and reactive intermediates. In reactions of phosphoryl-stabilized carbanions with aldehydes, several intermediates are formed reversibly. Less-hindered intermediates A and A’, which eliminate to E-alkene, exist in equilibrium with more-hindered intermediates B and B’ giving Z-alkene (Scheme 2). In our reactions, the stabilization of the negative charge in the deprotonated phosphonate is higher for 4 than for 3 due to conjugation of the negative charge with the SF5 group in the former case (σI(SF5) = 0.55, σR(SF5) = 0.11 [5]). Consequently, 4− is more stable and less nucleophilic than 3−, and therefore, in comparison to the A-to-B equilibrium the A’-to-B’ equilibrium is shifted more towards A’ providing only (E)-6 product (Scheme 2).
Table 3: HWE reactions of phosphonate 4 with aldehydes.
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-131-i8.svg?max-width=637&scale=1.0)
|
|||
| Entry | R (equiv) | t (min) | 6, Yield (%)a |
|---|---|---|---|
| 1 | 4-NO2C6H4 (1.2) | 30 | 6b, 97 |
| 2 | 4-ClC6H4 (1.1) | 80 | 6c, 85 |
| 3 | 4-MeOC6H4 (1.1) | 260 | 6d, 76 |
| 4 | n-C6H13 (1.1) | 90 | 6h, 84 |
aIsolated yield.
![[1860-5397-8-131-i2]](/bjoc/content/inline/1860-5397-8-131-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reactive intermediates involved in HWE reactions to alkenes 5 and 6.
Scheme 2: Reactive intermediates involved in HWE reactions to alkenes 5 and 6.
To demonstrate the versatility of this methodology leading to new carbon-substituted SF5-benzenes, several transformations of primary products 5 and 6 were performed. Reduction with hydrogen (1 atm) in the presence of catalytic Raney nickel did not provide full conversion to the respective anilines. Furthermore, the reaction mixtures contained products 7 and 8. At higher pressure (20 atm), complete nitro group and C=C reduction of stilbenes 5 and 6 to compounds 7 and 8, respectively, took place (Table 4). Several other conditions were tested, but no system for selective reduction of the nitro group was found.
Table 4: Hydrogenation of stilbene derivatives 5 and 6.
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-131-i9.svg?max-width=637&scale=1.0)
|
|||||
| Entry | 5 or 6 | X | Y | R | 7 or 8, Yield (%)a |
|---|---|---|---|---|---|
| 1 | 5a | SF5 | H | Ph | 7a, 87 |
| 2 | 6c | H | SF5 | 4-ClC6H4 | 8c, 64 |
| 3 | 6d | H | SF5 | 4-MeOC6H4 | 8d, 82 |
aIsolated yield.
Diazotization was carried out with the aim to prepare 1,2-diarylethane 9d from aniline 8d (Scheme 3). A combination of sodium nitrite and phosphoric acid was used. (With HCl, substitution of the amino function by a chlorine atom was observed, and the use of H2SO4 resulted in low solubility of the formed anilinium in water.) An ether cosolvent (Et2O or t-BuOMe) improved the yield compared to aqueous or aqueous/THF mixtures. The presence of reducing hypophosphorous acid provided a mixture of the expected 9d and the cyclized product 10d resulting from electrophilic aromatic substitution of the substituted phenyl cation intermediate (formed by the decomposition of the diazonium salt), to the electron-rich anisole ring in an unusual meta-position relative to the methoxy group. The more activated para-position is unavailable, and the reaction in ortho-positions would give too strained a product. The formation of the cyclized side product is not restricted to compounds with an electron-donor substituent on the aromatic ring. Similarly to 8d, the cyclic product was detected by GCMS in diazotization of compound 8c (product not isolated). Compound 10d was fully characterized by spectroscopic methods, and the yield was increased to 51% by performing the diazotization reaction in the absence of a reducing reagent (Scheme 3). Aromatization of 10d by oxidation using CAN was performed to give SF5-substituted phenanthrene 11d in good yield (Scheme 4).
![[1860-5397-8-131-i3]](/bjoc/content/inline/1860-5397-8-131-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Diazotization/reduction of 8d to 9d and the formation of unexpected cyclized product 10d.
Scheme 3: Diazotization/reduction of 8d to 9d and the formation of unexpected cyclized product 10d.
![[1860-5397-8-131-i4]](/bjoc/content/inline/1860-5397-8-131-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of substituted phenanthrene 11d.
Scheme 4: Synthesis of substituted phenanthrene 11d.
To avoid problems with alkene reduction and electrophilic aromatic substitution during nitro group removal, we decided to try a different approach to the general synthesis of SF5-containing stilbene derivatives, as demonstrated in the synthesis of 13d shown in Scheme 5. The nitro group in phosphonate 4 was removed by a reduction/diazotization/reduction sequence to give phosphonate 12 in good yield. The following HWE reaction with 4-methoxybenzaldehyde afforded 13d in good yield, exclusively as the E-isomer.
![[1860-5397-8-131-i5]](/bjoc/content/inline/1860-5397-8-131-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of phosphonate 12 and SF5-stilbene derivative 13d.
Scheme 5: Synthesis of phosphonate 12 and SF5-stilbene derivative 13d.
Conclusion
In conclusion, we have shown access to (E)-2-nitro-1-alkenyl-(pentafluorosulfanyl)benzenes from nitro-(pentafluorosulfonyl)benzenes by VNS reaction with diethyl chloromethylphosphonate followed by stereoselective HWE reaction with aldehydes. Reduction of (E)-2-nitro-1-alkenyl-(pentafluorosulfanyl)benzenes provided 2-(2-arylethyl)-(pentafluorosulfanyl)anilines, and the formation of (E)-1-alkenyl-4-(pentafluorosulfanyl)benzenes was demonstrated from diethyl 4-(pentafluorosulfanyl)benzylphosphonates.
Supporting Information
| Supporting Information File 1: Experimental details and characterization data for all new compounds. | ||
| Format: PDF | Size: 310.7 KB | Download |
References
-
Case, J. R.; Ray, N. H.; Roberts, H. L. J. Chem. Soc. 1961, 2066–2070. doi:10.1039/JR9610002066
Return to citation in text: [1] -
Ray, N. H. J. Chem. Soc. 1963, 1440–1441. doi:10.1039/JR9630001440
Return to citation in text: [1] [2] -
Merrill, C. I.; Cady, G. H. J. Am. Chem. Soc. 1961, 83, 298–300. doi:10.1021/ja01463a011
Return to citation in text: [1] [2] -
Dudley, F. B.; Cady, G. H.; Eggers, D. F., Jr. J. Am. Chem. Soc. 1956, 78, 1553–1557. doi:10.1021/ja01589a013
Return to citation in text: [1] [2] -
Kirsch, P. Synthesis of complex Organofluorine Compounds. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2004; pp 146–156. doi:10.1002/352760393X.ch2
Return to citation in text: [1] [2] -
Winter, R. W.; Dodean, R. A.; Gard, G. L. SF5-Synthons: Pathways to Organic Derivatives of SF6. In Fluorine-Containing Synthons; Soloshonok, V. A., Ed.; ACS Symposium Series, Vol. 911; American Chemical Society: Washington, DC, 2005; pp 87–118. doi:10.1021/bk-2005-0911.ch004
Return to citation in text: [1] -
Kirsch, P.; Röschenthaler, G.-V. Functional Compounds Based on Hypervalent Sulfur Fluorides. In Current Fluoroorganic Chemistry; Soloshonok, V. A.; Mikami, K.; Yamazaki, T.; Welch, J. T.; Honek, J. F., Eds.; ACS Symposium Series, Vol. 949; American Chemical Society: Washington, DC, 2007; pp 221–243. doi:10.1021/bk-2007-0949.ch013
Return to citation in text: [1] -
Aït-Mohand, S.; Dolbier, W. R., Jr. Org. Lett. 2002, 4, 3013–3015. doi:10.1021/ol026483o
Return to citation in text: [1] -
Dolbier, W. R., Jr.; Aït-Mohand, S.; Schertz, T. D.; Sergeeva, T. A.; Cradlebaugh, J. A.; Mitani, A.; Gard, G. L.; Winter, R. W.; Thrasher, J. S. J. Fluorine Chem. 2006, 127, 1302–1310. doi:10.1016/j.jfluchem.2006.05.003
Return to citation in text: [1] -
Bowden, R. D.; Greenhall, M. P.; Moilliet, J. S.; Thomson, J. The Preparation Of Fluorinated Organic Compounds. WO Patent WO1997005106, Feb 13, 1997.
Return to citation in text: [1] -
Bowden, R. D.; Comina, P. J.; Greenhall, M. P.; Kariuki, B. M.; Loveday, A.; Philp, D. Tetrahedron 2000, 56, 3399–3408. doi:10.1016/S0040-4020(00)00184-8
Return to citation in text: [1] [2] -
Chambers, R. D.; Spink, R. C. H. Chem. Commun. 1999, 883–884. doi:10.1039/A901473J
Return to citation in text: [1] -
Umemoto, T.; Garrick, L. M.; Saito, N. Beilstein J. Org. Chem. 2012, 8, 461–471. doi:10.3762/bjoc.8.53
Return to citation in text: [1] -
Umemoto, T.; Chika, J. Processes for preparing 1,3-dinitro-5-(pentafluorosulfanyl)benzene and its intermediates. U.S. Patent Appl. 2011/0301382 A1, Dec 8, 2011.
Return to citation in text: [1] -
Beier, P.; Pastýříková, T.; Vida, N.; Iakobson, G. Org. Lett. 2011, 13, 1466–1469. doi:10.1021/ol2001478
Return to citation in text: [1] -
Beier, P.; Pastýříková, T.; Iakobson, G. J. Org. Chem. 2011, 76, 4781–4786. doi:10.1021/jo200618p
Return to citation in text: [1] [2] -
Beier, P.; Pastýříková, T. Tetrahedron Lett. 2011, 52, 4392–4394. doi:10.1016/j.tetlet.2011.06.011
Return to citation in text: [1] -
Pastýříková, T.; Iakobson, G.; Vida, N.; Pohl, R.; Beier, P. Eur. J. Org. Chem. 2012, 2123–2126. doi:10.1002/ejoc.201200127
Return to citation in text: [1] -
Vida, N.; Beier, P. J. Fluorine Chem., in press. doi:10.1016/j.jfluchem.2012.04.001
Return to citation in text: [1] -
Crowley, P. J.; Mitchell, G.; Salmon, R.; Worthington, P. A. Chimia 2004, 58, 138–142.
Return to citation in text: [1] -
Kirsch, P.; Bremer, M.; Heckmeier, M.; Tarumi, K. Angew. Chem., Int. Ed. 1999, 38, 1989–1992. doi:10.1002/(SICI)1521-3773(19990712)38:13/14<1989::AID-ANIE1989>3.0.CO;2-K
Return to citation in text: [1] -
Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3064–3072. doi:10.1021/ja00875a006
Return to citation in text: [1] -
Maryanoff, B. E.; Reitz, A. B. Chem. Rev. 1989, 89, 863–927. doi:10.1021/cr00094a007
Return to citation in text: [1] -
Boutagy, J.; Thomas, R. Chem. Rev. 1974, 74, 87–99. doi:10.1021/cr60287a005
Return to citation in text: [1] -
Rein, T.; Pedersen, T. M. Synthesis 2002, 579–594. doi:10.1055/s-2002-23535
Return to citation in text: [1] -
Ando, K. J. Synth. Org. Chem., Jpn. 2000, 58, 869–876.
Return to citation in text: [1] -
Ando, K. J. Org. Chem. 1997, 62, 1934–1939. doi:10.1021/jo970057c
Return to citation in text: [1] -
Still, W. C.; Gennari, C. Tetrahedron Lett. 1983, 24, 4405–4408. doi:10.1016/S0040-4039(00)85909-2
Return to citation in text: [1]
| 1. | Case, J. R.; Ray, N. H.; Roberts, H. L. J. Chem. Soc. 1961, 2066–2070. doi:10.1039/JR9610002066 |
| 2. | Ray, N. H. J. Chem. Soc. 1963, 1440–1441. doi:10.1039/JR9630001440 |
| 3. | Merrill, C. I.; Cady, G. H. J. Am. Chem. Soc. 1961, 83, 298–300. doi:10.1021/ja01463a011 |
| 4. | Dudley, F. B.; Cady, G. H.; Eggers, D. F., Jr. J. Am. Chem. Soc. 1956, 78, 1553–1557. doi:10.1021/ja01589a013 |
| 13. | Umemoto, T.; Garrick, L. M.; Saito, N. Beilstein J. Org. Chem. 2012, 8, 461–471. doi:10.3762/bjoc.8.53 |
| 26. | Ando, K. J. Synth. Org. Chem., Jpn. 2000, 58, 869–876. |
| 27. | Ando, K. J. Org. Chem. 1997, 62, 1934–1939. doi:10.1021/jo970057c |
| 28. | Still, W. C.; Gennari, C. Tetrahedron Lett. 1983, 24, 4405–4408. doi:10.1016/S0040-4039(00)85909-2 |
| 10. | Bowden, R. D.; Greenhall, M. P.; Moilliet, J. S.; Thomson, J. The Preparation Of Fluorinated Organic Compounds. WO Patent WO1997005106, Feb 13, 1997. |
| 11. | Bowden, R. D.; Comina, P. J.; Greenhall, M. P.; Kariuki, B. M.; Loveday, A.; Philp, D. Tetrahedron 2000, 56, 3399–3408. doi:10.1016/S0040-4020(00)00184-8 |
| 12. | Chambers, R. D.; Spink, R. C. H. Chem. Commun. 1999, 883–884. doi:10.1039/A901473J |
| 5. | Kirsch, P. Synthesis of complex Organofluorine Compounds. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2004; pp 146–156. doi:10.1002/352760393X.ch2 |
| 8. | Aït-Mohand, S.; Dolbier, W. R., Jr. Org. Lett. 2002, 4, 3013–3015. doi:10.1021/ol026483o |
| 9. | Dolbier, W. R., Jr.; Aït-Mohand, S.; Schertz, T. D.; Sergeeva, T. A.; Cradlebaugh, J. A.; Mitani, A.; Gard, G. L.; Winter, R. W.; Thrasher, J. S. J. Fluorine Chem. 2006, 127, 1302–1310. doi:10.1016/j.jfluchem.2006.05.003 |
| 16. | Beier, P.; Pastýříková, T.; Iakobson, G. J. Org. Chem. 2011, 76, 4781–4786. doi:10.1021/jo200618p |
| 2. | Ray, N. H. J. Chem. Soc. 1963, 1440–1441. doi:10.1039/JR9630001440 |
| 3. | Merrill, C. I.; Cady, G. H. J. Am. Chem. Soc. 1961, 83, 298–300. doi:10.1021/ja01463a011 |
| 4. | Dudley, F. B.; Cady, G. H.; Eggers, D. F., Jr. J. Am. Chem. Soc. 1956, 78, 1553–1557. doi:10.1021/ja01589a013 |
| 5. | Kirsch, P. Synthesis of complex Organofluorine Compounds. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2004; pp 146–156. doi:10.1002/352760393X.ch2 |
| 6. | Winter, R. W.; Dodean, R. A.; Gard, G. L. SF5-Synthons: Pathways to Organic Derivatives of SF6. In Fluorine-Containing Synthons; Soloshonok, V. A., Ed.; ACS Symposium Series, Vol. 911; American Chemical Society: Washington, DC, 2005; pp 87–118. doi:10.1021/bk-2005-0911.ch004 |
| 7. | Kirsch, P.; Röschenthaler, G.-V. Functional Compounds Based on Hypervalent Sulfur Fluorides. In Current Fluoroorganic Chemistry; Soloshonok, V. A.; Mikami, K.; Yamazaki, T.; Welch, J. T.; Honek, J. F., Eds.; ACS Symposium Series, Vol. 949; American Chemical Society: Washington, DC, 2007; pp 221–243. doi:10.1021/bk-2007-0949.ch013 |
| 23. | Maryanoff, B. E.; Reitz, A. B. Chem. Rev. 1989, 89, 863–927. doi:10.1021/cr00094a007 |
| 24. | Boutagy, J.; Thomas, R. Chem. Rev. 1974, 74, 87–99. doi:10.1021/cr60287a005 |
| 25. | Rein, T.; Pedersen, T. M. Synthesis 2002, 579–594. doi:10.1055/s-2002-23535 |
| 17. | Beier, P.; Pastýříková, T. Tetrahedron Lett. 2011, 52, 4392–4394. doi:10.1016/j.tetlet.2011.06.011 |
| 19. | Vida, N.; Beier, P. J. Fluorine Chem., in press. doi:10.1016/j.jfluchem.2012.04.001 |
| 16. | Beier, P.; Pastýříková, T.; Iakobson, G. J. Org. Chem. 2011, 76, 4781–4786. doi:10.1021/jo200618p |
| 11. | Bowden, R. D.; Comina, P. J.; Greenhall, M. P.; Kariuki, B. M.; Loveday, A.; Philp, D. Tetrahedron 2000, 56, 3399–3408. doi:10.1016/S0040-4020(00)00184-8 |
| 20. | Crowley, P. J.; Mitchell, G.; Salmon, R.; Worthington, P. A. Chimia 2004, 58, 138–142. |
| 21. | Kirsch, P.; Bremer, M.; Heckmeier, M.; Tarumi, K. Angew. Chem., Int. Ed. 1999, 38, 1989–1992. doi:10.1002/(SICI)1521-3773(19990712)38:13/14<1989::AID-ANIE1989>3.0.CO;2-K |
| 22. | Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3064–3072. doi:10.1021/ja00875a006 |
| 15. | Beier, P.; Pastýříková, T.; Vida, N.; Iakobson, G. Org. Lett. 2011, 13, 1466–1469. doi:10.1021/ol2001478 |
| 14. | Umemoto, T.; Chika, J. Processes for preparing 1,3-dinitro-5-(pentafluorosulfanyl)benzene and its intermediates. U.S. Patent Appl. 2011/0301382 A1, Dec 8, 2011. |
| 18. | Pastýříková, T.; Iakobson, G.; Vida, N.; Pohl, R.; Beier, P. Eur. J. Org. Chem. 2012, 2123–2126. doi:10.1002/ejoc.201200127 |
© 2012 Iakobson and Beier; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)